

Abstract
We designed a class of small dimeric cyclic guanidine derivatives, which display potent antibacterial activity against both multidrug-resistant Gram-negative and Gram-positive bacteria. They could compromise bacterial membranes without developing resistance, inhibit biofilms formed by E. Coli, and exhibit excellent in vivo activity in the MRSA-infected thigh burden mouse model.
Table of Contents
A class of small dimeric cyclic guanidine derivatives was designed to display potent and broad spectrum antibacterial activity.
Antimicrobial resistance is an escalating threat in global public health,1–3 and requires consistent actions worldwide. Indeed, multidrug-resistant bacterial strains, include Gram-positive bacteria methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant Staphylococcus epidermidis (MRSE), Vancomycin-Resistant Enterococci (VRE), and Gram-negative bacteria Escherichia coli (E. Coli), Klebsiella pneumoniae (KP), and Pseudomonas aeruginosa (PA), have emerged to be the major cause of hospital and community-acquired infections.4, 5 As such, novel antibiotics that inhibit a panel of multidrug-resistant bacteria is in an urgent need.6–8
In recent decade, host-defense peptides (HDPs) have emerged as an alternative approach to combat bacteria resistance.9–11 Conventional antibiotics are known to target specific membrane or intracellular components of bacteria, however, HDPs preferentially interact with negatively charged bacterial membranes due to the intrinsic difference between bacterial and mammalian cell membranes,4, 12, 13 leading to the destruction of membrane integrity and bacterial cell death. The antimicrobial mechanisms of HDPs are complex, and a few models including barrel stave, carpet, toroidal pores have been proposed.4, 14 Nonetheless, all potential membrane-disrupting mechanisms could reduce the risk of resistance development15 as these interactions with bacteria are based on physical charge-charge interaction and lack defined membrane targets. Such mechanisms also confer HDPs with broad-spectrum bactericidal activity.4, 16 It should be noted that some HDPs do have defined intracellular targets besides their membrane- disruptive activity, nonetheless, these combined mechanisms of actions could indeed further synergize their antimicrobial activity.13 Despite enthusiasm, there are obstacles associated with antibiotic HDPs and HDP-mimicking oligomeric peptidomimetics,17–19 including difficulty in scale-up, low cost-effectiveness, potential immunogenicity and systematic toxicity. Therefore, recently there has been considerable interest in the search of small molecules which mimic mechanism of action of HDPs.16, 20–22
Bis-guanidine related compounds such as hexamidine have been used as antiseptics and disinfectant in past decades.23, 24 Recently it is suggested that cyclic guanidine compounds may be more potent antibacterial agents than linear guanidines, possibly due to their stronger electrostatic interaction with negatively charged bacterial membranes.7 For instance, a series of bis-cyclic guanidine compounds were recently obtained from combinatorial libraries and showed broad-spectrum antibacterial activity, however, their structures lack symmetry and rational design, and thus could face challenge in further optimization.25 Building upon these studies, we anticipated that bis-guanidine compounds bearing amphipathic structures could mimic mechanism of action of HDPs. Indeed, amphipathic xanthone derivatives bearing bis-arginine moieties recently demonstrated enhanced membrane selectivity, although they showed potent antimicrobial activity only against Gram-positive bacteria.26, 27 Brilacidin, a symmetric bis-guanidine investigational new drug candidate also designed to mimic the mechanism of action of HDPs, possesses an amphipathic structure to replicate the innate function of HDPs.28 As such, we envisioned that bis-cyclic guanidine compounds could be rationally designed to adopt cationic amphipathic structures, and thus capable of mimicking HDPs and revive as a promising approach to combat bacterial resistance.
Compared with HDPs which have large molecular weight (MW 1500‒3000 Da) and multiple cationic charges and hydrophobic groups, these small bis-cyclic guanidines could be facilely accessed and scaled-up without compromising antimicrobial activity and potential of combating drug resistance, thus they are envisioned to be more promising in antibiotic therapeutic development. We hypothesized that if bis-cyclic guanidine compounds are endowed with the ability of bacterial membrane action by incorporating hydrophobic residues, their overall structures would be amphipathic with positive charges. Therefore, they could interact with bacterial membrane effectively.
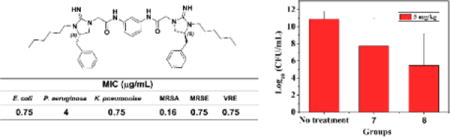
As shown above, the amphipathic bis-cyclic guanidine compounds could be conveniently designed by using a linker to dimerize the five-membered cyclic guanidine moiety bearing different lipophilicities. The molecules were synthesized in a straightforward manner (Scheme S1), allowing future optimization and development of this class of compounds at ease. To this end, a new series of bis-cyclic guanidine compounds (MW 600‒900 Da) were synthesized (Figure 2), and tested against a panel of multidrug resistant bacteria (Table 1).16 As expected, some compounds showed exceptional in vitro and in vivo activity. When R1 was kept as the phenyl group and R2 was just proton (no substituent), no activity was detected for 1 and 2 with aliphatic (C4H8) or aromatic (m-phenylene) linker under the tested condition. We reasoned that compounds without hydrophobic group on cyclic guanidine do not lead to strong hydrophobic interaction with bacteria membranes, even although they could reach on the surface of negatively charged bacteria through electrostatic interactions. We thus hypothesized that appending hydrophobic groups onto the cyclic guanidine ring would enhance the interaction of the compounds to associate with bacterial membranes. As anticipated, bearing an ethyl group on the cyclic guanidine ring, compounds 3 and 4 started to show excellent activity against Gram-negative bacteria E. coli with MICs of 1.5 and 3 μg/mL, respectively.
Fig. 2.
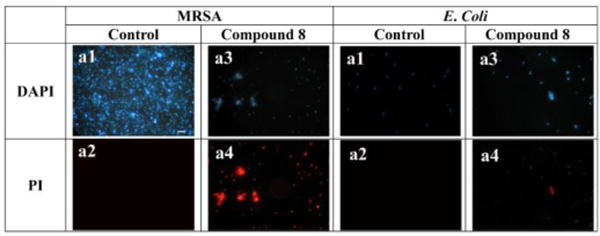
Fluorescence micrographs of MRSA and E. coli that were treated or not treated with 10 µg/mL of 8 for 2 h. a1, control, no treatment, DAPI stained; a2, control, no treatment, PI stained. a3, treatment with 8, DAPI stained; a4, treatment with 8, PI stained. Scale bar = 10 µm.
Table 1.
The antibacterial activity of compounds 1–9.
| Cpd | MIC (µg/mL)
|
HC50(µg/mL) | AI[b] | |||||
|---|---|---|---|---|---|---|---|---|
| Gram Positive | Gram Negative | |||||||
|
| ||||||||
| MRSA | MRSE | VRE | E. Coli | PA | KP | |||
| 1 | >25 | ND[a] | ND | >25 | ND | ND | ND | ND |
| 2 | >25 | ND | ND | >25 | ND | ND | ND | ND |
| 3 | 12 | >25 | >25 | 1.5 | >25 | >25 | ND | ND |
| 4 | 12 | >25 | >25 | 3.0 | >25 | >25 | ND | ND |
| 5 | 0.33 | 0.75 | 0.75 | 1.5 | 12 | 3.0 | 140 | 424 |
| 6 | 3.0 | 1.5 | 1.5 | 3.0 | 25 | 3.0 | >250 | >83 |
| 7 | 0.33 | 0.75 | 0.75 | 1.5 | 6.0 | 1.5 | >250 | >769 |
| 8 | 0.16 | 0.75 | 0.75 | 0.75 | 4.0 | 0.75 | >250 | >1534 |
| 9 | >25 | ND | ND | 3.0 | ND | ND | ND | ND |
| D[c] | 0.5 | 0.5 | 1.0 | >25 | >25 | >25 | ||
| C[d] | >25 | >25 | >25 | 0.5 | 1.0 | 0.5 | ||
ND, Not determined because compounds are not active.
AI, Activity Index, determined by HC50/MICMRSA.
D, Daptomycin, is included for comparison against gram-(+) bacteria.
C, Colistin, was included in the test as the positive control against gram-(−) bacteria.
Replacement of ethyl group in 3 and 4 with the 3-phenylpropyl group led to compounds 5 and 6, respectively. Intriguingly, with more hydrophobic and longer chains which were expected to better span the phospholipid bilayer, both 5 and 6 displayed exciting antibacterial activity with MICs less than 3.0 μg/mL for Gram-negative bacteria (except for P. aeruginosa), and less than 3.0 μg/mL against Gram-positive bacteria. It is particularly noted that the MIC of compound 5 was as potent as 0.33 μg/mL, which is better or at least comparable to any known bis-cyclic guanidine compounds. It also seemed p-phenylene and m-phenylene spacers do not impact activity intensively, as 3 and 4, and 5 and 6 exhibited similar activity.
The subsequent studies revealed that the aliphatic chain C6H13 as the R2 group (Table 1) could further enhance the antimicrobial activity. Both 7 and 8 exhibited potent and broad-spectrum activity against all tested bacterial strains. It was very encouraging that compound 8 had remarkable MICs of 0.75 μg/mL toward most strains, and MIC of 0.16 μg/mL against MRSA. In addition, both compound 7 and 8 were very selective, as their hemolytic activity are all more than 250 μg/mL, which demonstrated 769 and 1534 folds of selectivity against MIC values of MRSA, respectively. It is known that daptomycin29 and colistin30 are last-resort antibiotics and active against Gram-positive and Gram-negative bacteria, respectively. Compound 8 had almost same or even better activity against Gram-positive bacteria compared with daptomycin, meanwhile showed comparable activity against Gram-negative bacteria compared with colistin, suggesting its promising therapeutic potential. Further attempts using more hydrophobic 2-adamantylethyl (compound 9) as the R2 group did not yield compounds with more potent and broad-spectrum activity.
To test the hypothesis that our compounds could possess the mechanism of action of HDPs and interact with bacterial membranes, fluorescence microscopic studies were conducted to evaluate the ability of most potent compound 8 to compromise membranes of E. coli (Gram-negative bacterium) and S. aureus (Gram-positive bacterium). As well known, 4',6-diamidino-2-phenylindole (DAPI) could stain membranes of bacterial cells with blue fluorescence regardless of cell viability, whereas the red fluorescence of propidium iodide (PI) due to the DNA intercalation could only be observed in the presence of impaired membranes. As shown in Fig. 2, treatment of 8 gave rise to the red fluorescence under the PI channel in both S. aureus and E. coli groups, indicating that the membranes of these bacteria were disrupted. Moreover, significant aggregation of S. aureus occurred in the presence of 8, probably due to the loss of membrane potential upon membrane leakage.18 The amount of bacteria were significantly in the presence of 8, possible due to strong antimicrobial activity of 8.
We next conducted time-kill studies to investigate the bacterial killing kinetics of compounds 7 and 8 by using E. Coli. Both compounds could eradicate E. Coli very rapidly, as shown in Figure 3a and 3b. At the concentration of 12.5 or 25 μg/mL, compound 7 could completely remove all bacteria in 2 h, and at 50 μg/mL it could eradicate bacteria in just 0.5 h (Fig. 3a). Notably, Compound 8 is more potent as it could rapidly kill E. Coli completely in 0.5 h even at the concentration of 12.5 μg/mL (Fig. 3b). These data suggested that 7 and 8 could kill bacteria in a similar way to that of HDPs.
Fig. 3.
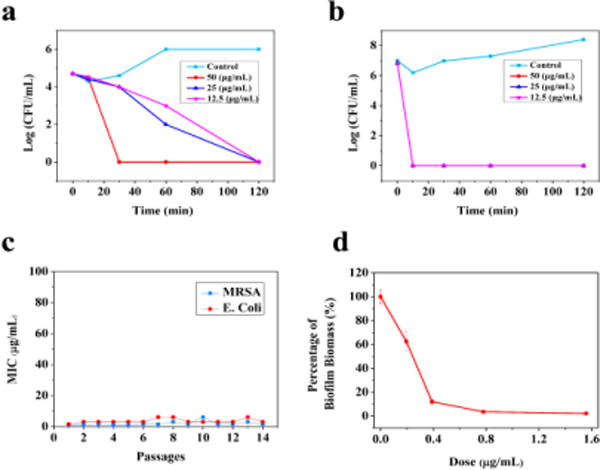
Time-kill curves of 7 (a) and 8 (b) for E. Coli. The killing activity was monitored for the first 2 h. (c) Drug-resistance study of 8 toward MRSA and E. Coli. (d) Biological activity of 8 on inhibition of biofilm by E. Coli.
One of the biggest concerns of current antibiotics is the bacterial resistance as described vide supra, while HDPs take advantage of no immediate drug resistance in bacteria due to lack of defined molecular targets as they disintegrate bacterial membranes. To assess the potential emergence of bacterial resistance toward these cyclic guanidines, the compound 8 was employed for further investigation. The compound was incubated with either MRSA or E. Coli in the well at the concentration of half-MIC every day and tested for their activity through 14 successive passages. As shown in Fig. 3c, MICs of 8 were virtually constant after 14 passages, indicating that they do not readily induce drug resistance in both MRSA and E. Coli. These outcomes suggest that our bicyclic guanidine compounds are not vulnerable in developing drug resistance, which is analogous to the mechanism of action of HDPs.
The bacterial biofilm is another severe problem because bacteria in biofilm generally tolerate antibiotic treatment and thus more difficult to eradicate than planktonic cells.31, 32 Moreover, the bacterial biofilm infection could contaminate medical devices,33 therefore, organ catheters and implants coated with biofilm-inhibiting antibacterial agents are needed as effective therapeutic methods.34 Biofilms formed by MRSA and E. coli have frustrated the treatment of persistent bacterial infections.35, 36 We thus sought to evaluate the compound 8 for its ability to inhibit biofilm formation of MRSA and E. coli. It is known that low concentrations of antibiotic agents should be used to study their anti-biofilm activity. As shown in Fig. 3d, at 0.19 μg/mL, 8 could inhibit 38% of biofilm formation of E. coli. At the concentration of 0.39 μg/mL, 8 could eradicate almost 90% of biofilm formation of E. coli. This analysis revealed that the bis-cyclic guanidine compound is an efficient biofilm formation inhibitor.
The development of membrane-active antibacterial peptides for treatment of bacterial infections has been suffered from difficulties with systematic toxicity and tissue distribution, thus few compounds have been reported with in vivo activity and advanced into clinical trials.37–39 We envisioned that as small molecules, our bis-cyclic guanidines may possess better therapeutic potential. The thigh burden model is a widely used animal model for evaluating preclinical antimicrobial activity of compounds.17, 40 We thus employed the thigh burden model to evaluate the in vivo anti-infective activity of compounds 7 and 8, in which the thigh muscle of neutropenic mice was inoculated with S. aureus, followed by intravenous (i.v.) administration of corresponding compounds. As shown in Fig. 4, significant activity was observed for both compounds at dose of 5 mg/kg when administered twice with a 6-hour interval between injections. A 3-log10 decrease in colony-forming unit (CFU) was observed for compound 7, while more significant decrease (5-log10 CFU) was observed for compound 8, indicating that compound 8 has better efficacy. The result suggested that our compounds provided significant protection against infection with S. aureus. Survival experiments in the future could provide more information.
Fig. 4.
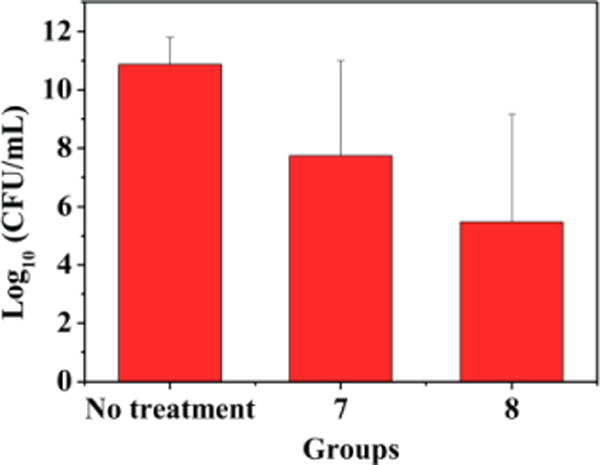
In vivo efficacy of the compounds 7 and 8 in thigh-infection mouse model. Neutropenic mice (n = 4 per group) were inoculated in the posterior thigh muscles with S. aureus ATCC 33591 at 1 × 106 CFU per thigh and then treated with 7 and 8 (5 mg/kg per dose) by i.v. bolus injection in the tail vein at 1 and 7 h after infection.
In summary, we have developed a new class of bis-cyclic guanidine-based small molecules (MW 600‒900 Da) starting from simply α-Phenylalanine. These molecules exhibit remarkable potency against a panel of multidrug-resistant Gram-positive and Gram-negative bacteria. Although other antimicrobial mechanisms cannot be excluded, our studies suggest that these compounds could kill bacteria rapidly by disrupting bacterial membranes, a mechanism analogous to that of HDPs. This is consistent to their amphipathic structures, with the one having proper balance of hydrophobicity and cationic charge showing the most potent antibacterial activity. The hypothesis is further supported by the fact that the susceptibility of MRSA bacteria to the lead compounds remained nearly unchanged even after 14 passages. Furthermore, antibiotic therapeutic potential of these molecules was confirmed in the MRSA-infected thigh burden mouse model. Our work illustrated the potential of bis-cyclic guanidines for the development of potent antimicrobial molecules with molecular masses in the range of 600‒900. Further studies on optimization of activity and selectivity, as well pharmacokinetic assessments are underway in our lab.
Supplementary Material
Fig. 1.
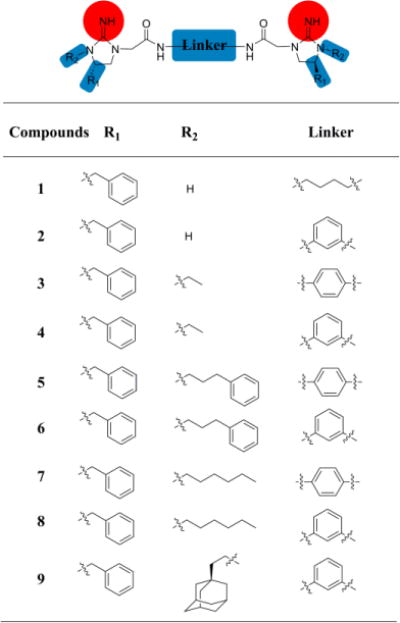
The structures of compounds 1–9.
Acknowledgments
This work was supported by NSF CAREER 1351265 and NIH 1R01GM112652-01A1. This work has been supported in part by the Mass Spectrometry and Peptide Facility, Department of Chemistry, University of South Florida.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Levy SB, Marshall B. Nat Med. 2004;122 doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 2.Doyle MP, Loneragan GH, Scott HM, Singer RS. Compr Rev Food Sci Food Saf. 2013;12:234. [Google Scholar]
- 3.Niu Y, Wang RE, Wu H, Cai J. Future Med Chem. 2012;4:1853. doi: 10.4155/fmc.12.111. [DOI] [PubMed] [Google Scholar]
- 4.Marr AK, Gooderham WJ, Hancock RE. Curr Opin Pharmacol. 2006;6:468. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 5.WHO. Antimicrobial resistance: global report on surveillance. 2014 [Google Scholar]
- 6.O’Daniel PI, Peng Z, Pi H, Testero SA, Ding D, Spink E, Leemans E, Boudreau MA, Yamaguchi T, Schroeder VA, Wolter WR, Llarrull LI, Song W, Lastochkin E, Kumarasiri M, Antunes NT, Espahbodi M, Lichtenwalter K, Suckow MA, Vakulenko S, Mobashery S, Chang M. J Am Chem Soc. 2014;136:3664. doi: 10.1021/ja500053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K. Nature. 2015;517:455. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoganathan S, Miller SJ. J Med Chem. 2015;58:2367. doi: 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown KL, Hancock REW. Curr Opin Immunol. 2006;18:24. doi: 10.1016/j.coi.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Hancock RE, Sahl HG. Nat Biotechnol. 2006;24:1551. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 11.Mercer DK, O’Neil DA. Future Med Chem. 2013;5:315. doi: 10.4155/fmc.12.213. [DOI] [PubMed] [Google Scholar]
- 12.Hancock REW, Brown KL, Mookherjee N. Immunobiology. 2006;211:315. doi: 10.1016/j.imbio.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Matsuzaki K. Biochimi Biophys ACTA Biomembr. 2009;1788:1687. doi: 10.1016/j.bbamem.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Sengupta D, Leontiadou H, Mark AE, Marrink SJ. Biochim Biophys Acta. 2008;1778:2308. doi: 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Tew GN, Scott RW, Klein ML, DeGrado WF. Acc Chem Res. 2010;43:30. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng P, Huo D, Nimmagadda A, Wu J, She F, Su M, Lin X, Yan J, Cao A, Xi C, Hu Y, Cai J. J Med Chem. 2016;59:7877. doi: 10.1021/acs.jmedchem.6b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. Proc Natl Acad Sci USA. 2009;106:6968. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu Y, Padhee S, Wu H, Bai G, Qiao Q, Hu Y, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. J Med Chem. 2012;55:4003. doi: 10.1021/jm300274p. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Wu H, Teng P, Bai G, Lin X, Zuo X, Cao C, Cai J. J Med Chem. 2015;58:4802. doi: 10.1021/acs.jmedchem.5b00537. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh C, Manjunath GB, Akkapeddi P, Yarlagadda V, Hoque J, Uppu DS, Konai MM, Haldar J. J Med Chem. 2014;57:1428. doi: 10.1021/jm401680a. [DOI] [PubMed] [Google Scholar]
- 21.Mensa B, Kim YH, Choi S, Scott R, Caputo GA, DeGrado WF. Antimicrob Agents Chemother. 2011;55:5043. doi: 10.1128/AAC.05009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Pachaiyappan B, Gruber JD, Schmidt MG, Zhang YM, Woster PM. J Med Chem. 2016;59:3140. doi: 10.1021/acs.jmedchem.5b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raulji CM, Clay K, Velasco C, Yu LC. J Pediatr Hematol Oncol. 2015;32:315. doi: 10.3109/08880018.2015.1013588. [DOI] [PubMed] [Google Scholar]
- 24.Grare M, Dibama HM, Lafosse S, Ribon A, Mourer M, Regnouf-de-Vains JB, Finance C, Duval RE. Clin Microbiol Infect. 2010;16:432. doi: 10.1111/j.1469-0691.2009.02837.x. [DOI] [PubMed] [Google Scholar]
- 25.Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina-Franco JL, Giulianotti MA, Houghten RA, Shaw LN. J Med Chem. 2015;58:3340. doi: 10.1021/jm501628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin S, Koh JJ, Aung TT, Lim F, Li J, Zou H, Wang L, Lakshminarayanan R, Verma C, Wang Y, Tan DT, Cao D, Beuerman RW, Ren L, Liu S. J Med Chem. 2017;60:1362. doi: 10.1021/acs.jmedchem.6b01403. [DOI] [PubMed] [Google Scholar]
- 27.Koh JJ, Lin S, Aung TT, Lim F, Zou H, Bai Y, Li J, Lin H, Pang LM, Koh WL, Salleh SM, Lakshminarayanan R, Zhou L, Qiu S, Pervushin K, Verma C, Tan DT, Cao D, Liu S, Beuerman RW. J Med Chem. 2015;58:739. doi: 10.1021/jm501285x. [DOI] [PubMed] [Google Scholar]
- 28.Kowalski RP, Romanowski EG, Yates KA, Mah FS. J Ocul Pharmacol Ther. 2016;32:23. doi: 10.1089/jop.2015.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weis F, Beiras-Fernandez A, Schelling G. Curr Opin Investig Drugs. 2008;9:879. [PubMed] [Google Scholar]
- 30.Yahav D, Farbman L, Leibovici L, Paul M. Clin Microbiol Infect. 2012;18:18. doi: 10.1111/j.1469-0691.2011.03734.x. [DOI] [PubMed] [Google Scholar]
- 31.Davies D. Nat Rev Drug Discov. 2003;2:114. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- 32.Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Nat Rev Microbiol. 2016;14:563. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 33.Padhee S, Li Y, Cai J. Bioorg Med Chem Lett. 2015;25:2565–2569. doi: 10.1016/j.bmcl.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 34.Bjarnsholt T, Ciofu O, Molin S, Givskov M, Hoiby N. Nat Rev Drug Discov. 2013;12:791. doi: 10.1038/nrd4000. [DOI] [PubMed] [Google Scholar]
- 35.Brauner A, Fridman O, Gefen O, Balaban NQ. Nat Rev Microbiol. 2016;14:320. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 36.Park SR, Tripathi A, Wu J, Schultz PJ, Yim I, McQuade TJ, Yu F, Arevang CJ, Mensah AY, Tamayo-Castillo G, Xi C, Sherman DH. Nat Commun. 2016;7:10710. doi: 10.1038/ncomms10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 38.Vallon-Eberhard A, Makovitzki A, Beauvais A, Latgé JP, Jung S, Shai Y. Antimicrob Agents Chemother. 2008;52:3118. doi: 10.1128/AAC.00526-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radzishevsky IS, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A. Nat Biotechnol. 2007;25:657. doi: 10.1038/nbt1309. [DOI] [PubMed] [Google Scholar]
- 40.Andes D, van Ogtrop ML, Peng J, Craig WA. Antimicrob Agents Chemother. 2002;46:3484. doi: 10.1128/AAC.46.11.3484-3489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.