

Abstract
In a recent paper in Science, Tomasetti et al. present an expanded model for cancer risk, which they claim demonstrates the relative contribution of mutations caused by replication errors, environment and heredity. The foundation of this model is the theory that the overwhelming driver of cancer risk is mutations. This perspective will present experimental evidence and evolutionary theory to challenge the basis of this underlying theory. An argument will be presented that the mutation-centric model of cancer suggests unrealistic solutions to cancer and distracts the research community from more promising approaches that consider tissue context.
Keywords: microenvironment, cancer risk, Somatic Mutation Theory, aging, oncogenesis
The recent paper from Tomasetti, Li and Vogelstein (1) begins with “It is now widely accepted that cancer is the result of the gradual accumulation of driver gene mutations that successively increase cell proliferation”. The authors are correct that this paradigm is widely accepted. Indeed, mutations are necessary for cancer development. However, by not considering the context-dependence of mutational effects on cellular fitness and the effects of aging and other carcinogenic factors on tissue microenvironments, this paradigm is inadequate to explain links between cancers and their causes. If this paradigm is fundamentally deficient, the model developed by Tomasetti et al. estimating the contributions of replication-associated mutations (R), environmental factors (E) and heredity (H) to cancer risk is called into question.
In their modeling, the impacts of R, E and H on cancer incidence are solely ascribed to their effects on mutation occurrence (Fig. 1-top). The idea that endogenous and environmental factors influence cancer development primarily by increasing the burden of mutations stands in opposition to 1) basic evolutionary theory and 2) substantial evidence to the contrary.
Fig. 1. Models connecting cancer to its causative contexts.
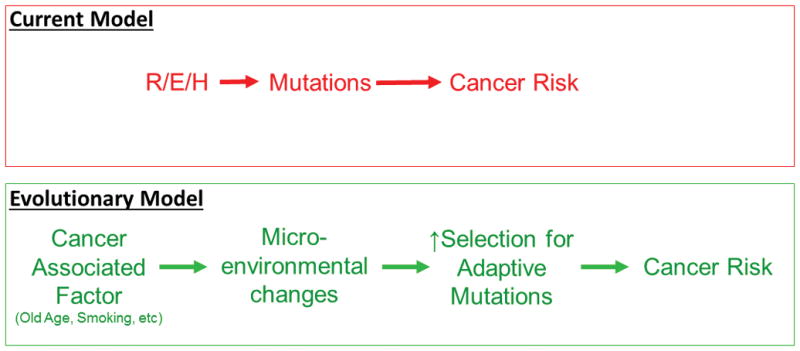
Top: According to the current paradigm that forms the foundation for the Tomasetti et al. model, the primary role of R, E and H in determining cancer risk is through increasing mutational burden in tissue stem cells. Bottom: An alternative model posits that the major impact for contexts like old age and environmental exposures on cancer risk is through alterations in tissue microenvironments that promote selection for adaptive mutations that contribute to cancer development. Adaptive mutations could emanate from R, E and/or H, and increases in mutational burden from R, E and/or H should increase cancer risk. However, the impact of cancer-associated factors on selection will have a much greater deterministic role.
1 – The authors are correct that many mutations result from normal cellular processes (e.g. errors during DNA replication and oxidative base damage). Whether engendered by intrinsic or extrinsic causes, base substitutions or modifications and insertions/deletions that escape DNA repair lead to mutations (2). The modeling by Tomasetti et al. rests on the premise that these mutations limit cancer etiology and thus cancer risk – who gets cancer and when. Of course, evolution requires mutations (including epigenetic changes for somatic evolution). But mutations are not the drivers of evolution – they are the substrate upon which natural selection acts. We know that the evolution of species does not result from the gradual accumulation of advantageous mutations, but instead reflects adaptation in response to intermittent environmental changes (3,4). Similarly, for somatic evolution, selection on mutations is highly dependent on tissue context. Mutations that are purged by stabilizing selection in young, healthy tissues can become positively selected late in life or in damaged tissue, due to adaptation to altered tissue environments (Fig. 1-bottom and Fig. 2) (e.g. (5)). In addition to the role for tissue changes in altering selective pressures for oncogenic events, the microenvironment can directly influence the phenotype of malignant cells without altering their genetic makeup (6). In fact, carcinoma cells and reprogrammed melanoma cells can even contribute to normal tissue architecture when incorporated into mouse embryos (7,8). Inherited genetic polymorphisms can also influence parameters that would be expected to affect malignant phenotypes and the fitness impact of oncogenic mutations, such as by modulating inflammation and immune function (often through complex interactions of many alleles) (e.g. (9)). By only accounting for mutation occurrence, the impacts of R, E and H on tissue microenvironments, immunity and other non-cell autonomous factors are not considered.
Figure 2. Changing roles of somatic selection in life.
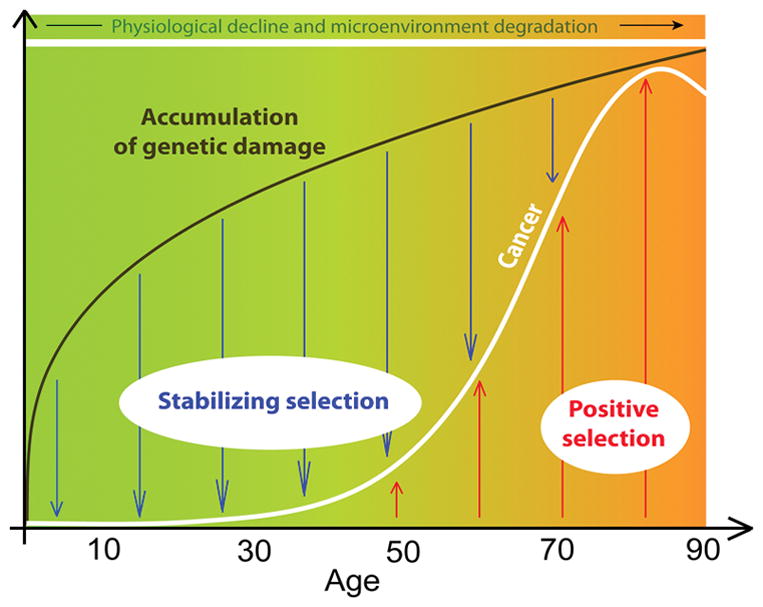
Mutations and epigenetic changes accumulate rapidly during ontogeny and body growth, and then more slowly post-maturation. Through periods of likely reproduction, stabilizing selection is dominant in healthy tissue microenvironments, leading to suppression of oncogenic clonal expansions. Late in life, the degradation of tissue microenvironments engenders positive selection for adaptive mutations, leading to oncogenic clonal expansions. The expansion of cell clones bearing oncogenes increases the risk of cancer.
2 - Numerous studies reveal that the increase in mutational burden brought about by aging, smoking and other carcinogenic exposures cannot explain most of the impact on cancer risk. First, roughly half of mutations and epigenetic changes occur by maturity (10–12), consistent with the much more rapid cycling of cells during ontogeny and body growth (13,14) (Fig. 2). Mutational load changes modestly from maturity to old ages (in well-powered studies, typically 2–3-fold) (10–12), and yet cancer risk rises exponentially in older ages. Even for smoking, mutations appear to account for only a fraction of the substantial increase in associated cancer risk. A recent report examined mutational burden in 13 cancers associated with smoking (15). Of these 13 cancers, only lung adenocarcinoma and larynx cancer showed a substantial smoking-dependent increase in mutational burden (4.5- and 2.5-fold, respectively). Liver and kidney cancers showed less than 1.5-fold increases, and 9 of the 13 cancers showed no significant differences in mutations. Notably, the smoking-associated increase in mutations for all cancers combined was only 1.15-fold. Thus, smoking-induced mutations alone cannot explain the increased risk (up to 110-fold) for various cancers.
Lifetime mutation accumulation in stem cells cannot explain varying cancer predisposition across tissues and species. A recent analysis of mutation accumulation in stem cells from human liver, small intestine and large intestine showed that very similar numbers of mutations (roughly 2500) accumulated per epithelial stem cell in each of these tissues by old age (16). Yet carcinoma incidence is about 5 and 30-fold higher for the large intestine relative to liver and small intestine, respectively. For mice, a similar study showed that stem cells in the large and small intestines accumulated roughly 250 and 500 mutations in a lifetime, respectively (17). Thus, each human intestinal stem cell accumulates 5–10-fold more mutations relative to mouse, likely resulting from the 20–30-times longer potential lifespans for humans compared to laboratory mice. If we consider that human intestines should have greater than 1000-fold more cells, humans should accumulate roughly 10,000-fold more mutations in this organ than mice in a lifetime. While similar comparisons are not available for stem cells in other tissues, given the ~2000-fold greater cellularity of a human, overall mutation load doubtfully explains the similar rates of malignancy development in mice and humans.
Instead, we need to consider how aging or carcinogens change tissue microenvironments to increase selection for particular oncogenic mutations. The role for microenvironmental change in altering selective pressures is not accounted for in the Tomasetti et al. model. Just as for organismal evolution, mutations (including epigenetic changes) are required for somatic evolution and cancer to occur. However, just as for organismal evolution, context-dependent alterations in the forces of selection (dependent on age and carcinogen exposure) dictate cancer timing and incidence (18,19).
A powerful experimental example of mutations not limiting cancer incidence comes from mice with heterozygous mutations in DNA polymerase δ that disable its proofreading function (20). The L604G and L604K mutations increase the rate of somatic point mutations by 5- and 4-fold and DNA rearrangements by 17- and 38-fold, respectively. Strikingly, the +/L604G mice exhibit lifespans and cancer incidence curves that are similar to wild-type mice. Thus, a substantial increase in mutational burden did not significantly increase cancer incidence. The second DNA polymerase δ mutation (L604K) results in earlier, but not increased, cancer occurrence, a shift that parallels the shortened lifespans of these mice. Physiological decline, rather than altered mutation frequency, better explains altered age-dependent cancer risk in +/L604K mice.
Additional arguments against the mutation-centric paradigm include that oncogenic mutations frequently reduce the somatic fitness of stem cells in healthy niches (21), that oncogenic mutations are detected in normal tissues at rates that far exceed the frequencies of associated cancers (e.g. (22)), that malignancies driven by very different numbers of oncogenic mutations (such as chronic myeloid leukemia compared to most carcinomas) display similar age-dependent incidence curves (23), and that larger animals like blue whales do not suffer more cancer than smaller ones like mice (24). In total, mutation load correlates poorly with cancer risk across a lifetime, between tissues, for genotypes within a species, and across species. Therefore, one cannot estimate the contributions of R, E and H to cancer incidence solely by considering their impact on mutations.
Still, since mutations are clearly necessary for cancer development, should not a model that estimates cancer risk based on contributions of R, E and H to mutations have predictive value? The value should depend on the extent to which mutation occurrence dictates who gets cancer, how cancer risk relates to known causes, which tissues it occurs in, and when the cancers develop in life. As argued above, mutation occurrence correlates poorly with cancer risk, and many contexts can greatly modulate cancer risk without similarly impacting mutations. Consider the associations of viruses with cancer risk. It is well accepted that oncogenes encoded by Epstein-Barr virus (EBV) directly contribute to the development of Burkitt lymphoma (25). However, 90% of humans are estimated to be infected chronically with EBV, and yet Burkitt lymphoma is very rare, requiring multiple other secondary factors (e.g. concomitant infection with Human Immunodeficiency Virus or malarial parasites). While it is useful to understand the role of EBV in Burkitt lymphoma pathogenesis, it would not be very useful to try to understand the risk of this lymphoma based on EBV infection. Analogously to EBV infection, humans and other animals (from mice to blue whales) will experience many oncogenic mutations throughout our lives, and yet only a fraction of us will develop cancer, likely late in life. Estimating cancer risk, whether per tissue or per individual, based solely on mutation causation neglects other key factors involved in cancer evolution to the extent that such estimates are likely to be misleading.
Adherence to the mutation-centric paradigm, in the face of abundant evidence opposing it, is likely hindering progress in cancer research and therapeutic development. Tomasetti, Li and Vogelstein propose that we should be able to limit the impact of cancers that they ascribe to R mutations by improving early detection and by reducing mutation occurrence, such as by improving DNA repair (1). While early detection methods continue to have a huge impact on cancer mortality (e.g. for colon cancer), improving DNA repair is doubtfully a realistic or productive strategy. Evolutionary theory may suggest more fruitful approaches. The evolution of large and long-lived animals did not involve reductions in mutation rates – for example, human somatic cells have higher mutation rates than yeast and bacteria (26). Thus, we do not need to lower mutation frequency to prevent or treat cancer. A more effective strategy for cancer prevention could be to alter tissue landscapes in order to limit selection for the inevitable oncogenic mutations. Therapeutic strategies could be used to alter the selective value of cancer phenotypes by increasing the fitness of more benign phenotypes and/or disfavoring malignant genotypes through modulating tissue parameters, as has been shown for pH (27), oxygen levels (28), cytokines (5,29), and the presence of senescent cells (30). In each case, treatments engendering a tissue microenvironment that better approached that of a young healthy individual, such as by ameliorating inflammation, reduced the selective value of malignant or aggressive genotypes and thus impaired cancer initiation and progression. While preventing replication errors is not currently feasible, as these examples illustrate, we do possess the ability to manipulate microenvironmental parameters to limit cancer risk or favor more benign cancer phenotypes.
Acknowledgments
Financial Support: J.D. is supported by NIH grant R01 CA180175.
Mark Johnston, Mark Gregory, Hannah Scarborough and Kelly Higa of the University of Colorado Anschutz Medical Campus provided valuable suggestions for the manuscript, and Andrii Rozhok prepared Figure 2.
References
- 1.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330–4. doi: 10.1126/science.aaf9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 2017;168:644–56. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward PD, Kirschvink JL. A new history of life : the radical new discoveries about the origins and evolution of life on earth. New York: Bloomsbury Press; 2015. pages cm p. [Google Scholar]
- 4.Eldredge N. Reinventing Darwin : the great debate at the high table of evolutionary theory. New York: Wiley; 1995. p. xi.p. 244. [Google Scholar]
- 5.Henry CJ, Casas-Selves M, Kim J, Zaberezhnyy V, Aghili L, Daniel AE, et al. Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. The Journal of clinical investigation. 2015;125:4666–80. doi: 10.1172/JCI83024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature medicine. 2011;17:320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA. 1975;72:3585–9. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hochedlinger K, Blelloch R, Brennan C, Yamada Y, Kim M, Chin L, et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes & development. 2004;18:1875–85. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KT, et al. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell. 2016;167:1099–110.e14. doi: 10.1016/j.cell.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 10.Vijg J, Busuttil RA, Bahar R, Dolle ME. Aging and genome maintenance. Annals of the New York Academy of Sciences. 2005;1055:35–47. doi: 10.1196/annals.1323.007. [DOI] [PubMed] [Google Scholar]
- 11.Horvath S. DNA methylation age of human tissues and cell types. Genome biology. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finette BA, Sullivan LM, O’Neill JP, Nicklas JA, Vacek PM, Albertini RJ. Determination of hprt mutant frequencies in T-lymphocytes from a healthy pediatric population: statistical comparison between newborn, children and adult mutant frequencies, cloning efficiency and age. Mutation research. 1994;308:223–31. doi: 10.1016/0027-5107(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 13.Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. The Journal of clinical investigation. 2006;116:2808–16. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozhok AI, Salstrom JL, DeGregori J. Stochastic modeling indicates that aging and somatic evolution in the hematopoietic system are driven by non-cell-autonomous processes. Aging. 2014;6:1033–48. doi: 10.18632/aging.100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–22. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–4. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–5. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature reviews Cancer. 2012;12:487–93. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rozhok AI, DeGregori J. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8914–21. doi: 10.1073/pnas.1501713112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatesan RN, Treuting PM, Fuller ED, Goldsby RE, Norwood TH, Gooley TA, et al. Mutation at the polymerase active site of mouse DNA polymerase delta increases genomic instability and accelerates tumorigenesis. Mol Cell Biol. 2007;27:7669–82. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2012;32:1869–75. doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surveillance, Epidemiology, and End Results (SEER) Program. ( www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER 9 Regs Research Data, Nov 2014 Sub (1973–2012)
- 24.Caulin AF, Maley CC. Peto’s Paradox: evolution’s prescription for cancer prevention. Trends Ecol Evol. 2011 doi: 10.1016/j.tree.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grywalska E, Rolinski J. Epstein-Barr Virus–Associated Lymphomas. Seminars in Oncology. 2015;42:291–303. doi: 10.1053/j.seminoncol.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 26.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–52. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibrahim-Hashim A, Robertson-Tessi M, Enrizues-Navas P, Damaghi M, Balagurunathan Y, Wojtkowiak JW, et al. Defining cancer subpopulations by adaptive strategies rather than molecular properties provides novel insights into intratumoral evolution. Cancer research. 2017 doi: 10.1158/0008-5472.CAN-16-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–51. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science. 1990;248:1656–60. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 30.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017;7:165–76. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]