

Abstract
Rationale:
Leiomyosarcoma (LMS) is a malignant sarcoma that can occur in different anatomic sites, including the bone, showing similar histological characteristics but heterogeneous clinical behavior and prognosis. Primary bone LMS was first described in 1965. It is a very rare sarcoma, accounting for <0.7% of all primary malignant bone tumors.
Patient concerns:
We report the case of a 52-year-old male with primary bone LMS who presented with a solitary osteolytic lesion with focal cortical destruction in the left clavicle, seen on an x-ray and subsequent computed tomography (CT) scan.
Diagnosis:
The multidisciplinary Osteoncology team of our institute planned a biopsy that revealed the presence of spindle and pleomorphic cells with a positive reaction for smooth muscle actin and desmin at immunohistochemical analysis, without the presence of cartilage or bone matrix. These results were consistent with a high-grade malignant LMS arising from the bone.
Interventions:
Complete surgical resection of the tumor was performed and a decision was made with the patient not to proceed with adjuvant chemotherapy or radiotherapy.
Outcomes:
After more than 1 year of surgery, the patient is well, with no evidence of recurrent or metastatic disease. Follow-up is ongoing.
Lessons:
Little is known about the biology and clinical behavior of bone LMS due to its extreme rarity. A multidisciplinary team in a specialized center is needed for the optimal management of the disease. Surgery with a curative intent is the cornerstone of treatment of localized disease. No data are available about chemotherapy in neoadjuvant, adjuvant, or advanced settings. Further research is needed to identify more effective therapies.
Keywords: bone, chemotherapy, leiomyosarcoma, multidisciplinary team, smooth muscle differentiation
1. Introduction
Leiomyosarcomas (LMS) represent one of the most common types of soft tissue sarcoma (STS), accounting for about 7% to 10% of all STS, involving different anatomic sites, especially the retroperitoneum, the genitourinary tract, and the extremities. LMS can also occur in the bone, as primary or secondary tumor localization from distant sites, although the former is fairly uncommon, with <0.7% incidence of all primary malignant bone tumors.[1] The clinical behavior of bone LMS is generally aggressive. Most of the published studies on bone LMS have reported poor prognosis with a 35% overall survival (OS) rate.[2]
LMS diagnosis should be performed in highly specialized centers and is established by the presence of morphologically typical spindle cells for smooth muscle differentiation and the positivity of a smooth muscle actin (SMA) and other muscle markers on tumor cells, such as desmin and h-caldesmon.[3] Furthermore, the specific characteristic of primary bone LMS is the absence of either osteoid or chondroid matrix.[4,5] The molecular pathogenesis and biological heterogeneity of LMS have not yet been clarified.
The optimal management of primary bone LMS should be performed by a multidisciplinary team of experts in specialized referral centers. Nowadays, although the wide surgical removal of the primary lesion is the cornerstone of treatment for the localized disease with the aim to obtain clear surgical margins with a curative intent, the role of chemotherapy is currently under discussion.[6,7] Chemotherapy is the principal treatment option with a palliative purpose in the metastatic setting, even though the optimal chemotherapy scheme is still to be defined and few chemotherapy agents have shown any activity against LMS due to the lack of data on this subtype of sarcoma.[8–10]
There is an urgent need for a better understanding of the molecular mechanisms in STS pathogenesis, considering the different anatomic variants, especially the most rare ones, including primary bone LMS. New therapies and dedicated clinical trials are thus required to improve the outcomes of STS patients. This paper provides an overview of the major primary bone LMS clinical and histopatologic characteristics and their management. We also report our experience of a patient with localized, surgically treated primary bone LMS in the left clavicle.
2. Case report
Ethics approval was not necessary for this work due to its design (Case Report). Written informed consent was obtained from our patient for the submission of this manuscript and accompanying images.
A 52-year-old male patient presented with a solitary bone lesion in the left clavicle. His past medical history was positive for diabetes mellitus treated with oral hypoglycemic agents. There was no history of smoking or alcohol consumption. He reported mild pain and a solitary mass in the left clavicle. Ultrasound and x-ray imaging of the clavicle showed the presence of an osteo-rarefaction area associated with bone fracture without apparent pathological features. The formation gradually increased in size with a worsening of the pain. A subsequent CT scan of the left clavicle showed the presence of osteolytic lesions with a focal cortical destruction possibly correlated to pathologic fracture. The lesion was located predominantly in the medullary cavity and presented a soft tissue extension from the bone with indistinct tumor margins.
The patient was evaluated at our Institute by an Osteoncology Multidisciplinary Team, composed of an orthopedist, an oncologist, a radiologist, a pathologist, a radiotherapist, a physiatrist, a palliative therapist, and a nuclear medicine physician. The team suggested a biopsy of the bone lesion. The patient initially refused the biopsy. Two months later, due to the persistence of the pain, he performed another CT scan of the chest that revealed an increase in the size of the bone lesion, macroscopically measuring 5 cm, with multiple pathologic fractures and pathologically associated tumor tissue and cortical destruction (Fig. 1). The patient accepted to undergo biopsy of the lesion, which revealed the presence of spindle cells with positive reaction for SMA and desmin at immunohistochemical analysis, without the presence of cartilage or bone matrix. These results were consistent with a high-grade malignant LMS arising from the bone. A total body CT scan was performed after the biopsy, which provided no evidence of metastatic disease or other primary tumors. The multidisciplinary team reviewed the patient clinical condition, the radiographic imaging, and the results of the biopsy, suggesting surgery of the bone LMS. Subsequently, the patient underwent surgery for removing the bone lesion in the left clavicle.
Figure 1.
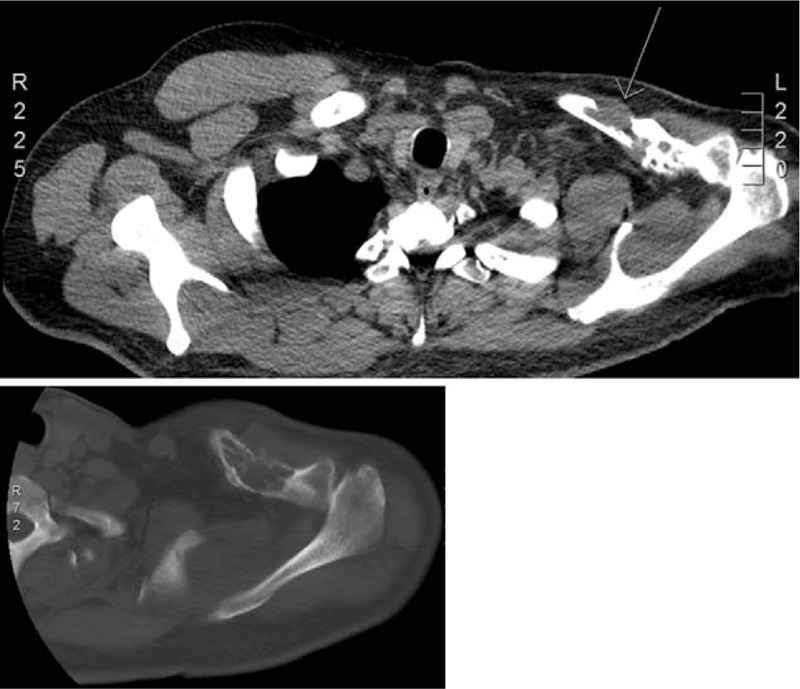
Axial CT scan images revealing the intraosseous lesion in the left clavicle with multiple pathologic fractures and pathologic-associated tumor tissue and cortical destruction.
Macroscopically, the histological examination of the resected bone lesion showed that the lesion was 3.5 cm wide and located in the medulla and cortex of the left clavicle with destruction of the cortex. Although there were no foci of necrosis, hemorrhage, or cystic changes, the presence of myofilaments and fibroblasts was detected. Microscopically, the tumor cells were morphologically related to LMS, and immunohistochemistry was positive for SMA, desmin, and caldesmon. Absence of staining for cytokeratin, S100 protein, or epithelial markers, and of osteoid or chondroid matrix was confirmed (Fig. 2). The surgical margins were negative. The neoplasm was classified according to the Enneking staging system as a high-grade extracompartimental tumor, stage IIB. The total body CT scan after surgery was negative, showing no tumor lesions in other anatomical sites.
Figure 2.
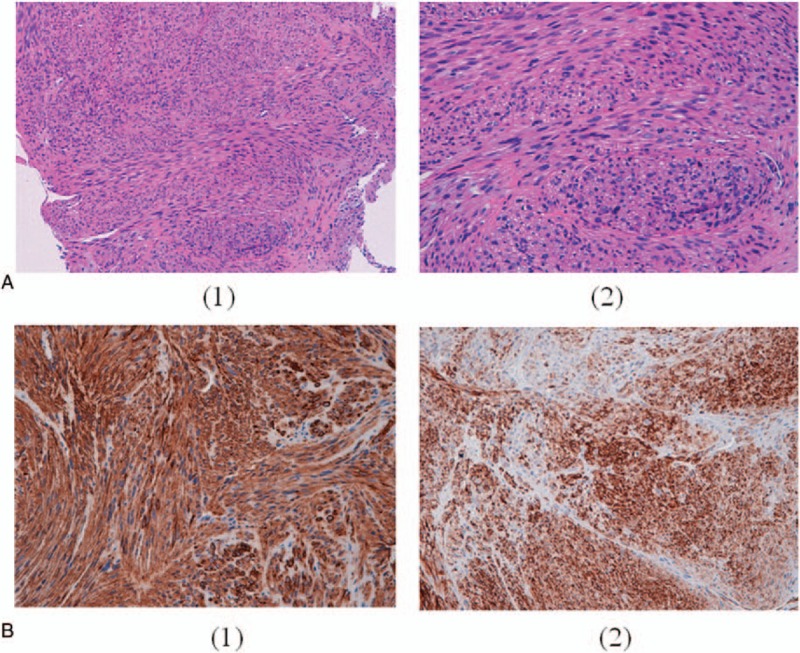
Histological aspects of the tumor tissue: (A) fascicles of spindle cells and pleomorphic cells with eosinophilic cytoplasm x 10 (1) and x 20 (2); (B) immunohistochemical results: expression of smooth muscle actin (1) and desmin (2).
As the postoperative course was uneventful, no adjuvant chemotherapy or radiotherapy was administered, after making shared decisions with the patient. A strict follow-up was planned with clinical and radiological evaluations every 3 months for the first 2 years. After 1 year of surgery, the patient is in good health condition, showing no symptoms and continuing the follow-up without evidence of recurrent or metastatic disease.
3. Discussion
LMS is an aggressive sarcoma that can arise in different anatomic sites, including the bone, showing similar histological characteristics, but heterogeneous clinical behavior and different prognosis. Primary bone LMS, first described in 1965, is a rare type of sarcoma, accounting for <0.7% of all primary malignant bone tumors.[1–3,11] Bone tumors are usually first diagnosed with radiographic imaging. The principal radiological feature consists in a solitary osteolytic lesion with indistinct margins and cortical destruction with no presence of bone matrix production. Rarely, osteolytic lesions can be associated with tumor tissue.[3–5] The most common symptoms are pain, swelling, and, occasionally, a palpable mass. In about 15% of cases, patients present with a pathological fracture.[12]
Histopathologic analysis in specialized centers represents the gold standard for the diagnosis of sarcomas, including LMS. The histopathologic characteristics of primary bone LMS are identical to those arising from other more common anatomic sites, showing the same morphological and phenotypic features as smooth-muscle differentiation.[13] The classic morphological pattern of LMS is represented by spindle cells, which are usually disposed in fascicles and intersect at perpendicular angles. Tumor cells show cytological atypia with cigar-shaped nuclei and abundant eosinophilic and fibrillar cytoplasm.[14] The immunohistochemistry expression of desmin, actin, and h-caldesmon represents the most significant element of a smooth muscle differentiation correlated to LMS diagnosis. None of these is absolutely specific for smooth muscle and for this reason the positivity of at least 2 of these markers provides a more confident diagnosis for LMS.[13]
Primary bone LMS diagnosis is characterized by the absence of either osteoid or chondroid matrix LMS. This type of tumor is usually circumscribed with the extensive infiltration of the trabecular bone. Areas of tumor necrosis or hemorrhage can be observed.[5,12] Primary bone LMS mostly presents a high histologic grade, showing many single or small, disorganized groups of spindle cells of different sizes showing nuclear pleomorphism, hyperchromasia, and prominent nucleoli. Low-grade tumors are often characterized by the prevalence of fascicles of spindle cells with relatively abundant dense fibrillar cytoplasm, elongated nuclei, and minimal nuclear atypia.[4] The main differential diagnoses include, among others, osteosarcoma, chondrosarcoma, and metastatic carcinoma. The lack of malignant osteoid or chondroid matrix and epithelial markers excludes the presence of osteosarcoma, chondrosarcoma bone sarcoma subtypes, and metastatic carcinoma, respectively.[5,12,15]
Clinical features of primary bone LMS and relevant prognostic factors are not well defined due to the few available data obtained mostly from retrospective analyses, case reports, and small case series. Potential risk factors have been associated with the pathogenesis of bone LMS, including Paget disease of the bone and orthopedic implant. A case of bone LMS in the femur secondary to bone infarction has also been reported, possibly due to a continuous reparative process and a vascular proliferation around the area of osteonecrosis.[16–18] Cases of intraosseous LMS secondary to radiation therapy, although very sporadic, have also been described.[19,20]
Antonescu et al[9] in 1997 tried to stratify the prognosis of 33 patients with primary bone LMS according to the histological grade, showing no significant difference in either recurrence or OS rates in low- and high-grade tumors. A case series evaluating the outcome in 8 patients with bone LMS showed that all patients developed metastasis within 12 months of primary diagnosis, regardless of the tumor grade.[15] A monocentric study evaluated the incidence, outcome, and prognostic factors associated with primary spindle cell bone sarcomas. A total of 196 patients with these types of bone sarcoma treated with the intent to cure, 30 of which were bone LMS, had similar outcomes compared with osteosarcoma patients treated at the same center at the same time. The most important prognostic factors associated with a decrease in survival were age >40 years, size >8 cm, the presence of a pathological fracture, amputation, involved margins, and a poor response to preoperative chemotherapy.[21,22] Our patient presented with progressive pain and a solitary mass in the left clavicle, which ultrasound and x-ray revealed to be an osteolytic formation with associated pathologic fracture and tumor tissue. A subsequent CT scan confirmed the radiological characteristics of the lesion. The clinical presentation and the radiographic imaging of the solitary intraosseous formation suggested the presence of a primary bone sarcoma.
The cellular origin of bone LMS has not been clearly established. Interestingly, there are 2 main hypotheses about the origin of primary bone LMS: the first suggests that it can arise from vascular smooth muscle cells in the bone; the second claims that it originates from intermediate cells, mostly fibroblasts, capable of smooth muscle differentiation.[23,24] As smooth muscle cells have been shown to synthesize connective tissue matrix, and myofilaments and fibroblasts can be present in various differentiation states, sarcomas originating from fibroblastic tissue were thought to possibly develop into a differentiation indistinguishable from LMS.[25] In our case, the presence of myofilaments and fibroblasts in the tumor area suggested the activation of these cells leading to LMS differentiation in the bone, indicating the importance of the microenvironment in the pathogenesis of this disease.
Local recurrence is relatively uncommon, while metastatic disease is more likely to develop early, generally in the first year, than after many years of initial diagnosis. Similarly to other types of STS, the most frequent site of metastasis is the lung; skeletal metastases, occurring mostly in the axial skeleton, are also frequent. Metastases to the liver, adrenal gland, kidney, and lymph nodes have a lower incidence.[26–28] Because of the low incidence of this type of bone sarcoma, the therapeutic benefit of the treatment is not well acknowledged, making the treatment of bone LMS very challenging. For this reason, the therapeutic decision for primary bone LMS should be made by an experienced multidisciplinary team. The radical surgical treatment of bone LMS with negative margins is similar to that of other primary malignant bone tumors, currently representing the only curative option. Although some authors have reported that complete surgical resection of the tumor is associated with longer OS, the outcome of patients with metastatic disease remains poor.[29]
Given the similar biological behavior and chemosensitivity of osteosarcoma and spindle cell sarcomas of the bone, including bone LMS,[21,22,30] it could be reasonable to extrapolate the treatment of osteosarcoma to other bone tumor types. No data, however, are available to support this approach. As the benefits of (neo-)adjuvant chemo- or radiation therapy of the treatment of bone LMS have not been established, it is unclear whether therapeutic approaches similar to those for extra-skeletal LMS or bone sarcomas are suitable also for bone LMS.[31–33]
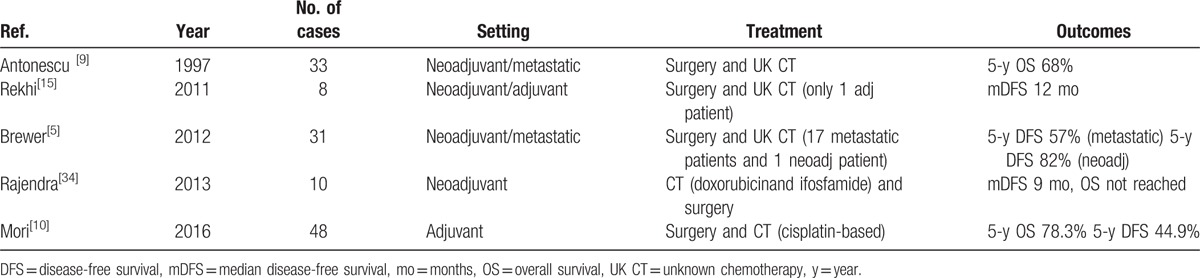
To date, few dedicated retrospective trials and case series have specifically evaluated the outcomes of patients affected by primary bone LMS (Table 1). A retrospective trial conducted by Antonescu et al[9] on 33 patients with primary bone LMS treated with chemotherapy and surgery showed a more aggressive behavior associated with the lesions with a prevalent osteolytic component. In this study, 21% of patients received neoadjuvant chemotherapy and no survival difference was reported between those treated with chemotherapy and without chemotherapy. Antonescu et al[9] showed that adjuvant radiotherapy was not associated with a benefit from the treatment of primary bone LMS, suggesting a possible resistance to radiotherapy. Another retrospective study was conducted on 31 bone LMS patients, of whom 18 received neo-adjuvant chemotherapy. This study reported no difference in the survival rate between patients treated with neoadjuvant chemotherapy and untreated patients, indicating that the clinicopathologic stage of the disease and the presence of metastasis at diagnosis were the only prognostic factors.[5] Rekhi et al[15] presented a case series of 8 patients affected by bone LMS, receiving neither adjuvant nor neoadjuvant, chemotherapy nor radiotherapy. All patients developed metastasis within 12 months of first diagnosis, with a clear indication of the poor prognosis of this disease.[15] A retrospective study conducted in Japan on 48 bone LMS patients showed that adjuvant chemotherapy, with cisplatin-based chemotherapy as the most common therapeutic regimen, was not correlated with an improvement in OS.[10] In a 2013 ASCO Annual Meeting abstract of a single-center study evaluating the role of neoadjuvant chemotherapy in the management of the localized disease, patients with high-grade primary bone LMS treated with neoadjuvant doxorubicin- and ifosfamide-based chemotherapy for 2 cycles and surgery were compared with patients with low-grade tumors treated with only surgical resection. The authors concluded that although surgical resection remains the mainstay treatment of bone LMS, the role of neoadjuvant chemotherapy requires further evaluation.[34] Another trial, the European treatment protocol for high-grade spindle cell bone sarcoma in patients >40 years (EUROBOSS Trial), which includes bone LMS, is currently ongoing to evaluate the role of multiagent chemotherapy, containing DOX, CDP, IFO, and MTX in these tumors and in osteosarcoma (NCT02986503).
Table 1.
Summary of principal published case series focused on bone LMS.
The management of metastatic bone LMS is challenging. Chemotherapy represents the principal approach in metastatic LMS, including in the skeletal variant, even though no clinical studies have yet been carried out specifically on the treatment of metastatic bone LMS. The efficacy of chemotherapy in metastatic sarcoma has been tested on all sarcoma histotypes, including LMS. Cisplatin, doxorubicin or doxorubicin-based chemotherapy, and dacarbazine constitute the most active agents for the treatment of bone sarcoma, and are generally used in the first-line metastatic setting.[35–37] A prospective study evaluating the role of doxorubicin and cisplatin in 37 patients with high-grade spindle cell sarcomas of the bone other than osteosarcoma or malignant fibrous histiocytoma, including few cases of bone LMS, showed a limited role of chemotherapy in the metastatic setting of these bone sarcomas.[38] The role of adjuvant and neo-adjuvant treatments in localized primary bone LMS and their effect on a long-term prognosis is still unclear. Due to the rarity of this disease, treatment of bone LMS remains highly personalized.
Our patient with localized high-grade primary bone LMS was treated with radical surgery alone following multidisciplinary evaluation and shared decision-making with the patient himself. He has been in good health with no evidence of recurrence or metastatic disease since March 2016. Although literature data would seem to suggest a higher risk of recurrence in the first years after surgery, there is still little information available on the prognosis and clinical course of primary bone LMS treated with surgery alone. In this scenario, it is essential to design translational and clinical trials focused on specific variants of rare tumors that often lack specific treatments, in order to improve the knowledge of the disease biology and to identify more effective “histology-driven” therapeutic agents.
Acknowledgment
The authors would like to thank Cristiano Verna and Veronica Zanoni for editing the manuscript.
Footnotes
Abbreviations: CT = chemotherapy, CT scan = computed tomography scan, LMS = leiomyosarcoma, RT = radiotherapy, SMA = smooth muscle actin.
Authorship: FR and TI conceived the idea for the paper.
AB, NR, RC, and VF performed the literature search.
FP furnished the histopathological report and the images.
ADV, LM, CL, CS, and GM were responsible for data analysis.
FR drafted the manuscript.
TI and DA revised the manuscript for important intellectual content.
All authors read and approved the final version of the manuscript for submission.
The authors declare no conflict of interest.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- [2].Stramare R, Orsatti G, Attar S, et al. Imaging features, differential diagnosis and management of leiomyosarcomas: case series and review of the literature. J Canc Sci Ther 2016;8:84–91. [Google Scholar]
- [3].Fletcher CDM, Unni KK, Mertens F. Pathology and Genetics of Tumours of Soft Tissue and Bone. World Health Organization. International Agency for Research on Cancer. Lyon: IARC Press; 2002. p. 427. [Google Scholar]
- [4].Myers JL, Arocho J, Bernreuter TW, et al. Leiomyosarcoma of bone: a clinicopathologic, immunohistochemical, and ultrastructural study of five cases. Cancer 1991;67:1051–6. [DOI] [PubMed] [Google Scholar]
- [5].Brewer P, Sumathi V, Grimer RJ, et al. Primary Leiomyosarcoma of Bone: Analysis of Prognosis. Hindawi Publishing Corporation Sarcoma. Volume 2012, Article ID 636849, 4 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wirbel RJ, Verelst S, Hanselmann R, et al. Primary leiomyosarcoma of bone: clinicopathologic, immunohistochemical, and molecular biologic aspects. Ann Surg Oncol 1998;5:635–41. [DOI] [PubMed] [Google Scholar]
- [7].Malawer MM, Link P, Donaldson SS. DeVita VT, Jr, Hellman S, Rosenberg SA. Sarcomas of bone. Cancer Principles and Practice of Oncology. Philadelphia, NY: Lippincott-Raven; 1997. 1789–852. 30. [Google Scholar]
- [8].Bathan AJ, Constantinidou A, Pollack SM, Jones RL. Diagnosis, prognosis and management of leiomyosarcoma: recognition of anatomic variants. Curr Opin Oncol 2013; 25:384–389. [DOI] [PubMed] [Google Scholar]
- [9].Antonescu CR, Erlandson RA, Huvos AG. Primary leiomyosarcoma of bone: a clinicopathologic, immunohistochemical, and ultrastructural study of 33 patients and a literature review. Am J Surg Pathol 1997;21:1281–94. [DOI] [PubMed] [Google Scholar]
- [10].Mori T, Nakayama R, Endo M, et al. Forty-eight cases of leiomyosarcoma of bone in Japan: a multicenter study from the Japanese musculoskeletal oncology group. J Surg Oncol 2016;114:495–500. [DOI] [PubMed] [Google Scholar]
- [11].Evans D, Sanarkin NG. Primary leiomyosarcoma of bone. J Pathol Bacteriol 1965;90:348–50. [DOI] [PubMed] [Google Scholar]
- [12].Khoddami M, Bedard YC, Bell RS, et al. Primary leiomyosarcoma of bone: report of seven cases and review of the literature. Arch Pathol Lab Med 1996;120:671–5. [PubMed] [Google Scholar]
- [13].Weiss SW, Goldblum JR. Enzinger FM, Weiss SW. Leiomyosarcoma. Soft Tissue Tumors 4th ed.St Louis, MO: The CV Mosby Co; 2001. 727–48. [Google Scholar]
- [14].Adelani MA, Schultenover SJ, Holt GE, et al. Primary leiomyosarcoma of extragnathic bone: clinicopathologic features and reevaluation of prognosis. Arch Pathol Lab Med 2009;133:1448–56. [DOI] [PubMed] [Google Scholar]
- [15].Rekhi V, Kaur A, Puri A, et al. Primary leiomyosarcoma of bone—a clinicopathologic study of 8 uncommon cases with immunohistochemical analysis and clinical outcomes. Ann Diagn Pathol 2011;15:147–56. [DOI] [PubMed] [Google Scholar]
- [16].Deyrup AT, Montag AG, Inwards CY, et al. Sarcomas arising in Paget disease of bone: a clinicopathologic analysis of 70 cases. Arch Pathol Lab Med 2007;131:942–6. [DOI] [PubMed] [Google Scholar]
- [17].Petra M, Gibbons CL, Athanasou NA. Leiomyosarcoma of bone arising in association with a bone infarct. Sarcoma 2002;6:47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Desai P, Perino G, Present D, et al. Sarcoma in association with bone infarcts. Arch Pathol Lab Med 1996;120:482–9. [PubMed] [Google Scholar]
- [19].Aoki T, Ozeki Y, Watanabe M, et al. Development of primary leiomyosarcoma of the sternum postirradiatin: report of a case. Surg Today 1998;28:1326–8. [DOI] [PubMed] [Google Scholar]
- [20].Korbi S, Meyer D, Skalli O, et al. Post-irradiation leiomyosarcoma. Case report with immunohistochemical studies. J Submicrosc Cytol 1987;19:365–9. [PubMed] [Google Scholar]
- [21].Pakos EE, Grimer RJ, Peake D, et al. The ’other’ bone sarcomas: prognostic factors and outcomes of spindle cell sarcomas of bone. J Bone Joint Surg Br 2011;93:1271–8. [DOI] [PubMed] [Google Scholar]
- [22].Stout AP, Lattes R. Tumors of the Soft Tissues. Atlas of Tumor Pathology, Second Series, Fascicle I. Washington, DC: Armed Forces Institute of Pathology; 1967. [Google Scholar]
- [23].Fornasier VL, Paley D. Leiomyosarcoma in bone: primary or secondary? A case report and review of the literature. Skeletal Radiol 1983;10:147–53. [DOI] [PubMed] [Google Scholar]
- [24].Overgaard J, Frederiksen P, Helmig O, et al. Primary leiomyosarcoma of bone. Cancer 1977;39:1664–71. [DOI] [PubMed] [Google Scholar]
- [25].Sanerkin NG. Primary leiomyosarcoma of the bone and its comparison with fibrosarcoma. A cytological, histological and ultrastructural study. Cancer 1979;44:1375–87. [DOI] [PubMed] [Google Scholar]
- [26].Toulmonde M, Le Cesne A, Mendiboure J, et al. Long-term recurrence of soft tissue sarcomas prognostic factors and implications for prolonged follow-up. Cancer 2014;120:3003–6. [DOI] [PubMed] [Google Scholar]
- [27].Goto T, Ishida T, Motoi N, et al. Primary leiomyosarcoma of the femur. J Orthop Sci 2002;7:267–73. [DOI] [PubMed] [Google Scholar]
- [28].Shen SH, Steinbach LS, Wang SF, et al. Primary leiomyosarcoma of bone. Skeletal Radiol 2001;30:600–3. [DOI] [PubMed] [Google Scholar]
- [29].Atalar H, Gunay C, Yildiz Y, et al. Primary leiomyosarcoma of bone: a report on three patients. Clin Imaging 2008;32:321–5. [DOI] [PubMed] [Google Scholar]
- [30].Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop 2006;30:445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jundt G1, Moll C, Nidecker A, et al. Primary leiomyosarcoma of bone: report of eight cases. Hum Pathol 1994;25:1205–12. [DOI] [PubMed] [Google Scholar]
- [32].Eady JL, McKinney JD, McDonald EC. Primary leiomyosarcoma of bone: a case report and review of the literature. J Bone Joint Surg Am 1987;69:287–9. [PubMed] [Google Scholar]
- [33].Young CL, Wold LE, McLeod RA, et al. Primary leiomyosarcoma of bone. Orthopedics 1988;11:615–8. [DOI] [PubMed] [Google Scholar]
- [34].Rajendra R, Pollack S, Rodler ET, et al. Outcome of Leiomyosarcoma of Bone: A Single Center Experience. 2013 ASCO Annual Meeting. May 31-June 4, 2013, Chicago, Illinois. [Google Scholar]
- [35].Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85. [DOI] [PubMed] [Google Scholar]
- [36].Lerner H, Amato D, Stevens C, et al. Leiomyosarcoma: the Eastern Cooperative Oncology Group experience with 222 patients. Proc Am Assoc Cancer Res 1983;24:142.(abstr C-561). [Google Scholar]
- [37].Judson I, Verweij J, Gelderblom H, et al. European Organisation and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft- tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 2014;15:415–23. [DOI] [PubMed] [Google Scholar]
- [38].Nooij MA, Whelan J, Bramwell VH, et al. European Osteosarcoma Intergroup. Doxorubicin and cisplatin chemotherapy in high-grade spindle cell sarcomas of the bone, other than osteosarcoma or malignant fibrous histiocytoma: a European Osteosarcoma Intergroup Study. Eur J Cancer 2005;41:225–30. [DOI] [PubMed] [Google Scholar]