

Abstract
The purpose of this research was to use next generation sequencing to identify mutations in patients with primary immunodeficiency diseases whose pathogenic gene mutations had not been identified. Remarkably, four unrelated patients were found by next generation sequencing to have the same heterozygous mutation in an essential donor splice site of PIK3R1 (NM_181523.2:c.1425 + 1G > A) found in three prior reports. All four had the Hyper IgM syndrome, lymphadenopathy and short stature, and one also had SHORT syndrome. They were investigated with in vitro immune studies, RT-PCR, and immunoblotting studies of the mutation's effect on mTOR pathway signaling. All patients had very low percentages of memory B cells and class-switched memory B cells and reduced numbers of naïve CD4+ and CD8+ T cells. RT-PCR confirmed the presence of both an abnormal 273 base-pair (bp) size and a normal 399 bp size band in the patient and only the normal band was present in the parents. Following anti-CD40 stimulation, patient's EBV-B cells displayed higher levels of S6 phosphorylation (mTOR complex 1 dependent event), Akt phosphorylation at serine 473 (mTOR complex 2 dependent event), and Akt phosphorylation at threonine 308 (PI3K/PDK1 dependent event) than controls, suggesting elevated mTOR signaling downstream of CD40. These observations suggest that amino acids 435–474 in PIK3R1 are important for its stability and also its ability to restrain PI3K activity. Deletion of Exon 11 leads to constitutive activation of PI3K signaling. This is the first report of this mutation and immunologic abnormalities in SHORT syndrome.
Keywords: PIK3R1 splice site mutations, Hyper IgM syndrome, lymphadenopathy, short stature, SHORT syndrome, mTOR pathway, next generation sequencing
Introduction
Many critical aspects of immune cell development, differentiation and function are controlled by phosphoinosidtide 3 kinases (PI3Ks) [1]. Information is accruing about the effects of human gene mutations affecting these molecules. Hyperactivation of the PI3K signaling pathway due to heterozygous gain-of-function mutations in the gene encoding PIK3CD has been found by several groups to result in defects in immune function [2–5]. The resulting clinical problems included respiratory infections, Epstein Barr virus and/or cytomegalovirus infections, antibody deficiency, lymphadenopathy and lymphoma susceptibility. More recently, dominant mutations in the gene encoding PIK3R1, the p85α regulatory subunit for PIK3CD, were also found to result in constitutive hyperactivation of that pathway and immunodeficiency [6–8]. Currently, the dominant PIK3R1 mutations linked to constitutive hyperactivation have been restricted to mutations of essential donor splice sites in intron 11, resulting in the exclusion of exon 11 (PIK3RIΔ434_475) [6, 7, 9]. Prior to these reports, a homozygous loss-of-function genotype in exon 6 of PIK3R1 was reported in a female patient characterized by agammaglobulinemia and absent B cells [10]. In addition, other dominant de novo mutations in exon 14 of PIK3R1 have been described among patients with SHORT syndrome characterized by short stature, hyperextensibility of joints, delayed bone age, hernias, low body mass index and a progeroid appearance [11–16]. No results of immune studies were recorded in the SHORT syndrome patients in the existing reports.
The purpose of this research was to use next generation sequencing to identify mutations in patients with primary immunodeficiency diseases whose pathogenic gene mutations had not been identified. Four unrelated patients were found by next generation sequencing to have the same de novo heterozygous mutation in an essential donor splice site of PIK3R1 (NM_181523.2:c.1425 + 1G > A) found in three prior reports [6–8]. All four had the Hyper IgM syndrome, lymphadenopathy and short stature, and one also had a clinical diagnosis of SHORT syndrome.
Patient, Materials and Methods
Patients
The patients from four unrelated families were referred to the Immunology Clinic at Duke University Medical Center (DUMC). All studies were performed with the approval of the DUMC Institutional Review Board and with the written informed consent of the patients' parents.
Patient 1
A 2.5 year old Caucasian male born to nonconsanguineous healthy parents began having recurrent otitis, sinusitis and dacryocystitis in early infancy. He also had problems with intermittent hypoglycemia. On examination, he was found to be symmetrically small with height and weight below the 5th percentile. He had enlarged tonsils and large cervical, axillary and inguinal lymph nodes. Family history was negative for immunodeficiency. On immune evaluation, he was found to have an IgG of 78, an IgA of 0, an IgM of 155 and an IgE of <1 and was started on monthly intravenous immunoglobulin infusions (Table 1). He is currently alive but with recurrent respiratory infections and lymphadenopathy.
Table 1. Clinical and Immunologic Findings in the Patients.
| Patient | 1 | 2 | 3 | 4 | |
| Gender | Male | Female | Male | Female | |
| Age at Presentation (yr) | 1 7/12 | 2 5/12 | 2 | 5 1/2 | |
| Growth Percentile | <5th | <10th | 5th | ⋘5th | |
| Viral Infections | – | – | RSV, H1N1 Parainfluenza | – | |
| Lymphoproliferation | 3+ | 3+ | 3+ | 2+ | |
| Autoimmunity | – | – | – | – | |
| Allergy | – | – | – | – | |
| Respiratory Infections | Ear, Sinus | Throat, Lung | Eye, Ear, Lung | Eye, Ear, Lung | |
| Other Features | – | – | – | Abnormal faciesa | |
| Ig Replacement | + | + | + | Normal Ranges | |
| IgGb mg/dl (Age in yr) | 78 (1 7/12) | 47 (2 5/12) | <60 (4 1/12) | 68 (5 ½) | 391–1047 |
| IgAb mg/dl (Age in yr) | 6 (1 7/12) | 8 (2 5/12) | <16 (4 1/12) | 0 (5 ½) | 15–95 |
| IgMb mg/dl (Age in yr) | 155 (1 7/12) | 209 (2 5/12) | 529 (4 1/12) | 197 (5 ½) | 49–202 |
| IgEb I.U./ml (Age in yr) | <10 (1 7/12) | 8 (2 5/12) | <10 (4 1/12) | 1 (5 ½) | 0–150 |
| Lymphocyte Studies (Yr) | 13 1/4 | 10 3/4 | 10 1/3 | 13 1/12 | |
| Absolute Lymphocyte Ct | 1280 | 1476 | 770 | 1254 | 1000–4800 |
| CD3+ T cells/μl | 891 | 1231 | 708 | 1157 | 631–4142 |
| CD3+ CD4 + T cells/μl | 321 | 344 | 324 | 658 | 340–2746 |
| CD3+ CD8 + T cells/μl | 392 | 773 | 181 | 433 | 154–1776 |
| Naïvec CD4 + T cells (%) | 14.2 | 31.1 | 11.1 | 14.2 | i 35.6–72.5 |
| Naïved CD8 + T cells (%) | 14.2 | 5.4 | 41.2 | 39.4 | i 36.1–74.2 |
| e CD3 + CD8 + PD-1+/μl | 56 | 325 | 20 | 22 | 12–141 |
| CD19+ B cells/μl | 182 | 38 | 16 | 29 | 51–821 |
| f Transitional B cells (%) | 52.7 % (102 abs) | 4.02 % (0 abs) | 48.8 % (5 abs) | 14.2 % (4 abs) | j 2.9 %–23.8 % (12–35 abs) |
| g Memory B cells (%) | 3.3 % (6 abs) | 11.9 % (1 abs) | 4.4 % (0 abs) | 1.2 % (0 abs) | 1.4–27.6 % (4-168abs) |
| h Switched Memory B (%) | 8.5 % (16 abs) | 11.9 % (1 abs) | 3.7 % (0 abs) | 2.4 % (1 abs) | 3.0–32.9 % (13-105abs) |
| CD3-CD56+ NK cells/μl | 175 | 155 | 16 | 36 | 12–864 |
| PHA Stimulated Cells (cpm) | 64,853 (1483) | 144,073 (2812) | 115,456 (981) | 137,276 (1914) | 117,512–235,792 |
| Anti-CD3 Stimulated (cpm) | 53,366 (1483) | 133,166 (2812) | 33,415 (981) | 27,782 (1528) | 59,808–186,006 |
| Candida Stimulated (cpm) | 1415 (888) | 41,566 (1450) | 6149 (4076) | 495 (545) | 6460–60,028 |
Abnormal values are in bold font
Triangular shape, deep-set eyes, thin nostrils, low-hanging columella, downturned small mouth, small chin and prominent forehead
Normal ranges for immunoglobulins are for 2 year old Caucasians
Naïve CD4+ T cells are CCR7 + CD45RA+/CD4+/CD3+
Naïve CD8+ T cells are CCR7 + CD45RA+/CD8+/CD3+
Senescent CD8+ T cells are CD57 + (PD-1)+/CD8+/CD3+
Transitional B cells are CD24++CD38++/IgD + CD27-/CD19+
Unswitched memory B cells are IgD + CD27+/CD19+
Class switched memory B cells are CD27 + IgD-/CD19+
Reference range provided by Dr. Alan Kirk of Department of Surgery, Duke University Medical Center
Reference range from R. van Gent et al.: Clin. Immunol. 133: 95–107, 2009
Patient 2
A 2.4 year old Caucasian female was born to nonconsanguineous healthy parents and was healthy until she began having recurrent otitis and pneumonia at 2.3 years of age. On examination, she was found to be symmetrically small with height and weight below the 10th percentile. She had large tonsils and massive cervical adenopathy and an elevated serum IgM of 209 mg/dl with very low levels of all of the other immunoglobulins (Table 1). She was started on monthly intravenous immunoglobulin infusions and is currently alive but with continued lymphadenopathy.
Patient 3
A 6 year old Caucasian male was born to nonconsanguineous healthy parents and began having recurrent otitis and dacryocystitis in infancy, followed by pneumonia on three occasions at age 4 years. On examination he was found to be symmetrically small, with height and weight below the 5th percentiles. Patient 3′s height was less than the fifth percentile for all visits; his mean parental height would predict that he should be at the 25th percentile for height as an adult. He had large tonsils, enlarged cervical, axillary and inguinal lymph nodes and splenomegaly. Immune evaluation at age 4 years revealed that he had undetectable serum IgA, IgG and IgE but an IgM of 529 mg/dl (Table 1). He is currently alive and receiving monthly IVIG with splenic peliosis but is otherwise well.
Patient 4
A 5 year old Caucasian female was born to nonconsanguineous healthy parents and began having recurrent pneumonias at age 3 years. She also had numerous bouts of otitis and had two sets of tympanostomy tubes. She has congenital dacryostenosis. She had large tonsils and adenoids and they were removed. Immune evaluation at age 5 years revealed undetectable serum IgA and IgE, an IgG of 68 mg/dl and an IgM of 197 mg/dl (Table 1). At age 12, she was diagnosed independently by the Genetics Division as having SHORT syndrome after ruling out multiple other genetic causes of short stature through whole exome sequencing in a commercial laboratory where the only abnormality detected was the PIK3R1mutation. Patient 4′s height was at the 0.01 percentile for all visits and her mean parental height would predict that she should be at the 25th percentile as an adult. She has a triangular facial shape with a prominent forehead, deep set eyes, thin nostrils, a low-hanging columella, a downturned small mouth, a small chin, a conductive hearing loss, hyperextensibility of the joints, delayed eruption of the secondary teeth, lack of subcutaneous fat and a learning disability. She is currently alive and receiving monthly IVIG.
Immunologic Phenotype Analysis
Flow cytometry of peripheral blood leucocytes was performed with labeled Abs to CD3ε, CD4, CD8, CD10, CD14, CD16, CD20, CD22, CD24, CD25, CD27, CD38, CD45, CD45RA, CD45RO, CD56, CD57, IgD, IgM, CD62L, CD197, CD279, TCRαβ, and TCRγδ. Samples were collected on a BD FACSCanto II cytometer and data analyzed using FACSDiva software. Lymphocyte proliferation was assessed by measuring [3H]thymidine incorporation into mononuclear cells following culture with optimal concentrations of the indicated stimuli as previously described [17].
Next Generation Sequencing, Alignment and Variant Calling
Next generation sequencing was carried out on the HiSeq2000 within the Genomic Analysis Facility in the Center for Human Genome Variation (Duke University). DNA samples from Patients 1 and 2 were analyzed using whole exome sequencing and from Patient 3 by whole genome sequencing. For the exome sequenced samples, sequencing libraries were prepared from primary DNA extracted from leukocytes of patients using either the KAPA Biosystem's (patient 1) or Illumina TruSeq (patient 2) library preparation kit following the manufacturer's protocol. For patient 1 and his parents, the Nimblegen SeqCap EZ V3.0 Enrichment kit (Roche NimbleGen, Madison, WI) was used to selectively amplify the coding regions of the genome according to the manufacturer's protocol. For patient 2, the 65-Mb Illumina TruSeq Exome Enrichment Kit (Illumina, San Diego, CA) was used to selectively amplify the coding regions of the genome according to the manufacturer's protocol. Patient 3 was whole-genome sequenced. Whole exome sequencing was performed initially at a commercial CLIA-certified laboratory for patient 4, and the mutation was confirmed by the authors.
Alignment of the sequenced DNA fragments to the Human Reference Genome (NCBI Build 37) was performed using the Burrows–Wheeler Alignment Tool (BWA) (version 0.5.10). The reference sequence used is identical to the 1000 Genomes Phase II reference and it consists of chromosomes 1–22, X, Y, MT, unplaced and unlocalized contigs, the human herpesvirus 4 type 1 (NC_007605), and decoy sequences (hs37d5) derived from HuRef, Human Bac and Fosmid clones and NA12878.
After alignments were produced for each individual separately using BWA, variant and genotype calling was performed using the Genome Analysis Tool Kit (GATK, version 1.6–11-g3b2fab9). SnpEff (version 3.3) was used to annotate the variants according to Ensembl (version 73) and consensus coding sequencing (CCDS release 14) and limited analyses to protein-coding or essential splice site (2 base pairs flanking an exon) mutations.
The sequence data for patients 1–3 were analyzed using established protocols that identify qualifying variants forming genotypes not observed in an in-house control database of 2357 samples or 60,706 sequenced samples made available by the Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org (accessed Jan., 2015, release 0.3). The PIK3R1 essential splice site de novo mutations were independently validated with Sanger sequencing for each trio.
Establishment of EBV Cell Lines and Cell Growth
Cyclosporine A, B95.8 culture EBV supernatant, and media (RPMI 1640 + PenStrep + L-glu + Hepes + 20%FBS) were added to 10 million peripheral blood leucocytes [18]. EBV-immortalized B-cell lines from patients and controls were cultured in RPMI 1640 medium (Sigma-Aldrich) supplemented with 20 % FBS (HyClone), 100 U/mL penicillin G, 100 U/mL streptomycin, 292 μg/mL of L-glutamine and 10 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid; pH 7.4). To determine cell expansion, 2 × 104 of EBV-transformed B-cell lines were plated in a 96-well plate in 0.2 mL media. Cell numbers were counted after cultured for 24, 48 and 72 h.
Immunoblot Analysis
EBV-transformed B cells were rested in PBS for 30′ at 37 °C followed by stimulation with an anti-CD40 antibody for 10 or 30 min or with human insulin (Sigma-Aldrich) for the indicated times and subsequently lysed in 1 % Nonidet P-40 lysis buffer containing freshly added protease and phosphatase inhibitor cocktails. Cell lysates were subjected to immunoblotting analysis using anti-p85α, phosphor-S6, S6 protein, phosphor-Akt serine 473, and β-actin antibodies according to published protocols [19].
RT-PCR and DNA Sequencing
Total RNAs from PBMCs lysed in the Trizol reagent were isolated according to the manufacturer's protocol. The first strand cDNA was made using the iScript Select cDNA Synthesis Kit (Bio-Rad). PIK3R1 cDNAwas amplified using primers huPIK3R1F 5′-TGGGAAATATGGCTTCTCTGA-3 and huPIK3R1R 5′-TCTTTCTCATTGCCTTCACG-3′ aligning to exon 10 and 12, respectively. PCR products were separated and visualized by agarose gel electrophoresis and further sequenced to identify aberrant splicing.
Glucose Uptake
To measure glucose uptake, cells were washed and resuspended in Krebs-Ringer-HEPES (at pH 7.4, 136 mM NaCl, 4.7 ml KCl,1.25 mM CaCl2,1.25 mM MgSO4, and 10 mM HEPES). 2-Deoxy-D-H3 glucose (2 μCi/reaction) was added, and the cells were incubated for 10 min at 37 °C The reactions were quenched by the addition of ice-cold 200 μM phloretin (Calbiochem, Gibbstown, NJ) followed by centrifugation through an oil layer (1:1 Dow Corning 550 Silicon fluid from Motion Industries, Birmingham, AL; and dinonyl phthalate from Sigma-Aldrich). The cell pellets were washed and solubilized in 1 M NaOH, and radioactivity was measured using a scintillation counter.
Results
Patients
All four patients have had recurrent upper and/or lower respiratory infections from early childhood, but only one had a history of viral infections and only with viral agents that cause upper respiratory illnesses (Table 1). They have not had recurrent fungal infections nor have they had opportunistic infections. Three have had recurrent bacterial dacryocystitis, and one has had intermittent hypoglycemia. All four are symmetrically small: three had heights and weights below the 5th percentile and patient 2′s height was below the 10th percentile. Three of the patients have had peripheral lymphadenopathy and the fourth had very large tonsils and adenoids that were removed. The lymph nodes are generally very large and are often soft and movable. Three patients have had lymph nodes excised and no lymph node has been found to be malignant. Lymph node histology in the three for whom it is available revealed hyperplastic lymphoid follicles with attenuated mantle zones (Fig. 1). Only one has splenomegaly and that is complicated by peliosis. None of the patients has autoimmunity or allergy. Only one patient is lymphopenic. Two of them have low numbers of B cells, and all have very low percentages of memory B cells and class-switched memory B cells. One of them has an elevated percentage and number of transitional B cells. They all have normal numbers of natural killer cells, although one has only 16 such cells. Their T cell counts are all within the normal range, but two of them have inverted CD4:CD8 ratios. All of them have low percentages of naïve CD4+ T cells, and two have low percentages of naïve CD8+ T cells. One of the patients has elevated CD8 senescent T cells (Table 1). T cell functional studies in the four patients suggest some T cell impairment in that two had low responses to PHA, three had low responses to immobilized anti-CD3 and three had no response to candida (Table 1).
Fig. 1.
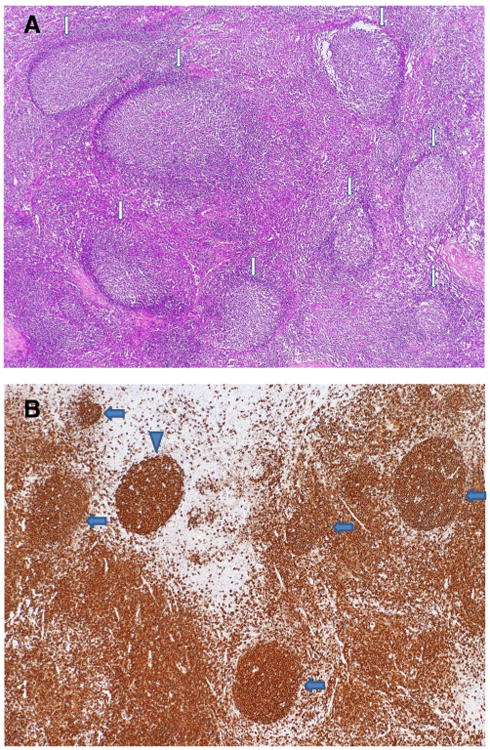
Histopathology of cervical lymph node biopsy of Patient 1. a A low magnification demonstrates reactive follicular hyperplasia with expanded germinal centers and attenuated mantle zones, as indicated by the arrows. Note the polarity of well-defined germinal centers. H&E stain, ×40. b Immunohistochemical stain for CD20 highlights well defined germinal centers with diminished mantle zones or absence of mantle zones, as indicated by the arrows. Note an apparently naked germinal center without mantle zone in upper left, as indicated by the arrowhead, and increased B-cells in the interfollicular area. Anti-CD20 stain, ×40
Mutation
Whole exome sequencing of patient 1 and his parents identified a putative de novo mutation in an essential donor splice site of PIK3R1 (NM_181523.2:c.1425 + 1G > A) (Supplementary Fig. 1). Among our set of 50 patients who have undergone next generation sequencing for immunodeficiency of unknown molecular type, we identified three other patients, patients 2, 3 and 4, with the exact same de novo PIK3R1 essential donor splice site mutation (NM_181523.2:c.1425 + 1G > A) (Supplementary Fig. 1). Variation at this essential splice site was not observed among 2357 samples sequenced for various projects at the Institute for Genomic Medicine, Columbia University (formerly the Center for Human Genome Variation, Duke University); nor was it observed among 59,413 samples among the ExAC consortia dataset that had at least 10-fold coverage at this precise site. For patients 1–3, the parents were also Sanger sequenced for the mutation and it was confirmed that the mutation arose de novo in those patients. For patient 4, who was clinically whole exome sequenced initially by a commercial CLIA-certified laboratory, the variant was Sanger confirmed in the child and confirmed to be absent in mother, but her father was unavailable (Supplementary Fig. 1). It is likely these de novo mutations arose in the germline cells; however, we have not ruled out the possibility that the mutations may be post-zygotic—limited to the hematopoietic system. In our patient population, we have not yet observed instances of the other two described PIK3R1 essential splice variants at the same site: NM_181523.2:c.1425 + 1G > C or NM_181523.2:c.1425 + 1G > T [6, 7].
Detection of Aberrant Splicing of Mutant mRNA
To determine if the mutation results in skipping of exon 11, we amplified PIK3R1 cDNA from total RNAs from patient 1 and his parents PBMCs using primers corresponding to exon 10 and 12 of the gene (transcript NM_181523.2). Both parents generated a single normal 399 base-pair (bp) band. The patient sample, however, produced both a 399 bp band and a unique 273 bp band (Fig. 2). Further sequencing of the PCR products revealed that the 273 bp band was produced by direct exon 10 to 12 splicing.
Fig. 2.
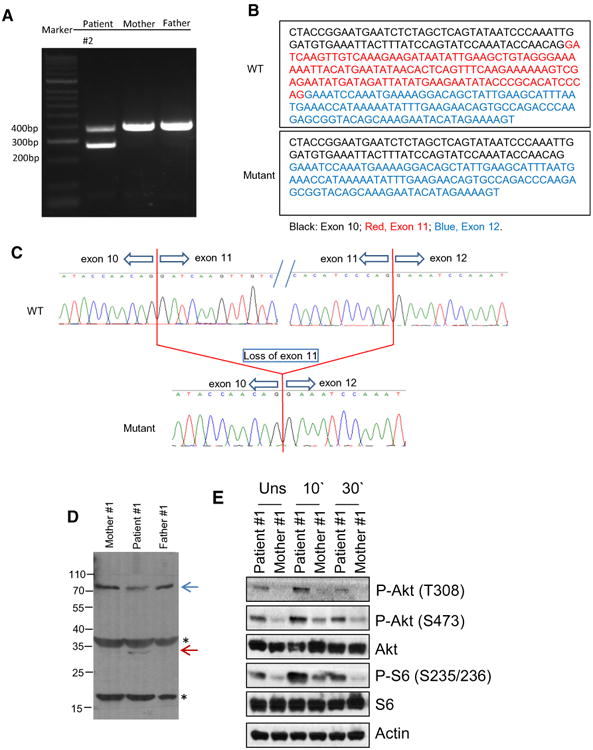
a–c Aberrant splicing in PIK3R1 mRNA in patient 1. a Detection of mutant form of PI3K1R mRNA by RT-PCR. b Sequence of normal and mutant allele cDNA. c Chromatographs showing aberrant exon 10 to 12 splicing in patient 1′s mutant PIK3R1 allele. d Detection of p85α (PIK3RI) protein in lysates from EBV-immortalized B cells from patient 1 and his parents. In the patient sample, the intensity of the band corresponding to p85α is decreased and there is a weak band (blue arrow) that could be vaguely seen right beneath the p85α band, which is likely the exon 11 deletion mutant of p85α. The band indicated with a red arrow is only seen in patient, which is likely a degraded product of the mutant p85α. The identities of the other two bands indicated by *, whether they are non-specific proteins detected by the anti-p85α antibody or degradation products of p85α, are unknown at present. e Assessment of S6 and Akt phosphorylation in EBV-B cells following anti-CD40 stimulation. EBV-B cells from patient 1 and her parents were rested in PBS at 37 °C for 30′ and then stimulated with an anti-CD40 antibody (10 μg/ml) for 10 or 30 min. Cell lysates were subjected to immunoblotting analysis with the indicated antibodies
Effects of p85αΔ434_475 Mutation on Signaling
To examine how p85αΔ434_475 mutation might affect p85α, we generated EBV-immortalized B cell lines from patient 1 and his parents. As shown in Fig. 2d, patient EBV-B cells expressed lower level of p85α compared with parent control. In addition, a small protein band of about 30 kDa was only observed in the patient's B cells but not parents. Thus, the mutant protein might be less stable than the WT protein. An important downstream event of PIK3 signaling is the activation of mTOR. Activated mTOR phosphorylates and activates S6 K1, leading to subsequent phosphorylation of the ribosomal protein S6. After resting in PBS at 37 °C for 30 min, EVB-B cells from both parents of patient 1 did not contain obvious S6 phosphorylation (mTOR complex 1 dependent event), Akt phosphorylation at serine 473 (mTOR complex 2 dependent event), and Akt phosphorylation at threonine 308 (PI3K/PDK1 dependent event). However, a low level but noticeable phosphorylation of these molecules was observed in patient 1′s EBV-B cells, suggesting constitutive activation of PI3K-mTOR signaling (Fig. 2e). Following anti-CD40 stimulation, patient 1′s EBV-B cells displayed a higher level of S6 and Akt phosphorylation than controls, suggesting elevated PI3K/PDK1 and mTOR signaling downstream of CD40. The same was true when they were stimulated with insulin for both patient 1 (top panel, Fig. 3a) and patient 4 (bottom panel, Fig. 3a). Moreover, patient EBV-B cells had increased uptake of 3H-2-deoxy-glucose following 1 h of stimulation with insulin, and were more responsive to insulin than the parent/control EBV-B cells (Fig. 3b). Altogether, these observations suggest that amino acids 435–474 in PIK3R1 are important for its stability and also restrain PI3K activity. Its deletion may lead to constitutive activation of PI3K/Akt/mTOR signaling, which is consistent with recent reports of patients with similar mutations [6–8].
Fig. 3.
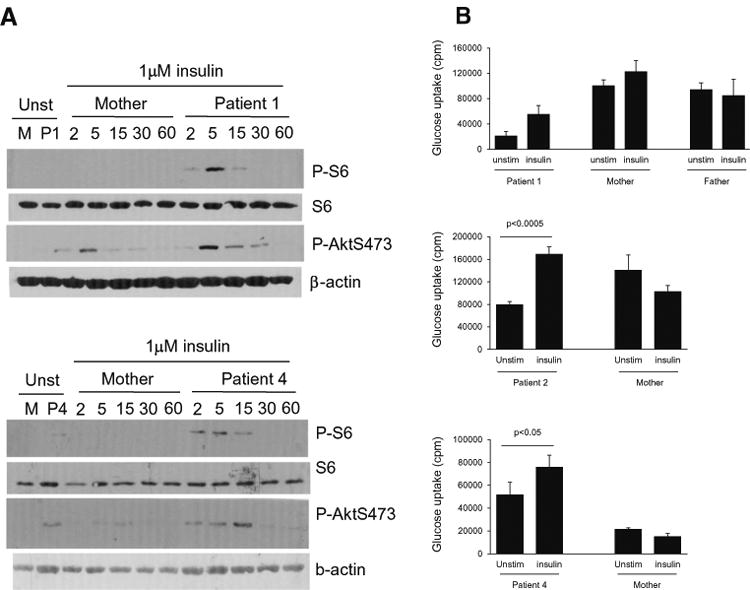
a Assessment of S6 phosphorylation in EBV-B cells following anti-CD40 stimulation. EBV-B cells from patients 1 and 4 and their mothers were rested in PBS at 37 °C for 30′ and then either unstimulated or stimulated with insulin (1 μM) for the indicated minutes. Cell lysates were subjected to immunoblotting analysis with the indicated antibodies. b Assessment of glucose uptake before and after insulin stimulation: EBV-transformed B cells from Patients1, 2 and 4 their parents (controls) were rested in PBS for 30 min followed by culture in the presence or absence of 1–2 μM insulin for 30–60 min. Glucose uptake was determined
Discussion
Including the 12 patients in the three earlier reports [6–8] all sixteen known cases of PIK3R1 immunodeficiency have had mutations at this essential donor splice site (NM_181523.2:c.1425 + 1G > A/C/T). Despite this apparent limited allelic heterogeneity for PIK3R1 immunodeficiency, the findings in the four patients presented here continue to highlight the heterogeneity of the clinical and immunologic abnormalities of the patients who have PIK3RI Δ434_475 essential splice mutations [6–8]. The clinical features of the four patients reported here are uniform and similar to some but not all of those described by Deau et al. [6] and Lucas et al. [7]. This could possibly be due to the younger ages of our patients when compared with those of Lucas et al. [7], but the ages at presentation of our patients were similar to three of the four patients described by Deau et al. [6] and all of the patients reported by Lougaris et al. [8] All four of our patients had short stature; poor growth was noted in the patients reported by Lucas et al. [7] and Lougaris et al. [8] but in only one of those reported by Deau et al. [6] Lymphadenopathy and/or tonsillar hypertrophy were noted in all four of our patients and in the four reported by Lucas et al. [7], and in three of the four reported by Lougaris et al. [8] but in only one of those reported by Deau et al. [6] None of our patients has had a diagnosed malignancy as yet and none has had problems with CMVor EBV. Our patients were each diagnosed clinically as having the Hyper IgM syndrome, but none of them had any of the known gene mutations reported in the Hyper IgM syndrome. Patient 1 had been found to have a common CD40L polymorphism, but his CD4+ Tcells displayed functional CD40L after activation [20]. Patient 4, with her diagnosis of SHORT syndrome, appears to be a unique case compared to other patients with this same mutation. While there may exist another mutation in Patient 4 that makes the presentation unique, it was not found in chromosomal microarray studies, fragile X studies, whole exome sequence analysis in the commercial lab setting, subsequent Sanger sequencing of PIK3R1 exon 14, or in deletion testing of the mitochondrial genome.
PIK3R1 is an established pleiotropic gene. In addition to immunodeficiency, SHORTsyndrome has been reported to be caused by dominant de novo mutations in exon 14 of PIK3R1 [11–16]. Immunologic studies were not reported on in those earlier SHORT syndrome publications. Thus, our Patient 4 with SHORT syndrome having the same mutation at this essential donor splice site (NM_181523.2:c.1425 + 1G > A/C/T) in intron 11 as well as the same immunologic findings as the other patients with PIK3R1 immunodeficiency, provides an important link within PIK3R1 mediated disease. The commercial lab reported Patient 4 as negative for additional PIK3R1, or other SHORT syndrome disease gene, variants. We also subsequently Sanger sequenced PIK3R1 exon 14 in our research lab setting and confirmed that both Patient 4 and her mother were not carriers of variants in exon 14. We performed glucose uptake studies because some but not all patients with SHORT syndrome have been insulin resistant. However, in keeping with the fact than none of the four patients reported here were clinically insulin resistant, the glucose uptake studies were normal (Fig. 3).
Our data suggest that p85αΔ434–475 mutation leads to elevated PI3K activity. The immunodeficiency observed in our patients is similar to those with gain-of-function mutations in PIK3CD, suggesting that this PIK3R1 mutation might cause elevated PIK3CD activity [21]. At present, it is still unclear how the p85αΔ434_475 mutation causes increased PI3K activity. Normal p85α stabilizes the p110 catalytic subunit but inhibits its lipid kinase activity in unstimulated cells. Under stimulating conditions, p85α recruits p110 to activated receptors or adaptor molecules via its SH2 domain, leading to activation of its kinase activity [22, 23]. Because the inter-SH2 domain of p85α is involved in p85α - p110 heterodimerization [24], it is possible that the p85αΔ434_475 mutant inefficiently inhibits its cognate partner p110 (p110δ in the hematopoietic cells and p110α in muscle and other tissues), leading to elevated PI3K signaling. Additionally, p85αΔ434_475 was under-presented compared with WT p85α in patients' samples, indicating that amino acids absent in the mutant in the inter-SH2 domain are important for its stability.
The concomitant observation of SHORT syndrome in patient 4 is interesting in that it confirms PIK3R1 as a hot spot of mutation for this syndrome. It also suggests that the p85αΔ434_475 mutation may affect other catalytic PI3K sub-units such as PIK3CA that are known to be important for insulin receptor signaling [25]. It is important to note that some SHORT syndrome patients with mutations in exon 14 [11–14] and a murine model mimicking one of the PIK3R1 mutations [26] displayed impaired PI3K signaling. Studies have demonstrated that chronic overactivation of signal pathways can lead impairment of receptor induced signaling [27]. It is possible that chronic overactivation of the PI3K pathway might trigger a negative feedback mechanism(s) that prevents normal insulin receptor signaling in some tissues, although we did not find abnormal signaling in patient 4′s blood lymphocytes.
The immunologic abnormalities in our patients were similar in many but not all respects to those previously reported [6–8]. Three of our patients have low numbers of B cells, and all have very low percentages of memory B cells and class-switched memory B cells. IgM was elevated in all four of our patients, in three of the four patients reported by Deau et al. [6], all of the patients reported by Lougaris et al. [8] but in only one of the patients reported by Lucas et al. [7] Most of our patients have low percentages of naïve CD4+ and CD8+ T cells, but only one had elevated CD8 senescent T cells as reported by Lucas et al. [7] T cell functional studies in our patients suggest some T cell impairment but this was not commented on in the three prior reports [6–8]. T cell dysfunction would not be unexpected, since PI3K signaling is required for the differentiation of T cells into subsets [1].
PIK3R1 has been identified as a gene intolerant to functional variation in the general human population. The Residual Variation Intolerance Score (RVIS) for PIK3R1 is −0.76, which corresponds to a ranking of 13.5 % most intolerant genes, genome wide (http://igm.cumc.columbia.edu/GenicIntolerance/). The intolerance of a gene to functional variation among the human population has been demonstrated to be highly predictive of disease-causing genes, particularly those causing disease through a dominant model [28]. Existing mouse knock-out work has also shown that disruptions to Pik3r1 result in a wide range of B cell phenotypes [29]. Moreover, Pik3r1 is considered an ‘essential’ gene, with mouse knock-outs resulting in complete perinatal lethality and partial postnatal lethality [29]. We have previously shown that the bioinformatics signature of a disruptive de novo mutation in an intolerant gene that is also known to be essential is highly linked to pathogenicity among children with undiagnosed genetic disorders [30].
Available mTOR inhibitors as well as non-selective PI3K inhibitors approved for use in other conditions already exist. A range of next generation PI3K inhibitors, which are highly selective for the delta isoform, are under development. These drugs could offer a pharmacological solution for these patients where existing treatment options are very limited and include extreme therapies such as bone marrow transplantation. The availability of highly selective orally available PI3Kdelta inhibitors has the potential to normalize the pathological mechanism in these patients.
Conclusions
Together, these observations suggest that amino acids 435–474 in PIK3R1 are important for its stability and also its ability to restrain PI3K activity. Deletion of exon 11 leads to constitutive activation of PI3K signaling. The clinical and immunologic manifestations of patients with dominant splice site mutations in PIK3R1 have been variable, with all having very low percentages of memory B cells and class-switched memory B cells and reduced percentages of naïve CD4+ and CD8+ T cells. The clinical manifestations in our patients were uniform, with all having the Hyper IgM syndrome, short stature and lymphadenopathy, except that one also had a clinical diagnosis of SHORT syndrome. This is the first report of immunologic abnormalities and this precise mutation in a patient ascertained for SHORT syndrome. The elucidation of the molecular mechanism in these patients results in the opportunity for new and potentially transformative therapeutic options.
Supplementary Material
Acknowledgments
This study was supported by the following grants: Baxter Healthcare Grant BT13-21694 to R.H.B. and NIH Grant R01 AI101206 to X-P. Z. Partial funding for this study was provided by UCB Celltech, including salary support for D.B.G M.A. and D.M. are employees of UCB. The other authors declare no conflict of interest. S.P is a National Health and Medical Research Council (NHMRC) CJ Martin Fellow. We thank our patients and their parents for participating in this study. We also thank B Krueger, J Bridges, Q Wang, N Ren, S Gewalt, S Kisselev, Y-F Lu, K Cronin, and N Walley for excellent technical support. We would like to acknowledge the following individuals for the contributions of control samples: W. B. Gallentine, E.L. Heinzen, A.M. Husain, K. N. Linney, M. A. Mikati, R. A. Radtke, and S. R. Sinha; J.P McEvoy, A. Need, J. Silver, and M Silver; D. H. Murdock and The MURDOCK Study Community Registry and Biorepository; G. Cavalleri, N. Delanty, and C. Depondt; J. Burke, C. Hulette, and K. Welsh-Bohmer; J. Milner; J. Hoover-Fong, N. L. Sobreira and D. Valle;E. J. Holtzman; W. L. Lowe; P. Lugar; S. M. Palmer; Z. Farfel, D. Lancet, E. Elon Pras; A. Poduri; M. Hauser; D. Marchuk; D. Koltai Attix, O. Chiba-Falek; E. T. Cirulli, V Dixon and J. McEvoy; K. Schmader, S. McDonald, H. K. White, M. Yanamadala, and the Carol Woods and Croasdale Retirement Communities; R. Gbadegesin and M. Winn; D. Daskalakis; Q. Zhao; A. Holden and E. Behr; R. Brown; and S. Kerns and H. Oster. The collection of control samples was funded in part by Bryan ADRC NIA P30 AG028377, the Ellison Medical Foundation New Scholar award AG-NS-0441-08, an award from SAIC-Frederick, Inc. (M11-074), funding from Biogen Idec, NIMH awards RC2MH089915, R01MH097971, R01MH099216, and K01MH098126, the Epi4K Gene Discovery in Epilepsy study (NINDS U01-NS077303) and the Epilepsy Genome/Phenome Project (EPGP - NINDS U01-NS053998), and the Center for HIV/AIDS Vaccine Immunology (“CHAVI”) under a grant from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (UO1AIO67854), NIAID (R01AI079088 and R01AI101206). Finally, the authors would also like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10875-016-0281-6) contains supplementary material, which is available to authorized users.
References
- 1.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–71. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. J Allergy Clin Immunol. 2014;134(1):233–6. doi: 10.1016/j.jaci.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol. 2014;34(3):272–6. doi: 10.1007/s10875-014-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2014;124(9):3923–8. doi: 10.1172/JCI75746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. 2014;211(13):2537–47. doi: 10.1084/jem.20141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol. 2015;159(1):33–6. doi: 10.1016/j.clim.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 9.Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. 2015;125(4):1764–5. doi: 10.1172/JCI81746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. J Exp Med. 2012;209(3):463–70. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013;93(1):158–66. doi: 10.1016/j.ajhg.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chudasama KK, Winnay J, Johansson S, Claudi T, Konig R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150–7. doi: 10.1016/j.ajhg.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141–9. doi: 10.1016/j.ajhg.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroeder C, Riess A, Bonin M, Bauer P, Riess O, Dobler-Neumann M, et al. PIK3R1 mutations in SHORT syndrome. Clin Genet. 2014;86(3):292–4. doi: 10.1111/cge.12263. [DOI] [PubMed] [Google Scholar]
- 15.Barcena C, Quesada V, De Sandre-Giovannoli A, Puente DA, Fernandez-Toral J, Sigaudy S, et al. Exome sequencing identifies a novel mutation in PIK3R1 as the cause of SHORT syndrome. BMC Med Genet. 2014;15:51. doi: 10.1186/1471-2350-15-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung BK, Gibson WT. Autosomal dominant PIK3R1 mutations cause SHORT syndrome. Clin Genet. 2014;85(3):228–9. doi: 10.1111/cge.12262. [DOI] [PubMed] [Google Scholar]
- 17.Buckley RH, Schiff SE, Sampson HA, Schiff RI, Markert ML, Knutsen AP, et al. Development of immunity in human severe primary T cell deficiency following haploidentical bone marrow stem cell transplantation. J Immunol. 1986;136:2398–407. [PubMed] [Google Scholar]
- 18.Sugden B, Mark W. Clonal transformation of adult human leukocytes by Epstein-Barr virus. J Virol. 1977;23(3):503–8. doi: 10.1128/jvi.23.3.503-508.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2011;117(15):4022–31. doi: 10.1182/blood-2010-08-300731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seyama K, Nonoyama S, Gangsaas I, Hollenbaugh D, Pabst HF, Aruffo A, et al. Mutations of the CD40 ligand gene and its effect on CD40 ligand expression in patients with X-linked hyper IgM syndrome. Blood. 1998;92(7):2421–34. [PubMed] [Google Scholar]
- 21.Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol. 2015;6 doi: 10.1111/cei.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci. 2015;40(2):88–100. doi: 10.1016/j.tibs.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Geering B, Cutillas PR, Vanhaesebroeck B. Regulation of class IA PI3Ks: is there a role for monomeric PI3K subunits? Biochem Soc Trans. 2007;35(Pt 2):199–203. doi: 10.1042/BST0350199. [DOI] [PubMed] [Google Scholar]
- 24.Chiu YH, Lee JY, Cantley LC. BRD7, a tumor suppressor, interacts with p85alpha and regulates PI3K activity. Mol Cell. 2014;54(1):193–202. doi: 10.1016/j.molcel.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21(10):589–98. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winnay JN, Solheim MH, Dirice E, Sakaguchi M, Noh HL, Kang HJ, et al. PI3-kinase mutation linked to insulin and growth factor resistance in vivo. J Clin Invest. 2016 doi: 10.1172/JCI84005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Zhang P, Krishna S, Wang J, Lin X, Huang H, et al. Unexpected positive control of NFkappaB and miR-155 by DGKalpha and zeta ensures effector and memory CD8+ T Cell differentiation. Oncotarget. 2016 doi: 10.18632/oncotarget.8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8):e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE. The mouse genome database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42(Database issue):D810–7. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–81. doi: 10.1038/gim.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.