

Abstract
Purpose
Current clinical parameters do not stratify indolent from aggressive prostate cancer (PCa). Aggressive PCa, defined by the progression from localized disease to metastasis, is responsible for the majority of PCa-associated mortality. Recent gene expression profiling has proven successful in predicting the outcome of PCa patients, however they have yet to provide targeted therapy approaches that could inhibit a patient’s progression to metastatic disease.
Experimental Design
We have interrogated a total of seven primary PCa cohorts (N = 1,900), two metastatic castration resistant PCa datasets (N = 293) and one prospective cohort (N = 1,385) to assess the impact of TOP2A and EZH2 expression on PCa cellular program and patient outcomes. We also performed immunohistochemical staining for TOP2A and EZH2 in a cohort of primary PCa patients (N = 89) with known outcome. Finally, we explored the therapeutic potential of a combination therapy targeting both TOP2A and EZH2 using novel PCa-derived murine cell lines.
Results
We demonstrate by genome-wide analysis of independent primary and metastatic PCa datasets that concurrent TOP2A and EZH2 mRNA and protein up-regulation selected for a subgroup of primary and metastatic patients with more aggressive disease and notable overlap of genes involved in mitotic regulation. Importantly, TOP2A and EZH2 in PCa cells act as key driving oncogenes, a fact highlighted by sensitivity to combination-targeted therapy.
Conclusions
Overall, our data supports further assessment of TOP2A and EZH2 as biomarkers for early identification of patients with increased metastatic potential that may benefit from adjuvant or neo-adjuvant targeted therapy approaches.
Introduction
Aggressive prostate cancer (PCa), as defined by the progression to metastatic disease after therapy for localized disease accounts for almost two thirds of prostate cancer deaths (1). Therefore, the need to more accurately identify lethal PCa, in an effort to personalize medicine for those in need, has led to a large-scale push for biomarker development in the field (2–4). With this, it is important to recognize, at the earliest time point, which patients are at a higher risk for developing aggressive disease and intercept their disease progression with appropriate therapies. We have previously demonstrated that combination therapy using an EZH2 inhibitor with the TOP2 poison etoposide, resulted in significantly increased therapeutic efficacy in models of lethal PCa associated with enhanced DNA damage (5). While each of these genes were independently associated with metastatic PCa (6–9), they have never been studied together in the context of early detection of patients with PCa that will relapse after localized therapy with curative intent.
The systematic study of TOP2A and EZH2 mRNA expression in primary PCa samples from two publicly available datasets (10,11) allowed us to confirm that overexpression of these two genes can select a subgroup of high-risk patients with a decreased time to biochemical recurrence and enriched for a mitotic gene signatures. Interrogation of additional datasets also revealed that PCa patients with high expression of both TOP2A and EZH2 mRNA and/or protein were more likely to progress to a metastatic disease and even die of their disease. In support, PCa metastatic datasets (12,13) also demonstrated that patients with increased TOP2A and EZH2 mRNA expression maintained enrichment of mitotic related gene signatures, suggesting that indeed TOP2A and EZH2 could provide early detection of primary tumors with metastatic potential. Altogether, our study supports the usefulness of TOP2A and EZH2 as valuable prognostic biomarkers to identify patients that are more likely to develop an aggressive, metastatic disease. Finally, using a novel cell line derived from a highly aggressive genetically engineered mouse models (GEMM) of PCa, we provide the rational that a targeted combination therapy against TOP2A and EZH2 in a subset of patients may have the potential to intercept progression to metastatic disease in the context of neo-adjuvant or adjuvant clinical trials.
Materials and Methods
Differentially Expressed Genes Analysis
Gene expression data were analyzed using Bioconductor 3.1 (http://bioconductor.org), running on R 3.1.3. To identify significant differences in gene expression in TOP2A+/EZH2+ and Other patients, moderated Student’s t-tests were performed using empirical Bayes statistics in the “Limma” package, and the resulting P-values were adjusted for multiple testing using the false discovery rate (FDR) Benjamini and Hochberg method; probe sets with adjusted P value FDR q < 0.05, and logFC > 1.5, were called differentially expressed (DEG).
Primary Prostate Cancer Analysis
Data from The Cancer Genome Atlas (10) was collected from multiple institutes from primary prostatectomies of patients with PCa. Samples then underwent RNA-sequencing on the Illumina HiSeq 2500 platform. Patient data (497 samples) was acquired as RSEM “counts” gene expression data from Firehose, a Broad Institute Software, with 52 matched normal samples. Data from The Memorial Sloan Kettering Cancer Center (11) set was collected from a single institute from primary prostatectomies of patients with PCa. Samples then underwent microarray analysis on the Affymetrix Human Exon 1.0 ST Array platform. Patient data (131 samples) was acquired as log2 transformed gene expression data from the Gene Set Omnibus (GEO) with 29 matched normal samples. Expression data for TOP2A and EZH2 was isolated for each patient separately, then split into high or low expression based on z-scores over normal expression, with the designations High as z > 1.5 and Other z < 1.5. Distributions for TOP2A and EZH2 were then overlapped, with patients in the high for both TOP2A and EZH2 being designated as TOP2A+/EZH2+ and all other samples being relegated to the “Other” groups. Samples were then pre-processed, by removing all genes consisting of read outs of 0 for more than 80% of samples. The data underwent scale normalization using the “Limma” package in R statistical software, and were then Voom Transformed. DEGs were isolated as described above. Those genes were then utilized to generate a scaled heatmap, constructed using the “Gplots” and “Heatmap.2” packages in R. Euclidian distances and hierarchal clustering was applied using “h.clust”, and further Principal Component Analysis was later conducted utilizing the “PrComp” package in R, to determine how similar samples were to one another.
Localized Prostate Cancer from Decipher GRID analysis
We used a total of 3,565 genome-wide expression profiles from PCa RP tissues from the Decipher Genomic Ressource Information Database (GRID) (Supplement Table 1). Expression profiling, specimen selection, RNA extraction and microarray hybridization were done in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory facility (GenomeDx Biosciences, San Diego, CA, USA) as previously described (14). Briefly, total RNA was extracted and purified using the RNeasy FFPE kit (Qiagen, Valencia, CA). RNA was amplified and labeled using the Ovation WTA FFPE system (NuGen, San Carlos, CA) and hybridized to Human Exon 1.0 ST GeneChips (Affymetrix, Santa Clara, CA). Quality control was performed using Affymetrix Power Tools and normalization was performed using the Single Channel Array Normalization (SCAN) algorithm (15). For this analyses, patients with expression of TOP2A and/or EZH2 above the 75th percentile in the cohort were categorized as TOP2A+ and/or EZH2+. To test associations between TOP2A expression, EZH2 expression and outcomes, we grouped patients into TOP2A+/EZH2+ (expression of both genes is greater than the 75th percentile), TOP2A−/EZH2− (expression of both genes is less than the 75th percentile) or patients with the expression of either gene greater than the 75th percentile (TOP2A+ or EZH2+). Then we conducted survival analysis using the Kaplan Meier method, with case-cohort reweighting when appropriate, across 4 case-cohort/cohort studies (Mayo Clinic Validation, N = 232; Thomas Jefferson University, N = 133; Johns Hopkins University post-RP, N = 262, Johns Hopkins University post-BCR, N = 213) to assess the prognostic impact of the four groups to predict metastasis outcome after RP or prostate cancer specific mortality (PCSM) if the data was available. P-values were calculated with the log-rank test. We also conducted univariate and multivariate analyses to associate subtypes with clinical outcome after adjusting for other clinicopathological variables including Gleason score, lymph node invasion, surgical margins, extracapsular extension, seminal vesicle invasion and pre-operative prostate specific antigen (PSA) levels in the Mayo Clinic Discovery (N = 545) case control cohort and a “Meta Cohort” from which we utilized genome-wide expression profiles of 751 patients with metastatic outcome follow-up from the Decipher GRID. These patients were pooled from four studies of either case-cohort or cohort design. Patients for these studies came from four institutes: John Hopkins University post-RP (N = 260)(16), Mayo II (N = 235)(17), Thomas Jefferson University (N = 139)(18) and Durham VA (N = 117)(19). A total of 120 non-randomly selected patients from case-cohort studies were removed before pooling the studies to avoid bias in estimating the hazard ratio. 631 patients were thus eligible for analysis, 70 of which developed metastasis. Median follow-up time for censored patients was 8 years and the median age at radical prostatectomy was 61 years. Finally, we used 2,293 prospective samples to associate the expression of TOP2A and EZH2 with metastatic outcome. But since we don’t have metastasis outcome, we used the Decipher test risk stratification as a surrogate for metastasis given the well validated evidence that Decipher is the strongest predictor of metastasis currently available (20).
Metastatic Prostate Cancer Analysis
Data from the Robinson et al. metastatic castration resistant prostate cancer (mCRPC) dataset was collected from 6 different institutes from metastatic lesions in patients diagnosed with prostate cancer (13). Samples then underwent RNA-sequencing on the Illumina HiSeq 2500 platform and patient data (N = 229) was acquired as transcripts per million (TPM). Data from Kumar et al. mCRPC dataset was collected from a single institute from metastatic lesions in patients diagnosed with prostate cancer (12). Samples then underwent microarray analysis on the Agilent-016162 PEDB Whole Human Genome Microarray 4×44K platform. Patient data (154 samples from 64 patients) was acquired from the Gene Set Omnibus (GEO) using “GEO2R” as log2 transformed values. Expression data for TOP2A and EZH2 was isolated for each patient separately, then split into quartiles with the designations high, intermediate high, intermediate low, and low. Quartile distributions for TOP2A and EZH2 were then overlapped, with patients in the highest quartile for both TOP2A and EZH2 being designated as TOP2A+/EZH2+ and all other samples being relegated to the “Other” group. Samples were then pre-processed, by removing all genes consisting of read outs of 0 for more than 80% of samples. The data underwent scale normalization using the “Limma” package in R statistical software, and were then Voom Transformed. DEGs were isolated as described above. Those genes were then utilized to generate a scaled heatmap, constructed using the “Gplots” and “Heatmap.2” packages in R. Euclidian distances and hierarchal clustering was applied using “h.clust”, and further Principal Component Analysis was later conducted utilizing the “PrComp” package in R, to determine how similar samples were to one another.
In silico Gene Set Enrichment Analysis
A rank list for each dataset was constructed by taking the gene name and the log2 fold change value for each of the DEGs in each set. The Gene Set Enrichment Analysis (GSEA) tool (http://www.broadinstitute.org/-gsea) was then used to analyze relationship of existing gene expression profiles (Hallmarks and Oncogenic Signatures) in the Molecular Signature Database (MSigDB) with the rank list generated. The GseaPreranked tool, with 1000 permutations, and a failure to collapse dataset to gene symbols, was used for GSEA analysis. All statistically significant data (P < 0.05 and FDR < 0.15) is provided (Supplement Tables 2 and 3). We acknowledge our use of the gene set enrichment analysis, GSEA software, and Molecular Signature Database (MSigDB; http://www.broad.mit.edu/gsea/) (21).
Tissue Microarray analysis of TOP2A and EZH2 (DFCI cohort)
Patients and Samples
A retrospective cohort of prostate cancer patients from a treatment facility of Dana Farber Cancer Institute (DFCI) Prostate Clinical Research Information Systems (CRIS) database was identified to include patients who had tissues available from radical prostatectomy (RP) or transurethral resections of the prostate (TURP) with 3 pre-defined tumor micro-array (TMAs). The IHC staining was performed and a multiplexed tyramide signal amplification method was performed on 4-μm sections of the TMA for detection of TOP2A and EZH2 proteins. TSA-plus fluorescence immunohistochemistry combined with spectral imaging was used to measure TOP2A and EZH2 expressions/cell positivity. The analysis cohort included 89 patient samples after quality control to ensure for proper assessment of marker values (Supplement Table 4). The analysis endpoints included time to biochemical recurrence (BCR) defined as time from RP to biochemical recurrence and time to lethal disease defined as time from RP to the development of metastases. The distribution of time to BCR or metastases according to TOP2A and EZH2 marker status (low vs high) in the combination 4 groups used Kaplan-Meier method. Cox proportional hazards model was used to assess the associations of time to events and marker status with estimate hazard ratio (HR), 95% CI. The multivariate model adjusted for clinical-pathologic factors of age, Gleason score, and pathological grade was also used. Additional details are provided in the Supplement Material and Methods.
Definition of TOP2A, EZH2 marker values
The case level positivity (= positive T cell #/total T cell #*100 %) for both markers, used a cutoff value on cell intensity readout level to define TOP2A and EZH2 positive cell. According to the cell data distribution, combined with representative images review, the top 90% cell intensity as chosen to be cutoff value for TOP2A and EZH2, respectively. The marker values were then dichotomized into low vs high categories using the pre-specified lower quartile level as cut-off, i.e., TOP2A- if <=lower quartile TOP2A cell positivity (%) or TOP2A+ otherwise; EZH2- if <=lower quartile EZH2 cell positivity (%) or EZH2+ otherwise. Subsequently 4 groups from the both markers were defined as: 1) TOP2A+/EZH2+ (reference group); 2) TOP2A-/EZH2-; 3) EZH2+; 4) TOP2A+.
Analysis of gene and protein expression in sKO and dKO murine prostatic tissues
Total RNA extraction was performed using prostate dorsal and lateral lobes of of Pten−/− (sKO) or Pten−/− ; Rb1−/− (dKO) PCa mouse models (22). Reverse transcription and quantitative real-time PCR (qRT-PCR) were done as previously described (5) and data was normalized according to Rpl32 levels. The oligonucleotides used for the analysis of gene expression were Top2a (Forward: AGGATTCCGCAGTTACGTGG; Reverse: CATGTCTGCCGCCCTTAGAA), Ezh2 (Forward: GCCAGACTGGGAAGAAATCTG; Reverse: TGTGCTGGAAAATCCAAGTCA) and Rpl32 (Forward: TTCCTGGTCCACAACGTCAAG; Reverse: TGTGAGCGATCTCGGCAC). Detection of TOP2A and EZH2 by fluorescence immunochemistry was done on formalin-fixed, paraffin embedded prostate tissues from a sKO and a dKO mouse and performed as described for the tissue microarray (Supplement Material and Methods).
sKO and dKO cell line establishment
A conditional reprogramming method was used to establish cell lines from sKO or dKO PCa mouse models (24). Briefly, tumor samples from the prostate of a 51-week-old sKO mouse or a 38-week-old dKO mouse was chopped into 2-mm fragments followed by digestion in a mixture of Collagenase/Hyaluronidase (Stem Cell Technologies, #7912) at 37 ⁰C for three hours. Cell clumps were then incubated with 0.25% trypsin for 1 hour on ice followed by Dispase (Stem Cell Technologies, #7913) and DNaseI (Sigma, #DN25) for 1 min. Cell suspensions were filtered through a 40-μm cell strainer, and then co-cultured with irradiated Swiss-3T3 feeder layer in F medium containing 10 μM Y-27632 (Enzo Life Sciences, #ALX-270-333) for two months. Epithelial colonies surrounded by irradiated fibroblast were harvested and trypsinized into single cells, plated in a 96-well plate. sKO and dKO cell lines were established from the homogeneous population in one single well and then continuously maintained in F medium (described previously (24)) with the supplement of 10 μM Y-27632 and 1nM R1881. Both cell lines were finally cultured using DMEM containing 10% fetal bovine serum.
Analysis of protein expression
Analysis of protein expression in sKO and dKO cell lines was performed as described previously described (5). Briefly cells were harvested and whole cell lysates was extracted using lysis buffer (PIPES 20 mM, NaCl 150 mM, EGTA 1 mM, MgCl2 1.5 mM, Triton X-100, pH 7.4) supplemented with both protease (Pierce, #88265) and phosphatase inhibitors (Pierce, #88667). Equal amount of denatured protein were resolved on a 4–15% SDS-PAGE Mini-PROTEAN® TGX™ gel (BioRad, #456-1083) and transferred to polyvinylidene difluoride membrane. Membranes were blocked (5% BSA), washed and then probed with the following rabbit monoclonal antibodies according to the manufacturer’s instructions: anti-EZH2 (Cell Signaling, #5246, 1:1000), anti-TOP2A (Abcam, #52934, 1:500), or anti-GAPDH (Cell Signaling, #2118, 1:10000).
Adherent Clonogenics
sKO and dKO were plated in a 6 well plate (5×104 cells/well). Following treatment as indicated with Etoposide (Sigma-Aldrich), EPZ6438, GSK126 (Xcess Biosciences Inc.) or DMSO (vehicle) for 24 or 72 hours, cells were trypsinized and replated either 500 or 1000 cells per well. Colony formation was assessed 10 days post plating by crystal violet staining.
Cell cycle analysis
sKO and dKO cells (5×104 cells/well) were seeded into 6-well plates (BD Bioscience), left to adhere, and treated as indicated. Following treatments, adherent and non-adherent cells were collected and washed in 1x PBS, and fixed in 50% ethanol at 4°C overnight. Cells were stained with propidium iodide solution containing RNase A (Sigma) for 15 minutes at 37°C. DNA content analysis was performed by using a FACS caliber cytometer.
Statistical analysis
Data are displayed as mean ±SEM. Differences were determined using two-tailed unpaired t-tests, using GraphPad Prism software. P values < 0.05 were considered statistically significant.
Results
We performed a cross-platform analysis to stratify high-risk prostate cancer patients (aggressive disease) from low-risk patients (indolent disease) within the primary disease setting. In The Cancer Genome Atlas (TCGA) (10) and Taylor, et al. (11) primary PCa datasets, samples were divided into patients with increased levels of both TOP2A+/EZH2+ and other. Unsupervised hierarchical clustering and principal component analysis (PCA) demonstrated that TOP2A+/EZH2+ primary PCa patients displayed a unique set of differentially expressed genes (DEG) (21) compared to other patients (Figure 1A,B; Supplement Figure 1A, B). In the TCGA dataset, high levels of TOP2A and EZH2 were associated with a shorter time to biochemical recurrence (Figure 1C), a feature that was also observed in the Taylor et al. dataset (Supplement Figure 1C). Comparing those patients’ tumors with increased levels of TOP2A and EZH2 to patients without increased levels of both genes in the TCGA and Taylor et al. datasets generated a DEG list, consisting of 269 and 362 genes respectively, with 86 of those genes shared between both primary tumor datasets (Figure 1D; Supplement Tables 5, 6). Gene Sets Enrichment Analyses (GSEA) using the Hallmark and Oncogenic Signatures gene sets revealed that DEG in patients with concurrent TOP2A+/EZH2+ expression in both datasets enriches for similar cell programs geared toward cellular proliferation (Figure 1E; Supplement Figure 2; Supplement Tables 2, 3). Additional interrogation revealed that TCGA TOP2A+/EZH2+ patients had statistically significant down regulation of CDKN1A, FGFR1 and PMP22, genes that were previously shown to be down regulated in lethal PCa (23) (Supplement Figure 3).
Figure 1. Concurrent TOP2A and EZH2 expression is associated with biochemical recurrence and a mitotic gene signature.
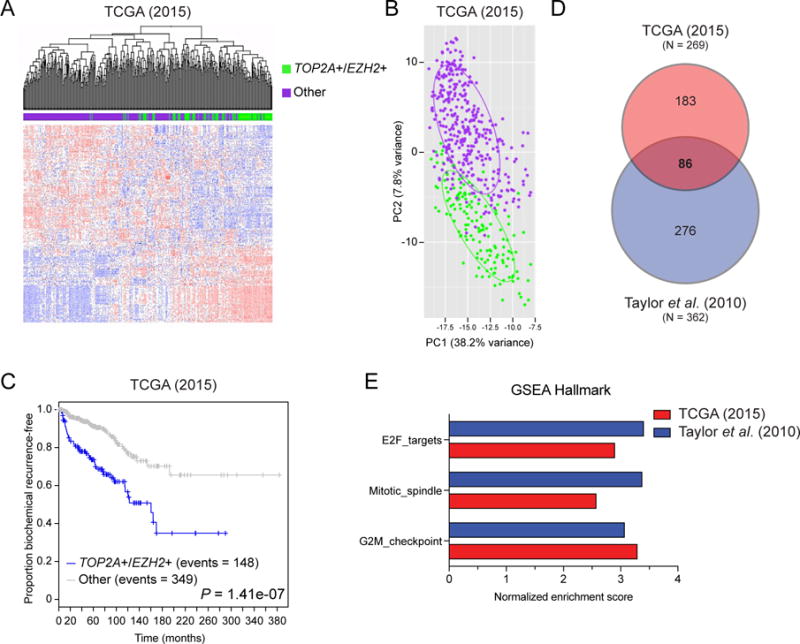
(A) Unsupervised clustering analysis of TCGA primary prostate cancer data demonstrates that patients with concurrent high TOP2A and EZH2 expression (TOP2A+/EZH2+; green) tightly cluster apart from other patients (purple) based on their differentially expressed genes (DEGs). (B) Unique DEGs between TOP2A+/EZH2+ and other patients was validated by principle component analysis (PCA). (C) Kaplan-Meier curves reveals that TOP2A+/EZH2+ patients have a faster progression to biochemical recurrence. (D) Comparison of DEGs in TOP2A+/EZH2+ patients versus other patients in TCGA (2015) and Taylor et al. (2010) independent primary datasets. (E) Gene Set Enrichment Analysis (GSEA) revealed statistically significant overlapping gene signatures involved in mitosis and E2F signaling (P < 0.05 and FDR < 0.15; Supplement Table 2).
Given that high levels of both TOPA2 and EZH2 expression in patients are associated to a poorly differentiated primary disease and a shorter time to biochemical recurrence, we hypothesized that those patients might be more prone to progress to an advance, metastatic disease. To test this hypothesis, we used 5 retrospective radical prostatectomy (RP) cohorts with outcome data (total N = 1,272) and one prospective cohort (N = 2,293) (Supplement Table 1) from the Decipher Genomic Ressource Information Database (GRID). In the Mayo Clinic Validation study (N = 232), a case-cohort consisting of high risk PCa patients (17) with a median follow-up of 7 years, we found that TOP2A+/EZH2+ patients had a faster progression to metastatic disease (Figure 2A). This finding was also confirmed in the Thomas Jefferson University dataset (N = 133), a cohort of PCa patients treated with radiation therapy (18) (Figure 2B) and in two Johns Hopkins University case-cohorts consisting of post-RP patients who received no therapy until the onset of metastasis (N = 262) (16) (Figure 2C) or of patients who had developed biochemical recurrence (post-BCR) and again received no therapy until the onset of metastasis (N = 213) (24) (Figure 2D). However, concurrent TOP2A and EZH2 expression in those datasets was not associated to a shorter time to biochemical recurrence (Supplement Figure S4). Importantly, TOP2A+/EZH2+ patients in both Johns Hopkins University cohorts were also more likely to die of their disease compared to patients with low levels of TOP2A and EZH2 expression (Both Low) as shown by a shorter time to PCa specific mortality (PCSM; Figure 2E, F). Strikingly, univariate and multivariate analyses using the Mayo Clinic Discovery dataset (N = 545) (25) demonstrate that concurrent TOP2A and EZH2 expression had a similar prognostic power than a high Gleason grade (> 8) in predicting the development of an aggressive, metastatic disease and outperformed all other clinicopathological parameters analyzed (Figure 2G, H). Additional univariate and multivariate analyses using a “Meta Cohort” (see the Materials and Methods for details) confirmed the prognostic power of TOP2 and EZH2 expression in predicting PCa progression (Supplement Tables 7 and 8). Finally, using 2,293 prospective RP patients whose PCa agressivness was stratified by the Decipher test, a validated surrogate of metastasis (20), we found that almost all patients with high expression of TOP2A and EZH2 were categorized as high risk of progressing to a metastatic disease (Figure 2I). Moreover, multivariate survival analysis of Decipher adjusting for TOP2A+/EZH2+ and other clinicopathologic risk factors revealed that both Decipher and TOP2A+/EZH2+ are significantly associated with metastasis and contain unique prognostic information (Supplement Table 9). Altogether, we found that high levels of TOP2A and EZH2 expression is consistently associated to the progression to a metastatic and lethal disease.
Figure 2. Concurrent TOP2A and EZH2 expression leads to faster progression to metastasis and prostate cancer specific mortality.

(A, B) Patients with concurrent high TOP2A and EZH2 expression in primary prostatic tumors (TOP2A+/EZH2+) have a shorter time to metastatic progression. (C, D) Progression to metastasis is also significantly faster for TOP2A+/EZH2+ patients who received no therapy after radical prostatectomy (RP) until the onset of metastasis (C) or for patients who develop biochemical recurrence (BR) and received no therapy until the onset of metastasis (D). (E, F) TOP2A+/EZH2+ patients have an increase prostate cancer specific mortality (PCSM) compared to TOP2A−/EZH2− patients. (G, H) Univariate and multivariate analyses revealed that high TOP2A and EZH2 expression is a strong prognostic factor for progression to metastasis compared to standard clinical parameters. (I) Patients with high TOP2A and EZH2 expressiong are mostly classified as high risk patients according to the Decipher test, a surrogate of PCa metastasis (Low N = 815; Intermediate N = 512; High N = 966; R = Pearson’s correlation). (Abbreviations: lymph node invasion, LNI; surgical margins, SM; extracapsular extension, ECE; seminal vesicle invasion, SVI; prostate specific antigen, PSA)
In order to evaluate if TOP2A and EZH2 protein levels could also discriminate patients bearing an indolent from those bearing an aggressive tumor, we performed immunohistochemical (IHC) staining utilizing a tissue microarray (TMA) of primary PCa patients who had a prostatectomy and either did or did not relapse with metastatic disease (Dana-Farber Cancer Institute cohort; Supplement Table 4). TOP2A and EZH2 protein expression was quantified and patients were then classified in one of four categories (TOP2A+/EZH2+, EZH2+, TOP2A+ or TOP2A-/EZH2-; Figure 3A). Kaplan Meier analysis showed that patients with high levels of both proteins had a higher rate and significantly shorter time to BCR and metastatic disease progression (Figure 3B, C). Moving forward, this initial finding requires external validation with a larger sample size, but our data shows that in addition to TOP2A and EZH2 gene expression levels, protein expression levels predict for patients who will develop metastatic/lethal prostate cancer with high accuracy in small tissue samples (Figure 3D).
Figure 3. Patients with concurrent TOP2A and EZH2 expression develop an aggressive, metastatic disease.
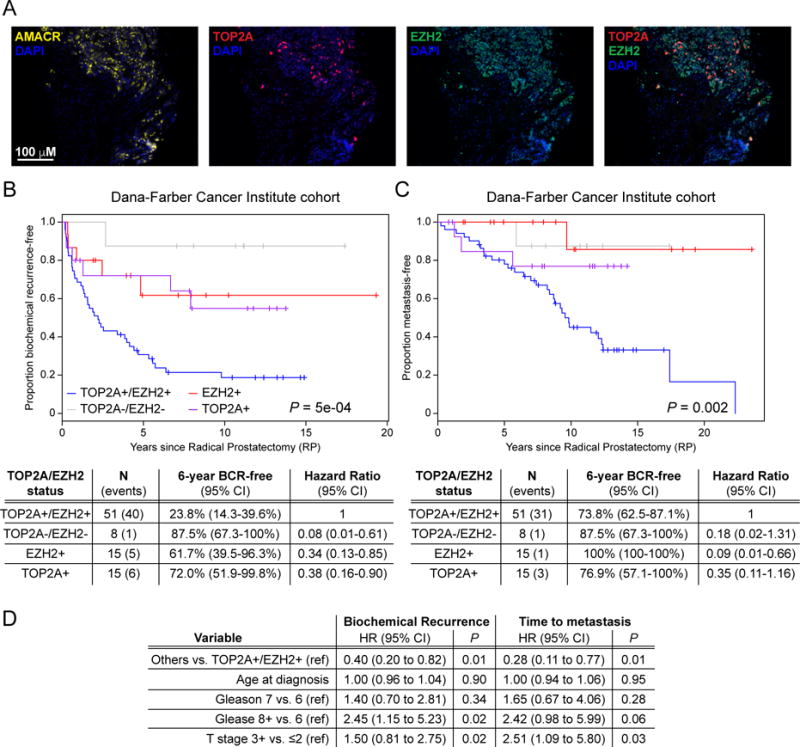
(A) Example of fluorescence immunohistochemistry for alpha-methylacyl-CoA racemase (AMACR; tumor epithelial cells) with 4′,6-diamidino-2-phenylindole (DAPI; cell nucleus), TOP2A and EZH2 on a representative TOP2A+/EZH2+ prostate tumor microarray core. (B, C) High expression of TOP2A and EZH2 proteins (TOP2A+/EZH2+) is associated to decreased time to BCR (B) and to metastatic event (C). (D) Multivariate CoxPH model of assessing the associations of marker groups (Others vs. TOP2A+/EZH2+) adjusting for clinical covariates.
With evidence that expression of TOP2A and EZH2 predict metastatic free survival from primary PCa samples, it was hypothesized that metastatic PCa samples harboring concurrent up-regulation of TOP2A and EZH2 would enrich for similar gene expression sets as identified in our primary PCa analysis. Using the Robinson et al. (13) and Kumar et al. (12) metastatic castration resistant prostate cancer (mCRPC) datasets, we observed that TOP2A+/EZH2+ patients expressed a unique gene signature as assessed by hierarchical clustering and PCA (Figure 4A, B). A generated DEG list, showed a total of 315 and 144 genes driven by high TOP2A/EZH2 expression in the Robinson et al. and the Kumar et al. datasets respectively, with a 107 gene overlap (Figure 4C; Supplement Tables 5, 6). Interestingly, as observed with primary PCa, GSEA using DEG characteristic of TOP2A+/EZH2+ patients revealed a consistent enrichment for gene sets related to mitosis in metastatic tumors (Figure 4D; Supplement Tables 2, 3), a hallmark that has been previously associated with aggressive disease (26,27).
Figure 4. Metastatic tumors with concurrent TOP2A and EZH2 expression maintain a set of differentially expressed genes that overlap with TOP2A+/EZH2+ primary PCa.
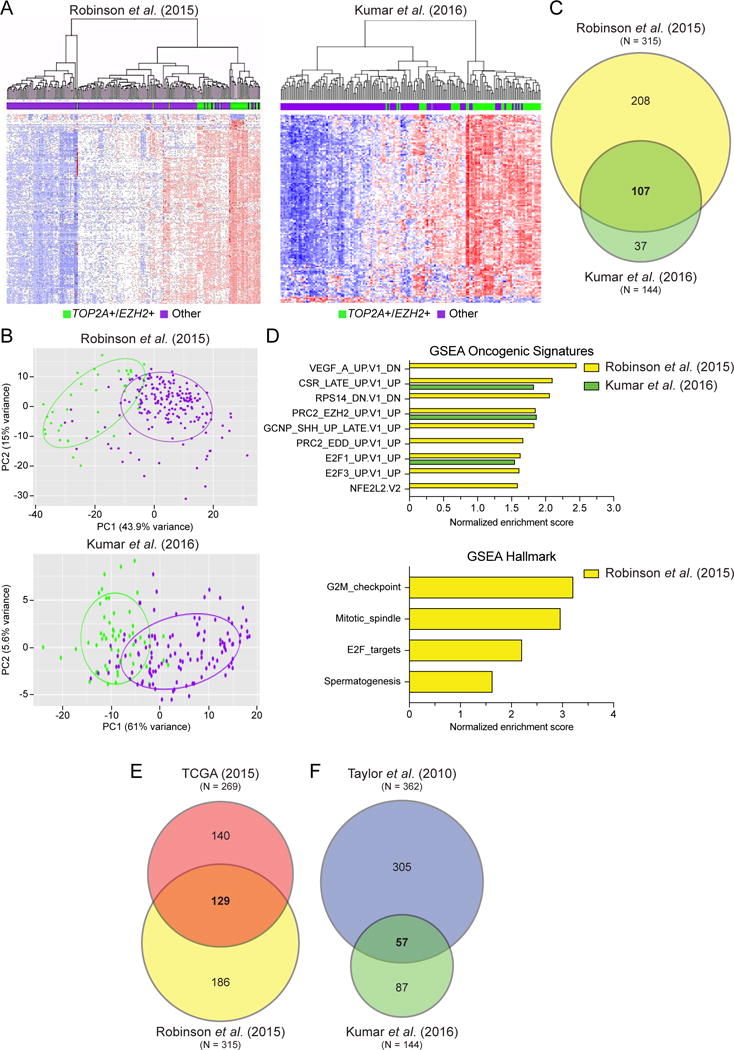
(A) Unsupervised clustering analysis of metastatic PCa data demonstrates that patients with concurrent high TOP2A and EZH2 expression (TOP2A+/EZH2+; green) tightly cluster apart from other patients (purple) based on their differentially expressed genes (DEGs). (B) Unique DEGs between TOP2A+/EZH2+ and other patients was validated by PCA. (C) An important proportion of DEGs in TOP2A+/EZH2+ metastatic prostate cancer patients are shared between Robinson et al. (2015) and Kumar et al. (2016) datasets. (D) GSEA revealed statistically significant enrichment of gene signatures involved mitosis and E2F signaling, P < 0.05 and FDR < 0.15. (E, F) Comparison of gene expression platforms including (E) RNA-seq and (F) microarray revealed that shared gene expression was maintained between TOP2A+/EZH2+ primary and metastatic PCa patients.
Next we compared primary and metastatic TOP2A+/EZH2+ patient gene signatures from RNA-sequencing and microarray platforms. The DEG lists provided a high level of overlap between the metastatic and primary PCa samples (Figure 4E, F; Supplement Table 6) with a core of 45 DEGs conserved in all four dataset (Supplement Table 6), as well as a high level of overlap in mitotic spindle deregulation and G2M/mitotic hallmarks (Figures 1F and 4D). This study and others demonstrate that biomarkers of aggressive PCa are often associated with mitotic deregulation (27–29). Mitotic deregulation and mitotic gene conversion is associated with therapy resistance and increased genome instability (30,31). This knowledge may give insight into underlying mechanisms as to why our identified patients with increased enrichment for mitotic related DEG progress more rapidly with BCR, metastatic disease and ultimately PCa specific mortality.
To test our biomarkers as targetable actions for therapeutic intervention we took advantage of newly generated preclinical model of highly aggressive GEMM of PCa (22). While prostate-specific deletion of Pten (sKO) is sufficient to initiate PCa development, the co-deletion of Pten and Retinoblastoma-1 (Rb1) (dKO) results in a robust PCa progression with spontaneous metastasis, a feature not observed in the sKO mice (22). Interestingly, dKO primary tumors show greater levels of TOP2A/EZH2 mRNA and protein compared to sKO GEMMs (Figure 5A, B), a feature that prompted us to generate cell line models (32) from primary tumors resected from those GEMM of PCa (Figure 5C). Importantly, the dKO cell line retained high levels of TOP2A and EZH2 expression compared to the sKO cell line (Figure 5D). Our previous work identified a significant increase in DNA damage and therapeutic efficacy by combining etoposide with EZH2 inhibitors (EZH2i) in pre-clinical models of lethal PCa (5). With this, we proposed that dKO cells would be more sensitive to combination treatment with etoposide and clinically relevant EZH2i, namely GSK126 and EPZ6438 (33,34). Both sKO and dKO cells were treated with either the vehicle control (DMSO) or combination therapy consisting of 1μM etoposide and 1μM or 5μM of EZH2i. Although intrinsically more proliferative than sKO cells, dKO cell lines lose their clonogenic ability upon combined inhibition of TOP2A and EZH2, a feature not observed in sKO cells (Figure 5E, Supplement Figure 5A). This hypersensitivity to TOP2A and EZH2 inhibition is likely related to the significant increase in accumulation of a SubG1 population (Figures 5F, Supplement Figure 5B), indicative of apoptosis associated with DNA damage as we have previously demonstrated (5).
Figure 5. Top2a and Ezh2 increased expression is associated with metastatic disease progression in genetically engineered mouse models of PCa and provide rationale for targeted therapy.
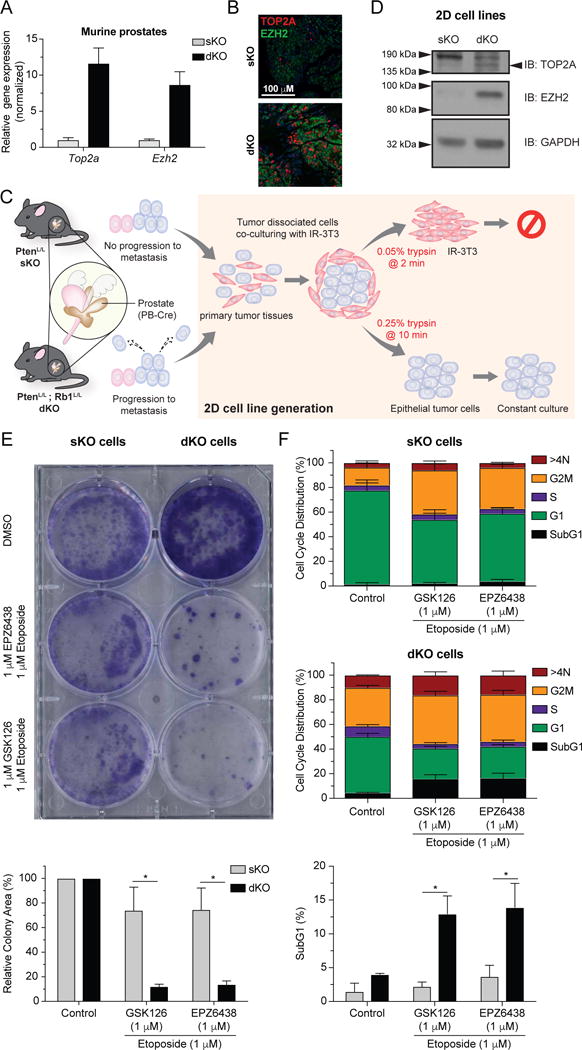
(A) Top2a and Ezh2 expression levels from mice with prostate specific deletion of Pten−/− (sKO) or Pten−/−; Rb1−/− (dKO) as assessed by qRT-PCR. (B) Fluorescence immunohistochemistry depicting high TOP2A and EZH2 expression in a dKO prostate tumor. (C) Schematic describing the generation of prostatic epithelial cell lines from sKO or dKO prostate tumors. (D) Confirmation that high expression level of TOP2A and EZH2 in dKO cell line was maintained following spontaneous immortalization. (E) Combination therapy targeting both EZH2 (GSK126 or EPZ6438, 1 μM) and TOP2A (Etoposide) activity induces loss of clonogenicity specific to dKO cells. (top: representative experiment; unpaired t test; * P < 0.05, triplicate, mean ±SEM). (F) Cell cycle analysis indicates that combination therapy induces apoptosis specifically in dKO cells as indicated by increased SubG1 accumulation (unpaired t test; * P < 0.05, triplicate, mean ±SEM).
Discussion
While an improved understanding of the molecular basis of PCa tumorigenesis has generated increased prognostic and predictive measures, the early identification of aggressive primary PCa is still not resolved. Genetic pathways and/or gene expression panels to predict PCa outcome and response to therapeutic interventions (35) are promising. Three recent gene expression panels have reported success in predicting PCa patient outcome, in terms of BCR, mortality, and metastatic progression (36–40). While these panels have indicated patient outcomes, they have yet to provide and/or validate targeted therapy approaches that could intercept a patient’s progression to metastatic disease. Provoked by this and our previous work (5,41), this current study presents data regarding two genes associated with metastatic PCa, which has never been considered as potential cooperating partners in the identification and treatment of aggressive primary PCa. Results within, convincingly demonstrate the ability to predict metastatic potential of primary PCa by expression analysis of TOP2A and EZH2. Further, in vitro PCa models demonstrate the ability to predict response to a combination therapy approach based on TOP2A and EZH2 gene or protein expression.
In addition, we found that high TOP2A and EZH2 gene expression in TCGA kidney cancer datasets (42–44) also selected for a majority of patients with worse progression-free survival involving progression to metastatic disease (Supplement Figure 6A, B). However, stratification of primary TCGA bladder cancer patients (45) by our methods did not predict metastatic potential or progression-free survival (data not shown). Overall, these data indicate that the potential of TOP2A and EZH2 expression to identify patients with metastatic potential can be extended to other genitourinary cancers and should be further explored in additional models.
While this study focused on the use of etoposide combination with EZH2 inhibitors, other targeted therapies maybe considered. Recently, exciting clinical results showed the dramatic impact of PARP inhibitors for treatment of patients with metastatic PCa with defects in DNA repair genes (46). Because our data highlight a subgroup of patients with increased mitotic DEGs and potential of increased genomic instability, PARP inhibitors may offer an alternative therapeutic opportunity. Overall these data could provide a novel direction for PCa risk stratification and the clinical management of primary disease (Figure 6).
Figure 6.
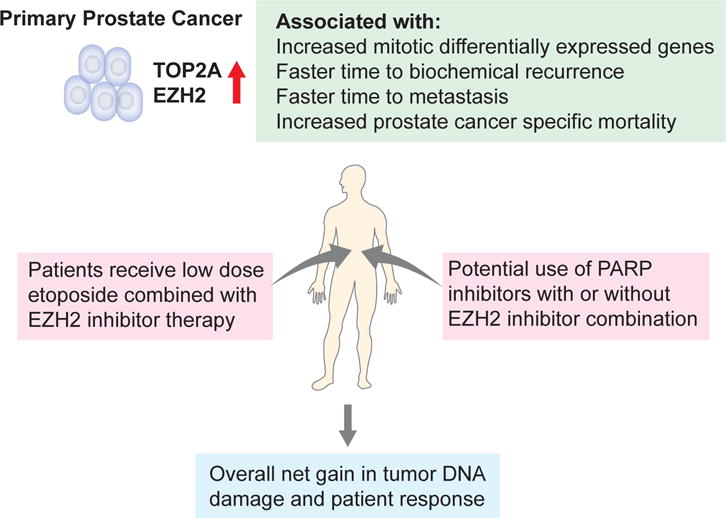
Schematic figure indicating the potential use of TOP2A and EZH2 for the early identification and treatment direction of aggressive primary PCa.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Metastatic PCa accounts for the majority of PCa specific mortality. However, the ability to distinguish primary prostate cancer with metastatic potential has not been achieved. Here, we show that primary human PCa with concurrent increased expression of TOP2A and EZH2 have a faster time to biochemical recurrence, are more susceptible to progress to a metastatic disease and thereby resulting in increased PCa specific death. Importantly, we demonstrate, using preclinical prostate cancer mouse cell lines models that concurrent increase of TOP2A and EZH2 result in a hypersensitivity to combination treatment with etoposide, a TOP2 poison, with inhibitors of EZH2. Together, our work provides substantial evidence for the utility of TOP2A and EZH2 as prognostic biomarkers and therapeutic targets to identify and intercept aggressive PCa progressing to a metastatic disease.
Acknowledgments
The authors thank Clyde Bango for technical assistance and Noriko Uetani for figure design and drawing.
Grant Support
D.P.L. is supported by a Prostate Cancer Foundation Young Investigator Award, a Cancer Research Society Next Generation of Scientist Scholarship, and a Canadian Institute of Health Research Fellowship. L.E. is supported by a Roswell Park Cancer Institute (RPCI) and a Dana-Farber Cancer Institute faculty start-up funds, and a Prostate Cancer Foundation Young Investigator Award. This study used shared resources supported by the RPCI Cancer Center Support Grant from the NCI (P30CA016056) and was supported by grants from the NIH (R21CA179907 to D.W.G. and R01CA207757 to D.W.G. & L.E.).
Financial support:
D.P.L. & L.E.: Prostate Cancer Foundation Young Investigator Award
D.W.G., K.H.E. & L.E.: NCI P30 (CA016056)
D.W.G. & L.E.: NIH R01 (CA207757)
Footnotes
Author’s Contributions
D.P.L. contributed to experimental design, data analysis and manuscript writing. C.J.S. edited the manuscript and provided vital reagents. M.Brown contributed to manuscript editing. S.R. performed experiments, analyzed data, and edited the manuscript. P.G. & K.M.W. performed experiments and analyzed data. S-Y.K. & D.W.G. provided vital reagents and contributed to manuscript editing. M.S., M.A., N.E, E.D., R.J.K., E.M.S, R.B.J., R.B.D., A.E.R., F.Y.F & D.E.S., analyzed and/or provided clinical data. M.Bowden, Y.H. & K.P.G. performed experiments and/or analyzed data. K.H.E. analyzed data. L.E. designed experiments, performed research, analyzed data, and wrote the manuscript.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Madu CO, Lu Y. Novel diagnostic biomarkers for prostate cancer. J Cancer. 2010;1:150–77. doi: 10.7150/jca.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prensner JR, Rubin MA, Wei JT, Chinnaiyan AM. Beyond PSA: the next generation of prostate cancer biomarkers. Sci Transl Med. 2012;4(127):127rv3. doi: 10.1126/scitranslmed.3003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzardi AE, Rosener NK, Koopmeiners JS, Isaksson Vogel R, Metzger GJ, Forster CL, et al. Evaluation of protein biomarkers of prostate cancer aggressiveness. BMC Cancer. 2014;14:244. doi: 10.1186/1471-2407-14-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirk JS, Schaarschuch K, Dalimov Z, Lasorsa E, Ku S, Ramakrishnan S, et al. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer. Oncotarget. 2015;6(5):3136–46. doi: 10.18632/oncotarget.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheville JC, Karnes RJ, Therneau TM, Kosari F, Munz JM, Tillmans L, et al. Gene panel model predictive of outcome in men at high-risk of systemic progression and death from prostate cancer after radical retropubic prostatectomy. J Clin Oncol. 2008;26(24):3930–6. doi: 10.1200/JCO.2007.15.6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Resende MF, Vieira S, Chinen LT, Chiappelli F, da Fonseca FP, Guimaraes GC, et al. Prognostication of prostate cancer based on TOP2A protein and gene assessment: TOP2A in prostate cancer. J Transl Med. 2013;11:36. doi: 10.1186/1479-5876-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karnes RJ, Cheville JC, Ida CM, Sebo TJ, Nair AA, Tang H, et al. The ability of biomarkers to predict systemic progression in men with high-risk prostate cancer treated surgically is dependent on ERG status. Cancer Res. 2010;70(22):8994–9002. doi: 10.1158/0008-5472.CAN-10-1358. [DOI] [PubMed] [Google Scholar]
- 9.Melling N, Thomsen E, Tsourlakis MC, Kluth M, Hube-Magg C, Minner S, et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis. 2015;36(11):1333–40. doi: 10.1093/carcin/bgv137. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–78. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass AG, Leo MC, Haddad Z, Yousefi K, du Plessis M, Chen C, et al. Validation of a Genomic Classifier for Predicting Post-Prostatectomy Recurrence in a Community Based Health Care Setting. J Urol. 2016;195(6):1748–53. doi: 10.1016/j.juro.2015.11.044. [DOI] [PubMed] [Google Scholar]
- 15.Piccolo SR, Sun Y, Campbell JD, Lenburg ME, Bild AH, Johnson WE. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics. 2012;100(6):337–44. doi: 10.1016/j.ygeno.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross AE, Johnson MH, Yousefi K, Davicioni E, Netto GJ, Marchionni L, et al. Tissue-based Genomics Augments Post-prostatectomy Risk Stratification in a Natural History Cohort of Intermediate- and High-Risk Men. Eur Urol. 2016;69(1):157–65. doi: 10.1016/j.eururo.2015.05.042. [DOI] [PubMed] [Google Scholar]
- 17.Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, et al. Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J Urol. 2013;190(6):2047–53. doi: 10.1016/j.juro.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Den RB, Feng FY, Showalter TN, Mishra MV, Trabulsi EJ, Lallas CD, et al. Genomic prostate cancer classifier predicts biochemical failure and metastases in patients after postoperative radiation therapy. Int J Radiat Oncol Biol Phys. 2014;89(5):1038–46. doi: 10.1016/j.ijrobp.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freedland SJ, Choeurng V, Howard L, De Hoedt A, du Plessis M, Yousefi K, et al. Utilization of a Genomic Classifier for Prediction of Metastasis Following Salvage Radiation Therapy after Radical Prostatectomy. Eur Urol. 2016;70(4):588–96. doi: 10.1016/j.eururo.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Spratt DE, Yousefi K, Deheshi S, Ross AE, Den RB, Schaeffer EM, et al. Individual Patient-Level Meta-Analysis of the Performance of the Decipher Genomic Classifier in High-Risk Men After Prostatectomy to Predict Development of Metastatic Disease. J Clin Oncol. 2017;35(18):1991–8. doi: 10.1200/JCO.2016.70.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irshad S, Bansal M, Castillo-Martin M, Zheng T, Aytes A, Wenske S, et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5(202):202ra122. doi: 10.1126/scitranslmed.3006408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benzon B, Zhao SG, Haffner MC, Takhar M, Erho N, Yousefi K, et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis. 2017;20(1):28–35. doi: 10.1038/pcan.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One. 2013;8(6):e66855. doi: 10.1371/journal.pone.0066855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y, et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin Epigenetics. 2015;7:40. doi: 10.1186/s13148-015-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, et al. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci U S A. 2015;112(47):E6544–52. doi: 10.1073/pnas.1518007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellis L, Loda M. Advanced neuroendocrine prostate tumors regress to stemness. Proc Natl Acad Sci U S A. 2015;112(47):14406–7. doi: 10.1073/pnas.1519151112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res. 2012;18(3):666–77. doi: 10.1158/1078-0432.CCR-11-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329(5987):82–5. doi: 10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Mao JH, Zhu W, Jain AK, Liu K, Brown JB, et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat Commun. 2016;7:12619. doi: 10.1038/ncomms12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 34.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842–54. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 35.Bostrom PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, Loeb S, et al. Genomic Predictors of Outcome in Prostate Cancer. Eur Urol. 2015;68(6):1033–44. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Cooperberg MR, Davicioni E, Crisan A, Jenkins RB, Ghadessi M, Karnes RJ. Combined value of validated clinical and genomic risk stratification tools for predicting prostate cancer mortality in a high-risk prostatectomy cohort. Eur Urol. 2015;67(2):326–33. doi: 10.1016/j.eururo.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooperberg MR, Simko JP, Cowan JE, Reid JE, Djalilvand A, Bhatnagar S, et al. Validation of a cell-cycle progression gene panel to improve risk stratification in a contemporary prostatectomy cohort. J Clin Oncol. 2013;31(11):1428–34. doi: 10.1200/JCO.2012.46.4396. [DOI] [PubMed] [Google Scholar]
- 38.Cullen J, Rosner IL, Brand TC, Zhang N, Tsiatis AC, Moncur J, et al. A Biopsy-based 17-gene Genomic Prostate Score Predicts Recurrence After Radical Prostatectomy and Adverse Surgical Pathology in a Racially Diverse Population of Men with Clinically Low- and Intermediate-risk Prostate Cancer. Eur Urol. 2015;68(1):123–31. doi: 10.1016/j.eururo.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 39.Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. The Lancet Oncology. 2011;12(3):245–55. doi: 10.1016/S1470-2045(10)70295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al-Deen Ashab H, et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016;76(17):4948–58. doi: 10.1158/0008-5472.CAN-16-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellis L. Determination of synthetic lethal interactions to provide therapeutic direction to end aggressive prostate cancer. Future Oncol. 2015;11(10):1451–4. doi: 10.2217/fon.15.61. [DOI] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research N. Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2016;374(2):135–45. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26(3):319–30. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373(18):1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.