

Abstract
Intravasation involves the migration of tumor cells across the local endothelium and escape into vessel flow. While tumor cell invasiveness has been correlated to increased intravasation, the details of transendothelial migration and detachment into circulation are still unclear. Here we analyzed the intravasation of invasive human breast cancer cells within a tissue-engineered microvessel model of the tumor microenvironment. Using live-cell fluorescence microscopy, we captured 2,330 hours of tumor cell interactions with functional microvessels and provide evidence for a mitosis-mediated mechanism where tumor cells located along the vessel periphery are able to disrupt the vessel endothelium through cell division and detach into circulation. This model provides a framework for understanding the physical and biological parameters of the tumor microenvironment that mediate intravasation of tumor cells across an intact endothelium.
Keywords: metastasis, intravasation, tumor vasculature, tumor microenvironment, invasion
INTRODUCTION
Metastasis involves a sequence of steps including invasion, intravasation, circulation, arrest, and extravasation that ultimately result in tumor cell dormancy and/or growth at a secondary site (1). Intravasation, the entry of tumor cells into circulation, is a critical step that occurs at both primary and secondary tumor sites (2). The discovery of circulating tumor cells years after primary treatment reveals that tumor cells continue to spread through the vasculature and is a negative prognostic indication for cancer relapse (2,3). Reducing intravasation may ultimately improve patient outcome; however, the details of these events are difficult to resolve, since it is a dynamic process that occurs at the interface between tumor cells and the local tumor microvasculature.
Our understanding of the dynamics of intravasation is derived largely from a relatively small number of intravital microscopy studies and few direct observations at the single cell level. For example, intravasation has been investigated in zebrafish and chick embryos (4,5), but only directly observed in mouse models (6,7). From these studies, we have learned that tumor associated macrophages (TAMs) enhance the invasion of breast cancer cells towards vessels through EGF and CSF1 chemotaxis (6,8) and local expression of VEGF causes down-regulation of endothelial cell-cell junctions, thus increasing the intravasation frequency. However, tumor cells over-secreting growth factors are also capable of intravasation across multiple in vivo systems without the assistance of TAMs (5,9,10). These studies suggest that there are multiple pathways for intravasation, but the lack of sufficient resolution has hampered our understanding of the mechanism of tumor cell transendothelial migration and detachment into circulation.
Recent advances in the development of in vitro microvessel models provide the tools to recreate the essential components of the tumor microenvironment and enable visualization of the details of the metastatic cascade (11–13). Here we set out to investigate the mechanism of intravasation of breast cancer cells and to address the question, how do tumor cells cross endothelial junctions to enter circulation? Using live-cell imaging in a tissue-engineered microvessel model of the tumor microenvironment, we analyzed over 2,330 hours of tumor cell interactions with functional microvessels and found that intravasation events were rare but predominately associated with mitosis. We quantified the deflection of peripheral tumor cells on the vessel endothelium and provide evidence for a model where mitotic single-cell rounding exerts a force on the endothelium that is sufficiently large to transiently open endothelial cell-cell junctions and expose the tumor cells to shear flow, which pulls the daughter cells into circulation. To confirm that this is the dominant mechanism of intravasation, we showed that tumor cells that extended protrusions across the interface did not intravasate. Similarly, tumor cells dividing in a larger perivascular space were unable to deflect the vessel endothelium and intravasate. These results demonstrate a simple, yet effective mechanism by which single tumor cells may undergo intravasation and provide a framework for understanding the physical and biological parameters that enable intravasation through this pathway.
MATERIALS AND METHODS
Device fabrication
The tumor-microvessel platform was fabricated as described previously (13). Briefly, high concentration rat tail collagen type I (Corning Inc., Tewksbury, MA) is diluted to 7 mg mL−l and neutralized with the manufacturer’s recommended amounts of DI water, 10x PBS, and 1 N sodium hydroxide. After neutralization, tumor cells are introduced into the collagen solution to a final concentration of 5×105 cells mL−1 and injected around a cylindrical template rod (diameter ~ 150 μm) within the polydimethyl siloxane (PDMS) housing of the platform (Supplementary Fig. S1). After collagen gelation at room temperature, the rod is removed, leaving behind a cylindrical channel within the collagen gel. The channel is subsequently coated with fibronectin (50 μg mL−1) to promote endothelial adhesion and spreading. Endothelial cells in suspension are introduced into the channel at a concentration of 5×106 cells mL−1 and allowed to settle and actively adhere to the channel walls. After the endothelial cells have spread for about 2 hours, normal growth media (NGM) is perfused through the vessel at a low applied shear stress (< 1 dyne cm−2) over-night. Devices were typically confluent after 1 day and were switched to higher shear stress (~4 dyne cm−2) conditions for at least 24 h before live-cell imaging.
Cell lines and culture conditions
Human umbilical vein endothelial cells (HUVEC) (Promocell, Heidelberg, Germany), human dermal microvascular endothelial cells (HMVEC) (Lonza, Walkersville, MD) and VeraVec HUVEC-TURBOGFP (HVERA-GFP) (cat no HVERA-UMB-202100) (Angiocrine Bioscience, New York, NY) were seeded in the cylindrical channel of the microvessel platform. Endothelial cells were grown in MCDB 131 (Caisson Labs, Carlsbad, CA) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Sigma, St. Louise, MO), 25 mg mL−1 endothelial mitogen (BT-203, Biomedical Technologies, Stoughton, MA), 2 U mL−1 herparin (Sigma), 1 μg mL−1 hydrocortisone (Sigma), 0.2 mM ascorbic acid 2-phosphate (Sigma), and 1% penicillin-streptomycin-glutamine (Life Technologies). Dual-labeled MDA-MB-231 breast cancer cells (BCCs) (AntiCancer Inc., San Diego, CA) were embedded within the collagen type I ECM around the microvessel (14). Cancer cells were grown in RPMI (Corning Inc) supplemented with 10% FBS and 1% penicillin-streptomycin (Life Tech). All culture conditions were in humidified environments with 5% CO2 at 37 °C. All cell lines were authenticated by their respective manufacturers and tested negative for mycoplasm.
Live-cell imaging
Fluorescence and phase-contrast images were obtained on a Nikon TE-2000 U microscope (Nikon Instruments Inc., Melville, NY). A 10x objective was used for live-cell, time-lapse imaging and permeability experiments. Images were acquired every 10 min at equally spaced intervals along the entire length of vessel, typically 12 locations, 1 mm apart. Images were focused along the top/bottom (i.e. poles) and middle (i.e. equator) of each segment of the vessel.
Immunofluorescence staining
CD133 (293C3, Miltenyi Biotech, San Diego, CA) mouse primary antibody was incubated at 5 μg mL−1 per the manufacturer’s recommended protocol. Mouse IgG2b isotype control (MA1-10427, Thermo Fisher Scientific) and mouse anti-CD31 (37-0700, Thermo Fisher Scientific) antibodies were incubated at the same concentration (i.e. 5 μg mL−1). All primary antibodies were incubated on separate samples. MDA-MB-231 and VeraVec were cultured on fibronectin coated glass coverslips for 2 days prior to fixing and staining. Cells were quickly washed with PBS and fixed with 3.7% formaldehyde for 3 min. Permeabilized samples were incubated in 0.1% Triton X-100 for 3 min. Cells were blocked in 10% donkey serum for 30 min. Primary and isotype control antibodies were incubated for 1 hr at room temperature. Secondary donkey anti-mouse IgG (H + L) antibody conjugated to Alexa Fluor 647 (A31571, ThermoFisher Scientific) was incubated for 1 hr. Cells were washed for 10 min, 3 times between fixing and incubation steps with PBS.
Image analysis
Intravasating tumor cells located at the ECM-vessel interface along the middle of the vessel (i.e. vessel equator) were manually traced in ImageJ (NIH, Bethesda, MD) at each time-lapse frame. Tumor cell circularity (i.e. shape factor, 4·π·area/perimeter2) was calculated as well as the maximum cell width perpendicular to the vessel wall. The latter was obtained by measuring the widths along the entire length of tumor cell perpendicular to the vessel wall and taking the maximum value with a custom written ImageJ plugin (program provided in Supplementary Materials). These values were plotted versus time. Cell motility descriptors (e.g. persistence, RMS speed, and directedness) were measured for tumor cells located at the interface (n = 38 cells continuously tracked for 12 – 17 h from 4 independent microvessels) as well as in the bulk ECM (n = 35 cells continuously tracked for 12 – 18 h from 7 independent microvessels). Cell locations were approximated in ImageJ from time-lapse images by manually tracing the nucleus of each tumor cell every hour. Only data from tumor cells tracked for a minimum of 12 hours were used, which reduced the variance of each cell’s average RMS speed, persistence, and directedness. Persistence is the shortest length connecting the start-to-end distance divided by the total path length acquired over the entire duration of cell tracking. An average RMS speed for each tumor cell was obtained by averaging the displacement over time for each one hour segment of a tumor cell’s migration; for example, a tumor cell tracked for 12 h will exhibit 12 individual RMS speeds that are combined as a batch average. Directedness is the cosine of the cell’s displacement angle, the angle between the cell’s start-to-finish vector acquired over the entire duration of tracking with respect to either the direction of flow (i.e. along the length of vessel) or towards the vessel (i.e. direction perpendicular to the vessel wall).
Frequency of intravasation, cell division, entry or exit events from the ECM-vessel interface
Tumor cell intravasation, cell division, and entry/exit events are rare (0 – 10 events per microvessel experiment) and individual tumor cells were tracked over varying amounts of time (e.g. 0.5 – 48 hours) because of cell migration into and out of the imaging focal plane; due to these constraints, we quantified the frequency of these events over aggregate cell hours obtained over 26 microvessel experiments rather than as a fraction of cells tracked. A tumor cell hour is the time spent by a single tumor cell at the location of interest (e.g. interface or ECM) for one hour. For example, five cells located at the ECM-vessel interface tracked for 10 hours results in a total of 50 cell hours. Tumor cells at the ECM-vessel interface or in the bulk ECM were tracked for a minimum of 30 min. All qualifying events (i.e. intravasation, cell division, entry or exit from the interface) were summed and divided by the total number of tumor cell hours.
Statistics
Values are reported as mean ± SEM. The principle statistical test used was a student t test (two-tailed assuming unequal variance). We considered a P value less than 0.05 to be statistically significant.
RESULTS
Tissue-engineered tumor-microvessel model
To enable high resolution imaging of the dynamics of intravasation into microvessels, human microvascular endothelial cells were cultured within 150 μm diameter cylindrical channels in a 3D collagen type I matrix. Dual-labeled MDA-MB-231 breast cancer cells (BCC) (GFP histone tag, RFP cytoplasm) were embedded within the collagen matrix as either single cells or clusters (Supplementary Fig. S1). After a continuous endothelial monolayer is formed, usually within 24 hours after seeding, vessels were maintained under a constant flow rate of 1 – 2 ml h−1 and time-lapse imaging was started the following day. The number of intravasating tumor cells has been positively correlated to the density of vessels ≥ 30 μm in diameter in tumor tissue (15), which suggests that vessels < 30 μm do not contribute significantly to intravasation. Tumor vasculature is formed quickly, typically lacking smooth muscle cells and/or pericytes, and exhibits irregular architecture often with incomplete endothelial lining (16,17); as a result, leaky tumor vessels will permit the heterogeneous extravasation of large particles and red blood cells into the perivascular space and exhibit a global permeability significantly higher than normal vasculature (12,17,18). Our vessels exhibit permeability values ranging from 10−5 to 10−6 cm s−1 for bovine serum albumin which is comparable to that of leaky tumor vessels (12,13). MDA-MB-231 is a human breast adenocarcinoma cell line from triple negative breast cancer that has been shown to readily metastasize to the brain in mouse models (19,20). MDA-MB-231 cells have been used in various in vitro assays, such as transwell invasion studies (21–23), microfluidic adhesion and extravasation experiments (24–27), and intravital microscopy (20,28).
Invasion
Our analysis is based on more than 2,330 hours of live-cell imaging of breast cancer cells (BCCs) across 26 individual microvessels (Fig. 1). Time-lapse images were acquired every 10 min along the length of the microvessel. When focused on the vessel equator, the endothelium is projected as a thin vessel wall perpendicular to tumor cells located in the ECM (Fig. 1A). Single BCCs in the matrix generally exhibited a spindle-like morphology and often displayed a rounded cell body with one or more protrusions at the leading edge while migrating, a characteristic of amoeboid migration (Fig. 1B) (13,29). Since the matrix is relatively dense, migration involves proteolytic degradation of the collagen ECM, which often creates etched tracks that are used by other cells to facilitate rapid directed migration (29–31) (Supplementary Video S1). Tumor cells in the ECM migrate at an average RMS speed of 0.20 ± 0.036 μm min−1 with a persistence of 0.32 ± 0.034 (n = 35 cells, 7 microvessels, tracked for 12 – 18 h). These BCC speeds are comparable to tumor cell invasion in dense collagen gels (6 – 10 mg ml−1) where proteolytic degradation is the limiting factor (13,29). Our persistence values are consistent with BCCs observed on 2D surfaces in the absence of a chemotactic gradient (32). Analysis of the angular orientation of BCC migration reveals no significant directedness (−0.028 ± 0.026) towards or away from the vessel; therefore approximately half of all tumor cells are randomly migrating towards the vessel. Similar analysis of BCC migration in the direction of flow, parallel to the vessel, shows no significant directedness (−0.057 ± 0.047). These results indicate that there is no apparent directional bias due to mechanotaxis (e.g. interstitial flow or pressure gradients) or chemotaxis (e.g. growth factor gradients from perfused media).
Figure 1.
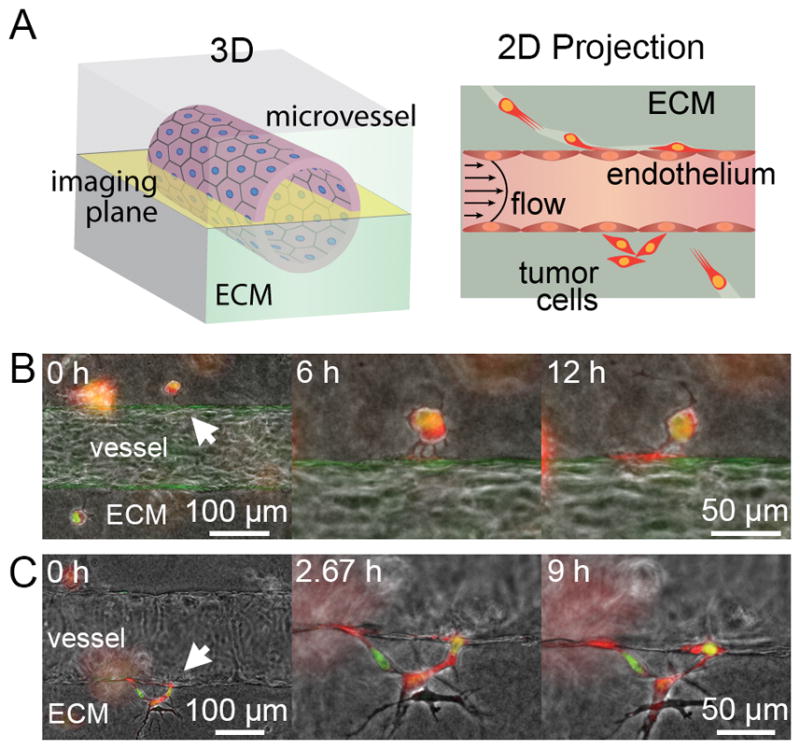
Tumor cell invasion in tissue-engineered microvessels. A, illustration of 3D microvessel and the resulting 2D projected image from widefield microscopy focused in the middle/equator of the vessel. B, single dual-labeled MDA-MB-231 breast cancer cell (BCC) extends multiple protrusions into the ECM-vessel interface over 6 hours, which coalesce into a larger protrusion at the interface over 12 hours. C, multiple BCCs invading into the ECM-vessel interface using amoeboid motility, as shown by the nuclear deformation observed at 2.67 hours. Flow is from left to right.
Insertion into the ECM-vessel interface
BCCs that migrate to the microvessel insert protrusions into the interface between the ECM and the vessel endothelium (Fig. 1B). Tumor cells that successfully invade into the ECM-vessel interface are typically within 50 – 100 μm of the vessel endothelium. Extended processes that make contact with the vessel endothelium are stabilized and exhibit flattened end-feet that progressively grow at the interface (Fig. 1B; Supplementary Video S2). The extended processes expand to a width of approximately 5 – 10 μm before the BCC is capable of squeezing into the interface (Fig. 1C). If the inserted BCC is part of a cluster, then additional tumor cells may collectively migrate into the ECM-vessel interface along etched tracks (Supplementary Video S1). Once inserted into the interface, the cells typically exhibit a flattened egg shape at the vessel poles (i.e. top/bottom) and appear elongated and spindle-like in the projected equatorial view (i.e. middle/sides) (Fig. 2A and B). The fraction of tumor cells at the ECM-vessel interface observed to migrate into the interface is 7.7 % (17 out of 221 interface BCCs) over the 2,330 cell hours. The majority of BCCs at the interface remain with only 3.8 % (8 out of 221 interface BCCs) migrating back into the ECM. Interfacial tumor cells were capable of invading into the ECM typically only if a predefined, etched track existed in the local ECM (Supplementary Video S3). The higher rate of tumor cell insertion into the interface versus returning to the ECM may be due to the lack of available etched tracks leading away from the interface; furthermore, tumor cells are confined to the vessel interface rather than invading into the ECM which requires proteolytic degradation and amoeboid motility.
Figure 2.

Imaging tumor cell invasion and intravasation at the ECM-vessel interface. A, illustration of a 3D microvessel and cross-section displaying the imaging focal planes at the vessel equator and pole. Below are representative images of a 3D projection and cross-section of a confocal z-stack of a HUVEC microvessel immunofluorescently stained for VE-cadherin (green) and nuclei (blue). B, illustration and representative images of tumor cells located at the vessel equator and pole where they appear spindle-like and flattened respectively.
Tumor cells at the ECM-vessel interface also exhibited an average RMS speed of 0.24 ± 0.019 μm min−1 with a persistence of 0.36 ± 0.034 (n = 38 cells tracked for 12 – 17 h from 4 independent microvessel studies). The RMS speed and persistence of these tumor cells are not statistically different from their values in the ECM (p = 0.34 and 0.42, respectively) despite being confined in two dimensions. Tumor cells in the interface exhibit no preferential directedness (−0.065 ± 0.044) with respect to flow.
Single cell intravasation
Analysis of intravasation was restricted to single cells at the equator of the microvessels to clearly distinguish the position and profile of tumor cells with respect to the ECM substrate and endothelium in the X-Y focal plane of imaging (Fig. 2A). Despite these restrictions, we observed a total of 2,330 cell hours from 221 breast cancer cells (BCCs) at the ECM-vessel interface across 26 separate microvessels. From this subpopulation of interfacial cells, we observed 8 intravasation events, thus measuring an intravasation rate of 3.4 × 10−3 h−1 (i.e. 8 events over 2,330 cell hours). All intravasation events displayed a characteristic rounding of the individual tumor cell, migration across the endothelium, and escape into circulation. In 4 of these 8 cases, intravasation was clearly associated with mitosis (Supplementary Fig. S2; Supplementary Videos S4 and S5), as identified by the visible organization of chromosomes and/or cell division prior to tumor cell detachment into flow. The remaining 4 cases were not definitively related to mitosis but clearly involved cell rounding (Supplementary Fig. S2; Supplementary Video S6). Tumor cells proliferate at a rate of 0.016 h−1 (i.e. 37 events over 2,330 cell hours) at the ECM-vessel interface, which is comparable to that in the bulk ECM of 0.014 h−1 (i.e. 19 events over 1,301 cell hours in n = 6 microvessels). In Figure 3A and B, two representative mitosis-mediated intravasation events show BCCs at the ECM-vessel interface undergoing mitosis, which leads to cell rounding, transendothelial migration (TEM), and escape into vessel flow.
Figure 3.

Intravasation of interfacial tumor cells within the tumor-microvessel model. A and B, representative time-series of intravasating dual-labeled MDA-MB-231 breast cancer cells (BCC). Notice the onset of mitosis and chromosomal organization during transendothelial migration across a VeraVec-GFP endothelium. Flow is from left to right. C, representative time-series of BCC division resulting in two daughter cells that remain at the interface. Flow is from left to right. D and E, cell circularity and maximum width vs. time for intravasating BCC featured in A. F and G, cell circularity and maximum width vs. time for intravasating BCC in featured in B. H and I, cell circularity and maximum width vs. time for non-intravasating BCC featured in C. J, maximum circularity and width of intravasating, n = 6 (red), and non-intravasating, n = 15 (blue), BCCs dividing at the ECM-vessel interface. K, X-Y location of a representative BCC invading along the ECM-vessel interface followed by cell rounding and transendothelial migration. L, illustration of invasion and mitosis-mediated intravasation where a tumor cell invades into the ECM-vessel interface and cell division results in deflection of the endothelium, transendothelial migration, and tumor cell escape into vessel flow.
While intravasation events are often preceded by mitosis, we also observed 33 tumor cell division events at the ECM-vessel interface that did not result in intravasation (Supplementary Fig. S3); cell shape descriptors could be analyzed for 15 of these events. In Figure 3C, a representative non-intravasating cell division event shows a single BCC rounding at the interface, which deflects the endothelium but does not result in TEM (Supplementary Video S7). Both resulting daughter cells resume a spindle-like elongated shape at the interface. In all 8 detected intravasation events, both daughter cells escape into circulation; however, we have observed unique cases where one daughter cell intravasates while the other returns to the interface (Supplementary Video S8). Additionally, it is possible for both daughter cells to return to the interface after TEM, possibly due to insufficient shear stress required to detach the cells completely from the ECM substrate (Supplementary Video S9).
As described above, BCCs imaged at the equator of microvessels and residing at the ECM-vessel interface appeared elongated with a low circularity. Cell shape descriptors (e.g. circularity and maximum cell width) were quantifiable in 6 of the 8 intravasation events (Supplementary Fig. S4). The onset of mitosis results in an increase in cell circularity, and the associated rounding imposes a deflection on the endothelium rather than on the stiffer ECM (Fig. 3D–I). If this deflection is large enough to disrupt the local endothelial cell-cell junctions, the tumor cell is exposed to the vessel lumen where the shear stress may pull it into circulation. All intravasation events were associated with deflections ≥ 20 μm, whereas cell-division events with smaller deflections did not result in TEM or intravasation (Fig. 3J). Intravasating cells, while rounding, exhibited an average maximum width of 30.3 ± 5.0 μm (n = 6), whereas dividing cells that did not intravasate exhibited a smaller average maximum width of 18.5 ± 1.5 μm (n = 15); these values are not statistically different (p = 0.067, two-tailed student’s t-test). During intravasation, tumor cell invasion along the ECM-vessel interface typically stopped during mitosis, and TEM was detected by the sudden translation of the tumor cell’s center of mass towards the vessel lumen, about half the maximum width (≈ 15 μm), before commencement of cell rolling and detachment (Fig. 3K). Our mitosis-mediated intravasation model is summarized in Figure 3L.
Supporting evidence for the physiological relevance of mitosis-mediated intravasation comes from the observations that: (1) tumor cell division potentiates detachment through down-regulation of adhesion proteins and results in monoclonal clusters of circulating tumor cells (33,34), (2) alternative attempts at TEM that involved extending cell protrusions into vessel flow were not successful (4), (3) and no intravasation was associated with cell division in a large perivascular space, but rather invasion and dispersal along the ECM-vessel interface.
Doxorubicin inhibits cell division and intravasation
To assess the role of cell division in intravasation of BCCs, microvessels were perfused with a mitotic inhibitor, doxorubicin, an intravenously administered chemotherapeutic that is used to treat a variety of cancer indications, such as breast and bladder cancers. As a small molecule anthracycline that passively diffuses across cell membranes, doxorubicin halts cell division by inhibiting topoisomerase II and intercalating with DNA (35). Here, we continuously introduced doxorubicin through functional microvessels at a pharmacologically relevant concentration of 0.2 μM in normal growth media (NGM) for 24 hours (36). After doxorubicin introduction for 8 hours, we measured the proliferation and intravasation rates of tumor cells imaged in the equatorial plane at the ECM-vessel interface. With a combined total of 617 cell hours from about 60 different tumor cells, we would predict approximately 8 cell division events and 2 intravasation events from previously measured division (0.016 h−1) and intravasation (0.0034 h−1) rates in NGM alone; however, we observed no proliferation or intravasation events in NGM supplemented with doxorubicin, which confirms that doxorubicin is a potent mitotic inhibitor and further supports mitosis at the ECM-vessel interface as a potential mechanism for tumor cell intravasation.
Tumor cell division enables detachment from clusters
We have observed intravasation from BCCs that collectively invade into the interface and overrun the vessel endothelium, thus bypassing TEM. Direct exposure to vessel flow, tumor cell overgrowth, and cell division all increase BCC susceptibility to shear forces and detachment (Supplementary Videos S10). This condition is similar to tumor vasculature that is incompletely lined by endothelial cells and/or partially lined by tumor cells, which is estimated to comprise up to 4% of the vascular surface area in colon carcinoma (16), thought to account for roughly 106 tumor cells per gram of tumor tissue shed per day, and thus permits the entry of both single and clusters of tumor cells (15,16,37). Here, the majority of exposed tumor cells detaching into flow are readily identified as undergoing cell division, and several having completed cytokinesis, appear as doublets, or two-cell clusters (Supplementary Video S11). While both monoclonal and polyclonal clusters of circulating tumor cells have been detected in mice (33,34), our mitosis-mediated intravasation model provides a plausible mechanism by which both single and two cell monoclonal clusters enter circulation.
Transendothelial migration without mitosis leads to blebbing
TEM exposes tumor cells to vessel flow, but does not enable detachment. We observe single tumor cells at the ECM-vessel interface that become directly exposed to vessel flow, but do not intravasate over 5 days (Supplementary Fig. S5), likely due to firm adhesion to the ECM substrate. Tumor cell division induces TEM by physically straining the overlying endothelium and reducing cell adhesion through the disassembly of focal adhesions (38). After being pulled into vessel flow, BCCs often leave behind protrusions anchored to the ECM substrate, which further indicates the competing role adhesion plays with shear stress-induced detachment (Supplementary Fig. S6). In the case where tumor cells at the interface extend protrusions across the endothelium and into vessel flow, the intraluminal shear stress and constriction from the surrounding endothelium typically results in shed portions or blebs from the tumor cells (Supplementary Video S12) and does not lead to intravasation over multiple days. This observation is consistent with that in zebrafish inoculated with MDA-MB-435 melanoma cells where blebs lost from tumor cell membranes extended into microvessels did not lead to full cell intravasation (4).
Perivascular space reduces mitosis-mediated disruption of the endothelium
In our model, intraluminal pressure is one of several factors (e.g. cell size, ECM stiffness) that dictate the force applied by a dividing tumor cell on endothelial cell-cell junctions. Microvessels are maintained under positive intraluminal pressure through relatively high vessel flow rates (1 – 2 ml h−1), thus resembling arterioles where the endothelium is in direct contact with the ECM. We can mimic post-capillary venules by reducing the flow rate, thus decreasing the intraluminal pressure and allowing the vessel endothelium to contract; this results in a gap or perivascular space between the vessel wall and ECM. The perivascular space is a common feature among post-capillary venules in the brain where immune cells preferentially extravasate and tumor cells can achieve greater dispersal by migrating along the interface (39,40). Under these conditions, dividing tumor cells within the perivascular space exert a smaller force on the overlying endothelium and are less likely to physically disrupt the endothelium during TEM (Supplementary Video S13).
To assess the time required for mitosis-mediated intravasation, we determined the time period between the initiation of cell rounding, exhibited when the tumor cell circularity surpassed 0.6, and tumor cell escape into circulation. Based on this criterion, the intravasation time varied from 20 minutes to 9.3 hours (Supplementary Figure S4). This range is consistent with intravasation times on the order of hours observed by IVM in mouse models (6), and here, the duration likely reflects variation in the cohesive strength of endothelial cell-cell junctions and remaining tumor cell focal adhesions to the ECM. Since at least half of all intravasation events were directly preceded by mitosis, this implies that mitosis-mediated intravasation is faster than other mechanisms, at least in our model.
DISCUSSION
Using a tissue engineered microvessel model (13), we have imaged and characterized invasion and intravasation of single MDA-MB-231 breast cancer cells (BCCs). The predominant mechanism that facilitated transendothelial migration (TEM) and detachment was tumor cell division at the ECM-vessel interface. Alternative methods for TEM, such as extending protrusions across the endothelium into vessel flow, typically resulted in shedding portions of the BCC and did not lead to intravasation. To confirm the important role of mitosis in physically disrupting endothelial junctions at the ECM-vessel interface, we observed that dividing BCCs in a perivascular space do not deflect the endothelium or intravasate. While our main focus is on intravasation across functional microvessels, we also observe intravasation from BCCs directly exposed to vessel flow after displacing the endothelium; under these conditions, tumor cell mitosis leads directly to cell detachment due to increased exposure to vessel flow and reduced adhesion to the ECM and/or nearby tumor cells. Since detaching tumor cells are in the process of dividing, a significant fraction are observed to have completed cytokinesis and will be circulating doublets, which is consistent with in vivo reports of monocolonal CTC clusters (33).
During a total of 2,330 cell hours imaged at the ECM-vessel interface, we observed 8 intravasation events. All 8 events involved tumor cell rounding and 4 events were definitively associated with mitosis (Supplementary Fig. S2). Cell rounding resulted in deflection of the overlying endothelium rather than the stiffer ECM substrate. The stiffness and height of adherent cells are known to transiently increase during cytokinesis while focal adhesions disassemble and integrin-mediated signaling is inactivated (38,41). If the force on the endothelium is sufficient to disrupt cell-cell junctions, then the tumor cell body becomes exposed to vessel flow, which initiates cell rolling, loss of cell adhesion to the ECM substrate, and tumor cell escape into circulation (Fig. 3L). The observations that: (1) all intravasating cells exhibited a maximum width ≥ 20 μm, and that (2) the majority of dividing cells less than 20 μm in maximum width (10 out of 15) failed to intravasate are consistent with a threshold force necessary to disrupt endothelial cell-cell junctions (Fig. 3J).
From the number of intravasation events imaged at the vessel equator (8 events over of 2,330 cell hours), we obtain an intravasation rate constant of 3.4 x 10−3 h−1. Although we limit our quantification to events occurring at the vessel equator, which captures about 10% of the vessel’s surface area, our intravasation rate is normalized to observed cell hours, and thus is applicable to all BCCs at the ECM-vessel interface. This rate is approximately 4-fold less than the intravasation frequency of 10−2 h−1 reported for the MMTV-PyMT mouse model of mammary carcinoma using intravital microscopy (IVM) where nearly all intravasation events occurred within close proximity of tumor associated macrophages (TAMs) along the local microvasculature (6). TAMs were shown to transiently down-regulate endothelial barrier properties through the secretion of VEGF, which enabled the intravasation of single tumor cells (6). Tumor cells have also been shown to utilize TAM-independent mechanisms of intravasation through the over-expression of growth factors such as HBEGF that down-regulate endothelial barrier properties (10). The lower intravasation rate observed in this model may be attributed to the lack of TAMs, which is consistent with the finding that the intravasation rate is increased by the presence of macrophages in a separate in vitro model (42). The inverse of our intravasation rate suggests that BCCs spend on average approximately 294 hours, or 12 days at the interface before intravasating across an intact vessel; however, the average time for mitosis-mediated intravasation to occur is 4.37 ± 1.5 h (n = 6) (Supplementary Fig. S4) and is consistent with IVM observations (6).
Tumor cell phenotype can influence the intravasation rate in a number of ways. For example, tumor cell speed at the ECM-vessel interface determines the sampling rate at which the tumor cell can find local disruptions or triple point junctions in the endothelium where there is an increased probability of successful TEM. The tumor cell’s proliferation rate determines the frequency of cell rounding and hence the opportunities for TEM due to force generated on the endothelium. The rate of tumor cell proliferation at the ECM-vessel interface is 0.016 h−1 (37 events over 2,330 cell hours), which is comparable to the tumor cell division rate within the ECM of 0.014 ± 0.0047 h−1 (19 events over 1,301 cell hours in n = 6 microvessels). Roughly 10% of dividing cells at the ECM-vessel interface were able to complete intravasation (i.e. TEM and detachment) under normal growth media conditions. Introducing doxorubicin, a potent mitotic inhibitor and commonly used chemotherapeutic for triple negative breast cancer, abolished tumor cell division after 8 hours of exposure; no intravasation events were observed over the recorded 617 cell hours acquired from 61 different tumor cells, which further suggests a link between proliferation and single/doublet tumor cell intravasation. Drawbacks to using mitotic inhibitors to assess metastatic potential include secondary effects on tumor cell function. For example, doxorubicin is implicated in disrupting cytoskeletal function, focal adhesions, and thus tumor cell invasion and potentially intravasation (43); however, ≈5% (3 out of 61 interface BCCs) of interfacial BCCs were observed invading into the ECM-vessel interface during 8 – 24 hours of doxorubicin perfusion, which is comparable to the 7.7% (17 out of 221 interface BCCs) under normal growth media conditions and suggests that the pharmacologically relevant doses of doxorubicin were not affecting tumor cell invasion and EMT to a large degree during the exposure.
Additionally, a larger tumor cell size will generate a greater deflection on the endothelium during mitosis, resulting in higher stress and possible separation of endothelial cell-cell junctions. The tugging force between endothelial cell-cell junctions (e.g. adherens and tight junctions) has been measured at approximately 0.5 – 8 nN μm−2 of junctional area, and the maximum force between two cells begins to saturate above 70 nN (44). Therefore, the separation force necessary for TEM between endothelial cells depends on both junctional area and integrity. Weakened endothelial junctions due to matrix stiffening has been correlated to increased vessel permeability and leukocyte trafficking (45). Interestingly, junctional strength between endothelial cells increases over the time-scale of minutes to compensate for exogenously applied forces (44); therefore, sudden deflection forces, such as tumor cell rounding/mitosis, may be more likely to break cell-cell junctions at the ECM vessel interface.
Similarly, the structure and function of the endothelium plays a key role in regulating intravasation. The cohesive force of local cell-cell junctions must be overcome to enable TEM (46), which has been studied in the TEM of leukocytes (39,45). Factors that down-regulate barrier function, either locally secreted by tumor cells or due to inflammation, are likely to promote intravasation (6). While factors such as EGF and VEGF increase the concentration of circulating tumor cells in tumor-bearing mice (6,7,10), it has not been established whether this is due solely to down-regulation of vessel barrier function or from modulation of tumor cell phenotype.
Accumulating evidence suggests that subpopulations of tumor cells, specifically cancer stem cells (CSC) and/or progenitor cells, exhibit a higher metastatic potential due to their increased capacity for self-renewal, invasion, and survival (47,48). Furthermore, CSCs may participate in the formation of functional tumor lined vessels, termed vasculogenic mimicry, that promote neoplastic growth and tumor progression (49). The mechanism by which CSCs intravasate is not well known; however, vasculogenic mimicry precludes the need for TEM during intravasation and the detection of circulating CSCs in the blood of patients with colorectal cancer is a negative prognostic indicator (50). The positive expression of CD133, a cancer stem cell marker detected in subpopulations of various tumor types (e.g. prostate, breast, colorectal, etc.) (50,51), on MDA-MB-231 BCCs is inconsistently reported (52,53). The MDA-MB-231 BCCs used in this study did not overtly express CD133 in any subpopulation of cells grown on fibronectin coated glass interrogated by immunofluorescence staining (Supplementary Fig. S7). The lack of CD133 positive cells may be due to clonal variation, but does not discount the role of CSCs on intravasation. This tumor-microvessel model may be uniquely capable of investigating the intravasation potential of CSCs by substituting a CSC rich population within the ECM. In our mitosis-mediated intravasation model, an asymmetric CSC division event at the interface would enable a CSC population to be maintained at the interface while shedding differentiated tumor cells (54); these released cells can be collected downstream and compared to those at the interface.
In addition to tumor cell phenotype, our results demonstrate that the probability a dividing tumor cell will intravasate depends on specific properties of the tumor microenvironment, such as matrix stiffness, vessel barrier function, intraluminal pressure, and matrix adhesion. ECM stiffness increases the mechanical force a tumor cell is able to exert on the overlying endothelium when undergoing cell division. High stiffness in breast tumor tissue is a negative prognostic indicator for vascular invasion (55). Stronger endothelial cell-cell junctions will improve the ability of the vessel endothelium to remain intact despite the underlying force exerted by a dividing tumor cell. Furthermore, if the intraluminal pressure decreases, then the endothelium as a whole may compress inward and the force of the dividing tumor cell is minimized (Supplementary Video S13). However, if a tumor cell has disrupted the endothelium and is exposed to intraluminal flow, its adhesion to the vessel basement membrane may oppose the shear stress and resist complete detachment and escape (Supplementary Video S9).
If we consider the rate constant for intravasation (kin) as a processes that requires an energy barrier (kT) to be surmounted, we can write kin = A exp(−EA/kT) where A is the attempt frequency and EA is the activation energy. The attempt frequency is simply the proliferation rate and the Boltzmann factor, exp(−EA/kT), represents the probability that a mitotic tumor cell is able to exert sufficient force to overcome the endothelial cell-cell junctions at that location. Therefore, by taking our intravasation rate (3.4 × 10−3 h−1) over our proliferation rate (0.014 h−1), we obtain the probability that a dividing tumor cell at the interface intravasates is approximately 0.24. The energy barrier for intravasation, kT, likely depends on a variety of factors mentioned above, such as the strength of endothelial cell-cell junctions, stiffness of the ECM, and tumor cell width, which in principle, could be deduced from additional physical measurements.
In summary, the use of a tissue-engineered microvessel model of the tumor microenvironment has revealed a novel mitosis-mediated mechanism of intravasation. Tumor cell division at the ECM-vessel interface exerts a positive force on the vessel endothelium where a larger tumor cell size and stiffer ECM substrate will increase the deflection of the vessel wall and stress on individual endothelial cell-cell junctions. Mitosis in this environment reduces a tumor cell’s adhesion to the ECM while increasing its size perpendicular to the interface and susceptibility to vessel shear stress and detachment. Since we have shown that mitosis is correlated to intravasation, tumor cells at the ECM-vessel interface with a higher proliferation rate will have an increased likelihood of TEM and escape into vessel flow. Strategies that target tumor cell proliferation, improve endothelial barrier properties, and reduce intraluminal pressure and ECM matrix stiffness will address mitosis-mediated TEM and detachment and may ultimately decrease the rate of intravasation.
Supplementary Material
Acknowledgments
Financial support: NCI Provocative Questions (NIH R01CA170629)
Footnotes
Conflicts of interest: none
References
- 1.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 3.Hayes DF, Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Miller MC, et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:4218–24. doi: 10.1158/1078-0432.CCR-05-2821. [DOI] [PubMed] [Google Scholar]
- 4.Stoletov K, Montel V, Lester RD, Gonias SL, Klemke R. High-resolution imaging of the dynamic tumor cell-vascular interface in transparent zebrafish. P Natl Acad Sci USA. 2007;104:17406–11. doi: 10.1073/pnas.0703446104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zijlstra A, Mellor R, Panzarella G, Aimes RT, Hooper JD, Marchenko ND, et al. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer Research. 2002;62:7083–92. [PubMed] [Google Scholar]
- 6.Harney AS, Arwert EN, Entenberg D, Wang YR, Guo P, Qian BZ, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2(hi) Macrophage-Derived VEGFA. Cancer Discov. 2015;5:932–43. doi: 10.1158/2159-8290.CD-15-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 8.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11:573–87. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deryugina EI, Quigley JP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. 2008;130:1119–30. doi: 10.1007/s00418-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou ZN, Sharma VP, Beaty BT, Roh-Johnson M, Peterson EA, Van Rooijen N, et al. Autocrine HBEGF expression promotes breast cancer intravasation, metastasis and macrophage-independent invasion in vivo. Oncogene. 2014;33:3784–93. doi: 10.1038/onc.2013.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katt ME, Placone AL, Wong AD, Xu Z, Searson PC. In vitro tumor models: advantages, disadvantages, variables, and selecting the right platform. Frontiers in Bioengineering and Biotechnology - Tissue Engineering and Regenerative Medicine. 2016;4:e12. doi: 10.3389/fbioe.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogorad MI, DeStefano J, Karlsson J, Wong AD, Gerecht S, Searson PC. Review: in vitro microvessel models. Lab Chip. 2015;15:4242–55. doi: 10.1039/c5lc00832h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong AD, Searson PC. Live-Cell Imaging of Invasion and Intravasation in an Artificial Microvessel Platform. Cancer Res. 2014;74:4937–45. doi: 10.1158/0008-5472.CAN-14-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman RM. The multiple uses of fluorescent proteins to visualize cancer in vivo. Nature reviews Cancer. 2005;5:796–806. doi: 10.1038/nrc1717. [DOI] [PubMed] [Google Scholar]
- 15.Liotta LA, Kleinerman J, Saidel GM. Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res. 1974;34:997–1004. [PubMed] [Google Scholar]
- 16.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci U S A. 2000;97:14608–13. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–6. [PubMed] [Google Scholar]
- 19.Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS One. 2009;4:e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee TH, Avraham H, Lee SH, Avraham S. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. The Journal of biological chemistry. 2002;277:10445–51. doi: 10.1074/jbc.M107348200. [DOI] [PubMed] [Google Scholar]
- 22.Liang M, Zhang P, Fu J. Up-regulation of LOX-1 expression by TNF-alpha promotes trans-endothelial migration of MDA-MB-231 breast cancer cells. Cancer letters. 2007;258:31–7. doi: 10.1016/j.canlet.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Ling Y, Chen Y, Li CL, Feng F, You QD, et al. Flavonoid baicalein suppresses adhesion, migration and invasion of MDA-MB-231 human breast cancer cells. Cancer letters. 2010;297:42–8. doi: 10.1016/j.canlet.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 24.Chen MB, Whisler JA, Jeon JS, Kamm RD. Mechanisms of tumor cell extravasation in an in vitro microvascular network platform. Integrative Biology. 2013;5:1262–71. doi: 10.1039/c3ib40149a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser SP, Ozerlat-Gunduz I, Onkal R, Diss JK, Latchman DS, Djamgoz M. Estrogen and non-genomic upregulation of voltage-gated Na+ channel activity in MDA-MB-231 human breast cancer cells: Role in adhesion. Journal of cellular physiology. 2010;224:527–39. doi: 10.1002/jcp.22154. [DOI] [PubMed] [Google Scholar]
- 26.Jeon JS, Zervantonakis IK, Chung S, Kamm RD, Charest JL. In vitro model of tumor cell extravasation. PloS one. 2013;8:e56910. doi: 10.1371/journal.pone.0056910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang M, Zhang P, Fu J. Up-regulation of LOX-1 expression by TNF-α promotes trans-endothelial migration of MDA-MB-231 breast cancer cells. Cancer letters. 2007;258:31–7. doi: 10.1016/j.canlet.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, et al. Visualizing extravasation dynamics of metastatic tumor cells. Journal of cell science. 2010;123:2332–41. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nature cell biology. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 30.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nature cell biology. 2007;9:1392–400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 31.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. The Journal of cell biology. 2009;185:11–9. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophysical journal. 2013;104:2077–88. doi: 10.1016/j.bpj.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A. 2016;113:E854–63. doi: 10.1073/pnas.1508541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. The Journal of pharmacy and pharmacology. 2013;65:157–70. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 36.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clinical pharmacokinetics. 2003;42:419–36. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 37.Bockhorn M, Jain RK, Munn LL. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? The Lancet Oncology. 2007;8:444–8. doi: 10.1016/S1470-2045(07)70140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamakita Y, Totsukawa G, Yamashiro S, Fry D, Zhang X, Hanks SK, et al. Dissociation of FAK/p130(CAS)/c-Src complex during mitosis: role of mitosis-specific serine phosphorylation of FAK. The Journal of cell biology. 1999;144:315–24. doi: 10.1083/jcb.144.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. Journal of neuropathology and experimental neurology. 2008;67:1113–21. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- 40.Burden-Gulley SM, Qutaish MQ, Sullivant KE, Lu H, Wang J, Craig SE, et al. Novel cryo-imaging of the glioma tumor microenvironment reveals migration and dispersal pathways in vivid three-dimensional detail. Cancer Res. 2011;71:5932–40. doi: 10.1158/0008-5472.CAN-11-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matzke R, Jacobson K, Radmacher M. Direct, high-resolution measurement of furrow stiffening during division of adherent cells. Nature cell biology. 2001;3:607–10. doi: 10.1038/35078583. [DOI] [PubMed] [Google Scholar]
- 42.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci U S A. 2012;109:13515–20. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Millerot-Serrurot E, Guilbert M, Fourre N, Witkowski W, Said G, Van Gulick L, et al. 3D collagen type I matrix inhibits the antimigratory effect of doxorubicin. Cancer cell international. 2010;10:26. doi: 10.1186/1475-2867-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, et al. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci U S A. 2010;107:9944–9. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, et al. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Science translational medicine. 2011;3:112ra22. doi: 10.1126/scitranslmed.3002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–70. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 47.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea--a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. discussion 95–6. [DOI] [PubMed] [Google Scholar]
- 48.Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell stem cell. 2010;6:603–15. doi: 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Yao XH, Ping YF, Bian XW. Contribution of cancer stem cells to tumor vasculogenic mimicry. Protein & cell. 2011;2:266–72. doi: 10.1007/s13238-011-1041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iinuma H, Watanabe T, Mimori K, Adachi M, Hayashi N, Tamura J, et al. Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes’ stage B and C colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1547–55. doi: 10.1200/JCO.2010.30.5151. [DOI] [PubMed] [Google Scholar]
- 51.Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. Journal of cell science. 2004;117:3539–45. doi: 10.1242/jcs.01222. [DOI] [PubMed] [Google Scholar]
- 52.Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013;32:544–53. doi: 10.1038/onc.2012.85. [DOI] [PubMed] [Google Scholar]
- 53.Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. Journal of cellular and molecular medicine. 2009;13:2236–52. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 55.Evans A, Whelehan P, Thomson K, McLean D, Brauer K, Purdie C, et al. Invasive breast cancer: relationship between shear-wave elastographic findings and histologic prognostic factors. Radiology. 2012;263:673–7. doi: 10.1148/radiol.12111317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.