

Abstract
The anti-androgen bicalutamide is widely used in the treatment of advanced prostate cancer (PCa) in many countries, but its effect on castration resistant PCa (CRPC) is limited. We previously showed that resistance to bicalutamide results from activation of mechanistic target of rapamycin (mTOR). Interestingly, clinical trials testing combinations of the mTOR inhibitor RAD001 with bicalutamide were effective in bicalutamide-naïve CRPC patients, but not in bicalutamide-pre-treated ones. Here we investigate causes for their difference in response. Evaluation of CRPC cell lines identified resistant vs sensitive in-vitro models, and revealed that increased eIF4E(S209) phosphorylation is associated with resistance to the combination. We confirmed using a human-derived tumor-xenograft mouse model that bicalutamide pre-treatment is associated with an increase in eIF4E(S209) phosphorylation. Thus, AR suppressed eIF4E phosphorylation, while the use of anti-androgens relieved this suppression, thereby triggering its increase. Additional investigation in human prostatectomy samples showed that increased eIF4E phosphorylation strongly correlated with the cell proliferation marker Ki67. SiRNA-mediated knock-down of eIF4E sensitized CRPC cells to RAD001+bicalutamide, while eIF4E overexpression induced resistance. Inhibition of eIF4E phosphorylation by treatment with CGP57380 (an inhibitor of MAPK interacting serine-threonine kinases Mnk1/2, the eIF4E upstream kinase) or inhibitors of ERK1/2, the upstream kinase regulating Mnk1/2, also sensitized CRPC cells to RAD001+bicalutamide. Examination of downstream targets of eIF4E-mediated translation, including survivin, demonstrated that eIF4E(S209) phosphorylation increased cap-independent translation whereas its inhibition restored cap-dependent translation which could be inhibited by mTOR inhibitors. Thus, our results demonstrate that while combinations of AR and mTOR inhibitors were effective in suppressing tumor growth by inhibiting both AR-induced transcription and mTOR-induced cap-dependent translation, pre-treatment with AR antagonists including bicalutamide increased eIF4E phosphorylation that induced resistance to combinations of AR and mTOR inhibitors by inducing cap-independent translation. We conclude that this resistance can be overcome by inhibiting eIF4E phosphorylation with Mnk1/2 or ERK1/2 inhibitors.
Keywords: eIF4E, mTOR, castration resistant prostate cancer, Mnk, androgen receptor
INTRODUCTION
The androgen receptor (AR) is known to play central roles in development and progression of prostate cancer (PCa). Patients with advanced PCa are treated with androgen-deprivation therapy (ADT) (1), but often develop resistance to ADT, resulting in castration-resistant PCa (CRPC). Multiple studies reported that ADT increases activation of mechanistic target of rapamycin (mTOR) (2, 3). Our laboratory demonstrated coordinated activation of mTOR and AR in PCa (4), and that the immunosuppressant rapamycin (sirolimus), a mTOR inhibitor, but not the unrelated immunosuppressant tacrolimus, reduced serum levels of the AR transcriptional target prostate specific antigen (PSA) in male kidney transplantation patients (5). However, rapamycin or its more-bioavailable analog RAD001 (everolimus) were ineffective as single agents in CRPC patients (6). We demonstrated that this is due to an increase in AR transcriptional activity caused by mTOR inhibition (7) and that inhibition of both pathways by combination of RAD001 and the anti-androgen bicalutamide caused growth arrest in some CRPC models.
Bicalutamide is used in many countries following ADT resistance. Based on our results, a Phase II clinical trial to test the efficacy of RAD001 and bicalutamide in bicalutamide-naive ADT-resistant PCa patients was carried out; showing significant efficacy (PSA decline≥50% in 62.5% patients) (8). However, in another trial with a bicalutamide-resistant population, RAD001 and bicalutamide was ineffective (9). The goal of the present study was to identify causes of resistance to RAD001+bicalutamide combination that may explain these differences.
mTOR can act in complex with raptor (mTORC1) or rictor (mTORC2) with entirely different functions (10). mTORC1 increases mRNA translation by phosphorylation of downstream molecules p70S6 kinase (p70S6K) and eukaryotic initiation factor 4E (eIF4E) binding protein-1 (4E-BP1), while mTORC2 regulates survival by phosphorylation of Akt(Ser473) and PKCα. mTORC1-activated p70S6K phosphorylates the 40S-ribosomal protein S6 (11). 4E-BP1 binds to and inactivates eIF4E; while 4E-BP1 phosphorylation by mTOR releases eIF4E which associates with eIF4G to form the translational initiation complex eIF4F. eIF4G-associated eIF4E binds to m7G cap at the 5’-end of eukaryotic mRNAs, initiating translation (12). Significantly, eIF4E(S209) phosphorylation by mitogen-activated protein kinase (MAPK)-interacting kinase1/2 (Mnk1/2) is important for tumorigenicity (13, 14), but not for normal mammalian growth (15, 16). Mnk1/2 knock-down inhibited eIF4E(S209) phosphorylation and suppressed PTEN loss-induced tumorigenesis (14). Further, knock-in mice expressing phospho-resistant eIF4E(S209A) prevented oncogenic transformation by Ras-activation or PTEN-loss in a prostate-specific model (17). Previous studies had indicated that AR transcriptional activity is decreased with PTEN-loss, while PI3K inhibition activates AR signaling (18). AR inhibition activates Akt signaling and combined pharmacological inhibition of PI3K and AR signaling caused regression in a PTEN-deficient PCa model (18).
Increased eIf4E(S209) phosphorylation has been associated with altered affinity to m7G-cap. Mnk1-induced eIF4E phosphorylation is associated with increased internal ribosome entry-site (IRES)-dependent translation (19). However, inhibition of Mnks reduced polysomal recruitment of terminal oligopyrimidine messenger RNA (TOP mRNA), which are targets of cap-dependent translation (20). Thus there is a discrepancy in the literature on the role of eIF4E phosphorylation regarding regulation of cap-dependent translation, which was pursued in the current project.
Here, we demonstrate that resistance of CRPC tumors to RAD001 and bicalutamide correlates with eIF4E phosphorylation. Both bicalutamide and the AR-inhibitor enzalutamide stimulate eIF4E(S209) phosphorylation, which prevents further treatment with combinations of AR and mTOR inhibitors. These results may have implications in ongoing and future clinical trials of AR antagonists (including enzalutamide and the androgen synthesis inhibitor abiraterone acetate, both used for patients with aggressive CRPC) with mTOR inhibitors.
RESULTS
High eIF4E(S209) phosphorylation associated with resistance of CRPC cells to combination of mTOR inhibitor and AR antagonist
We established models of response vs no-response to RAD001+bicalutamide combination, using CRPC lines that did not respond to bicalutamide alone (C4-2, PC346C, CWR-R1 and 22Rv1) (Figure 1A). Sensitivity was defined as >50% loss of viability by MTT assay at physiological doses of 1nM RAD001 and 10μM bicalutamide over 7-days. C4-2 cells were most sensitive to this combination with 69% viability loss (p=0.003) and PC346C most resistant (24% reduction; p=0.004). Similarly, 22Rv1 cells were sensitive to the combination (68.7% viability reduction, p=0.026) while another related line CWR-R1 was less sensitive (39.5% reduction; p=0.0002). Protein characteristics were investigated by western blotting to identify mechanisms of resistance vs sensitivity (Figure 1B). Cell lines sensitive to the combination of RAD001 and bicalutamide (C4-2, 22Rv1) expressed higher levels of phosphorylated Akt(S473) and 4E-BP1(S65) and lower levels of PTEN and phosphorylated eIF4E(S209) compared to resistant lines (PC-346C, CWR-R1).
Figure 1. Correlation between the responses of CRPC cells to bicalutamide (Casodex) and RAD001 and the expression and phosphorylation of various proteins.
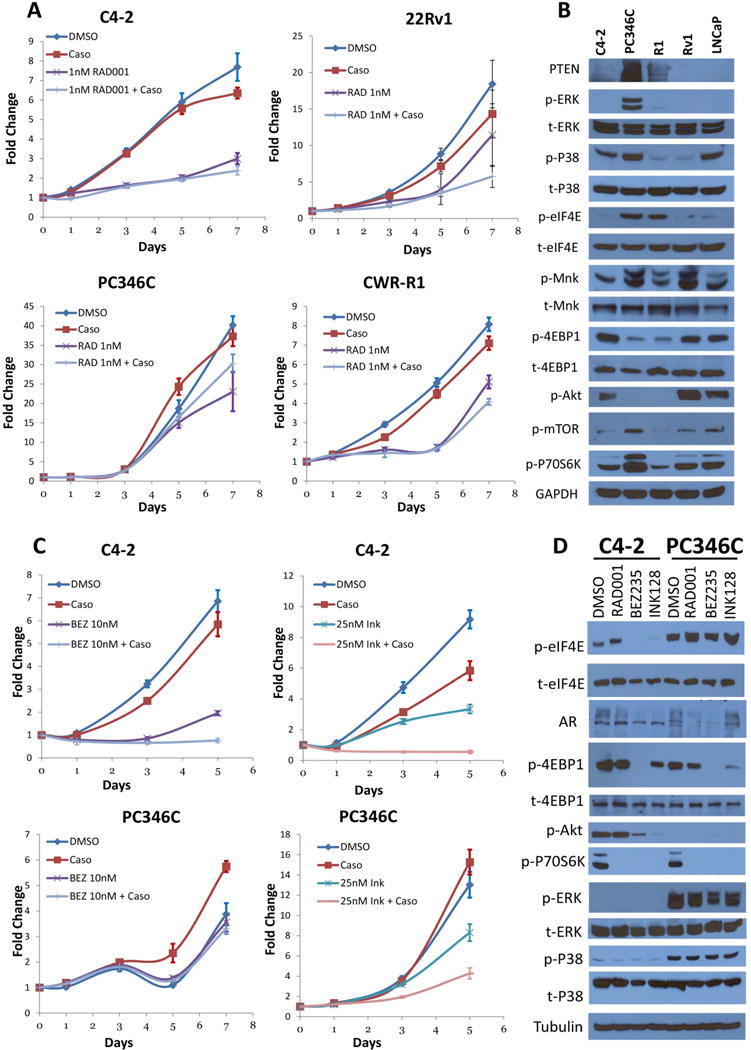
(A) MTT viability assay of PCa cell lines C4-2, 22Rv1 (Rv1), PC346C (not related to AR-null PC-3) and CWR-R1 (R1) treated with RAD001 (1nM) alone or in combination with 10 μM Bicalutamide (Caso) for up to 7 days. In C4-2 cells, bicalutamide and RAD001 individually reduced viability by 17% (p=0.065) and 61% (p-0.002) respectively, compared to control. However, the combination reduced viability by 69% (p=0.002), therefore improving the effects of both. Similarly, in 22Rv1 cells, neither bicalutamide (22%, p=0.26) nor RAD001 (38%, p=0.192) alone causes a significant viability reduction compared to control. However, the combination reduced viability by 68.7% (p=0.0259). Even in the resistant line CWR-R1, bicalutamide (12% decrease, p=0.004) improved the effects of RAD001 alone (36% decrease, p=0.0003) to 49.5% (p=0.0002); however, in PC-346C, bicalutamide made the effects of RAD001 (42.5%, p=0.023) worse (24.6%, p=0.004). Bicalutamide sensitive LNCaP cells were used as positive control. (Sensitivity to the drug combination was defined as >50% loss of viability). Results are shown in absorbance fold change to first day of treatment. (B) Western blots of C4-2, PC346C, CWR-R1, 22Rv1 and LNCaP cells without any treatment to show and compare basal levels of protein expression and phosphorylation between these cell lines. (C) MTT viability assay of C4-2 and PC346C cells challenged with the rapalogs BEZ235 (10nM) or INK128 (25nM) alone or in combination with Bicalutamide (Caso) for up to 7 days. Similar to RAD001, only C4-2, but not PC-346C, responded to the combination of BEZ235 and bicalutamide (89% reduction for C4-2, p=0.0008; 14.5% for PC-346C, p=0.123); however, both cell lines responded, at least partially, to the combination of INK128 and bicalutamide (94% reduction for C4-2, p=0.00013; 67.2% for PC-346C, p=0.006). (D) Western blots showing the mTOR pathway molecular profile of C4-2 and PC346C treated with RAD001, BEZ235 or INK128 for 48 hours.
We also tested by MTT the viability of C4-2 and PC-346C against the mTOR/PI3K inhibitor BEZ235 and the dual mTORC1/C2 inhibitor INK128, either alone or in combination with bicalutamide (Figure 1C). Similar to RAD001, C4-2 responded to both BEZ235 (reduced viability by 71.5% (p=0.002)) and INK128 (reduced by 63.5% (p<0.0001)). In contrast, PC-346C cells showed no significant response to BEZ235 (8.3%, p=0.351), and a small one to INK128 (36.3%, p=0.006). Even in combination with bicalutamide, the response in C4-2 cells were much stronger compared to PC-346C. Similar effects were observed when enzalutamide, a stronger AR inhibitor, was used in combination with mTOR inhibitors (Suppl.Fig. 1). PC-346C cells expressed negligible phosphorylated Akt and high phosphorylated ERK and p38MAPK (upstream regulators of eIF4E phosphorylation) compared to C4-2 (Figure 1D). eIF4E phosphorylation was attenuated by both BEZ235 and INK128 in C4-2 cells, where they suppressed growth, but not in PC-346C cells where they had no effect. Based on these results eIF4E phosphorylation correlated with resistance to mTOR and AR inhibitors in CRPC cell lines.
Bicalutamide treatment increases phosphorylation at eIF4E(S209), but not at Akt(S473) or 4E-BP1(S65), in CWR22 xenograft tumors in immunocompromised mice
We also tested the effect of the parent mTORC1 inhibitor rapamycin in combination with bicalutamide in an animal model. Since our clinical trial indicated that the combination is more effective in a bicalutamide-naive population (8), we selected the bicalutamide sensitive CWR22 patient-derived xenograft to test the effects of the drugs. Mice were subcutaneously implanted with suspensions of CWR22 cells. When palpable tumors were observed, animals were treated with (a) vehicle (b) 50mg/Kg bicalutamide (estimated intratumoral concentration 10μM) (c) 8mg/Kg rapamycin (estimated intratumoral concentration 1nM) or (d) rapamycin+bicalutamide (n=6 per arm). Since the effect of this combination on tumor growth is reported (21, 22), we investigated expression of factors identified above - phosphorylated Akt, 4E-BP1, eIf4E, etc. (Table 1). Serum was analyzed for PSA levels (as CWR22 is a human-derived tumor-line), showing that serum PSA was lower in combinations of rapamycin+bicalutamide compared to bicalutamide alone, in support of clinical observations (8) (Figure 2A).
TABLE 1.
Expression of various proteins in mice treated with (i) vehicle, (ii) 50 mg/kg bicalutamide, (iii) 4 mg/kg rapamycin and (iv) bicalutamide + rapamycin. P-value denotes difference between the groups. Significant differences are denoted in bold.
| CONTROL (n = 6) |
BICALUTAMIDE (n = 6) |
RAPAMYCIN (n = 6) |
RAPAMYCIN+BICALUTAMIDE (n = 6) |
P-Value (Kruskal- Wallis Test) |
|
|---|---|---|---|---|---|
| 4E-BP1 | 0.154 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 0 (0–1) | 1 (0–1) | 0 (0–1) | 0.5 (0–1) | |
| Phospho-4E-BP1(ser65) | 0.086 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 2 (2–2) | 2 (1–3) | 1.8 (1.5–2) | 1.5 (1–2) | |
| P70S6 Kinase | 0.007 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 2.5 (1–3) | 2 (2–3) | 1.2 (0–2) | 1.5 (0–2) | |
| Phospho-p70S6K | 0.035 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 0.5 (0.5–1) | 1 (0–1) | 1 (0–2) | 1.5 (1–2) | |
| Phospho-mTOR | 0.078 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 2 (1–2) | 2.5 (2–3) | 2.5 (1–3) | 2 (1–2) | |
| Akt | 0.375 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 1.2 (1–3) | 2 (1–3) | 2.2 (1–3) | 1 (1–3) | |
| Phospho-Akt (Ser 473) | 0.031 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 0.5 (0–1) | 0 (0–1) | 1 (0–3) | 1 (1–2) | |
| eIF4E | 0.139 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 2 (2–2) | 1.5 (1–2) | 2 (2–3) | 2 (1–3) | |
| Phospho-eIF4E | 0.036 | ||||
| N | 6 | 6 | 5 | 6 | |
| Median (Range) | 0.8 (0.7–1) | 1.1 (1–1.5) | 1 (0.8–1.2) | 1 (0.7–1.2) | |
| Phospho-ERK | 0.318 | ||||
| N | 6 | 6 | 6 | 6 | |
| Median (Range) | 0.4(0.0–1) | 0.0(0.0–0.5) | 0.5(0.0–0.5) | 0.5(0.0–1) |
Figure 2. Effect of bicalutamide and the mTORC1 inhibitor rapamycin on PSA levels and the phosphorylation of Akt and eIF4E in immunocompromised mice bearing CWR22 tumors.

(A) Boxplot showing comparison of PSA scores of serum collected from mice in each arm of the study (Bicalutamide alone vs. Bicalutamide in combination with Rapamycin) from weeks 1 and 2 (n=6 per arm). (B) Left - Immunohistochemistry staining of CWR22 xenografts from nude mice treated with DMSO, Rapamycin, Bicalutamide or the combination (n=6 per arm). After end of treatment, mice were euthanized and the tumors excised. Formalin fixed paraffin-embedded tumor specimens were stained with anti-phospho-Akt (Ser473) antibody. Right – Boxplot of phospho-Akt in CWR22 xenografts from different treated mice demonstrating a slight but not significant increase in akt phosphorylation in rapamycin treated mice when compared to vehicle alone and a significant increase in phospho-akt in the dual treated mice when compared to control (p=0.025) and bicalutamide alone (p=0.022) (n=6 per arm). (C) Left – Immunohistochemistry staining of CWR22 xenografts from nude mice with the same treatments as above now blotted against phospho-eIf4E (S209). Right – Boxplot of phospho-eIF4E scores in CWR22 xenografts showing a sharp increase in phosphorylation of eIF4E in Bicalutamide treated mice. Investigators were not blinded to the outcomes in this experiment.
Immunohistochemistry revealed a trend to higher Akt phosphorylation in rapamycin-treated mice compared to controls, and in dually treated tumors compared to vehicle- or bicalutamide-treated ones (Figure 2B). Both p70S6K and phospho-p70S6K, but not phospho-4EBP1, were differentially expressed between the groups (Table 1). Additionally, bicalutamide increased eIF4E phosphorylation (p=0.009) (Figure 2C), indicating that AR-antagonism stimulate eIF4E phosphorylation associated with resistance to AR and mTOR inhibitors.
AR, but not PTEN, negatively affects eIF4E(S209) phosphorylation
In both C4-2 and PC346C, bicalutamide increased p70S6K phosphorylation after 48 hours of treatment, which was prevented by addition of RAD001 (Figure 3A,B) (effect of RAD001 alone is shown in Suppl.Fig. 2). In C4-2 we observed a small increase in phospho-Akt with bicalutamide alone (2.22-fold), but not for the combination (Figure 3A), while in PC-346C, phospho-Akt increased up to 72 hours after treatment (9.79-fold for bicalutamide alone and 14.2-fold for the combination) (Figure 3B). Phosphorylation of eIf4E increased over time in both C4-2 (4.8-fold after 72 hours) and PC-346C cells (3.76-fold after 72 hours) with bicalutamide alone (Figure 3A,B), whereas the addition of RAD001 attenuated this increase in C4-2 cells but not in PC-346C. The increase in eIF4E phosphorylation upon 48-hour bicalutamide treatment is further demonstrated by immunofluorescence in C4-2 cells (Figure 3C).
Figure 3. The AR is a negative regulator of eIF4E phosphorylation at S209.

Western blots of C4-2 (A) and PC346C (B) cells treated with either bicalutamide alone or in combination with RAD001 and collected in different time points from 1 minute to 4 days. Different proteins of the mTOR pathway were blotted to show changes in expression and phosphorylation when treated with the drugs. It is noticeable an increase in P70S6K phosphorylation in both cell lines when treated with Bicalutamide alone, which goes in hand with previous findings from our laboratory where AR antagonists increase mTOR activity. The combination of an mTOR inhibitor with Bicalutamide ceases that activation as shown in both cells. We can also notice that phosphorylation of eIF4E remains the same overall in C4-2 but it’s increased in PC346C in both treatments. (C) Immunofluorescence of C4-2 cells treated for 48 hours with either vehicle (DMSO) or Bicalutamide and stained for phospho-eIF4E (FITC) or nuclear marker DAPI and both stains results overlapped (right) demonstrating increase in eIf4E phosphorylation in Bicalutamide treated cells. (D) Western blots of the normal prostate cell line PRSN1-1 and CRPC cells C4-2, C4-2b and PC346C demonstrating inverse correlation between AR expression and eIF4E phosphorylation. (E) MTT viability assay of PRNS1-1 (upper) or PRNS1-1 cells that were stable transfected with AR mutant T877A (lower) treated with different mTOR inhibitors up to 7 days showing no effect on viability of this cell line by any of the mTOR inhibitors unless AR was expressed. (F) Western blots of mTOR pathway proteins in PRNS1-1 cells transfected with empty vector, WT-AR or T877A mutant demonstrating decrease in phospho-eIF4E when AR is present in cells.
We next compared these cells to C4-2B, another LNCaP derivative, and pRNS-1-1, a PTEN-positive normal prostate-derived cell line immortalized by SV-40 transformation, that subsequently lost the expression of AR in culture (23–26), stimulating eIF4E phosphorylation (Figure 3D). Parental pRNS-1-1 cells were resistant to all three mTOR inhibitors tested (Figure 3E upper). Transfection of AR (T877A), the mutant AR found in LNCaP cells and its derivatives, rendered these cells sensitive to mTOR inhibitors (Figure 3E lower). Significantly, AR(T877A) (also wild-type AR, (Suppl.Fig. 3)) suppressed eIF4E phosphorylation (Figure 3F), supporting a role for eIF4E phosphorylation in resistance to mTOR inhibitors. The effect of AR on eIF4E phosphorylation was independent of PTEN status of the cell, and AR overexpression did not affect PTEN expression (Figure 3F). Further, expression of PTEN in C4-2 cells (Suppl.Fig. 4A) caused a decrease in Akt phosphorylation, but did not significantly affect eIF4E phosphorylation. As a result, although PTEN expression suppressed cell growth in C4-2 cells, it did not affect the response of these cells to the combination of RAD001 and bicalutamide (Suppl.Fig. 4B,C). Taken together, these data demonstrate that AR, but not PTEN, negatively affects levels of eIF4E(S209) phosphorylation and that increased eIF4E phosphorylation in CRPC can result from inhibition of AR activity during ADT.
Figure 3F also shows that expression of wild-type AR, but not mutant AR, suppresses phosphorylation of both Akt and p70S6 kinase, while 4E-BP1 phosphorylation is negatively proportional to AR expression in these cells, irrespective of AR mutation. On the other hand, eIF4E phosphorylation was suppressed by AR, irrespective of its level or mutational status. Both p70S6 kinase (27, 28) and Akt (29, 30) requires phosphorylation by both mTOR and PDK1 for full activation. In contrast, the phosphorylation of 4E-BP1 is mainly dependent on mTORC1 (31), while that of eIF4E, is dependent on Mnk/MAPK (13, 14). We conclude that in pRNS-1-1 cells, wild-type AR, but not mutant AR (T877A), has a strong negative effect on PDK1/PI3K. In contrast, both wild-type and mutant AR equally affects mTOR in these cells, thereby enabling AR-level but not AR-mutation-dependent 4E-BP1 phosphorylation. However, the effect of AR on MAPK is both mutation- and level-independent.
eIF4E(S209) phosphorylation correlates strongly with proliferation rates in human PCa specimens
Previous studies demonstrated that eIF4E levels and phosphorylation were increased in hormone refractory tissues compared to hormone sensitive ones (17), however, the effect of eIF4E phosphorylation on proliferation in human tumors was unknown. Sections from prostate tumors of 78 patients were stained against total and phospho-eIF4E(S209), PTEN, phospho-Akt, phospho-p70S6K, phospho-mTOR, phospho-4E-BP1, AR, and the cellular proliferation marker Ki67, and scored by a Pathologist who was blinded to the outcomes and cross-references. In support of previous reports (17), we observed a significant increase in both eIF4E (p=0.001) (Figure 4A) and phospho-eIF4E(S209) (p=0.033) (Figure 4B), cytoplasmic AR (p<0.001) and nuclear Ki67 (p<0.001) levels in matched tumors compared to surrounding non-tumor tissues (n=78) (Table 2). In tumors, eIF4E correlated significantly with PTEN, phospho-Akt(S473), phospho-p70S6 kinase(T389) and AR, while phospho-eIF4E correlated with phospho-mTOR(S2481) and with phospho-4E-BP1(S65) (Table 3). We determined significant correlation between phospho-eIF4E and Ki67 (Spearman correlation coefficient =0.42, p=0.012) (Figure 4C), but not between eIF4E levels and Ki67 (not shown). This ascertained that phosphorylation of eIf4E, and not its expression, play a role in proliferation in human PCa.
Figure 4. Correlation between eIF4E phosphorylation at S209 and Ki67 levels in PCa patient tissue obtained by prostatectomy.

(A,B) left -Formalin fixed paraffin-embedded human localized PCa specimens obtained by prostatectomy were arranged in a tissue microarray and stained with either anti-eIF4E (A) or anti-phospho-eIf4E (B) antibody. All stains other than Ki67 were analyzed based on scores from 0–3, where 0 represents no staining and 3 represents 100% staining, whereas Ki67 staining was scored based on the actual number of nuclei within a specified area staining for the antigen. Right - Boxplots depicting the intensity of eIf4E or phospho-eIF4E in the different tissue samples demonstrate that the expression of eIf4E and phospho-eIF4E is more prominent in tumor samples when compared to benign tissue samples. (C) Left - IHC of tissue microarray from localized PCa specimens from two different patients stained with either anti-phospho-eIF4E or the proliferation marker anti-Ki67. Right – Boxplot depicting the intensity of phospho-eIF4E vs. the Median Ki67 demonstrating positive correlation between eIF4E phosphorylation at Serine 209 and cell proliferation.
TABLE 2. Patient Characteristics.
Characteristics of 78 patients with primary PCa, who underwent radical retropubic prostatectomy from 1999–2002. The patients were selected based on (i) availability of patient outcome data and (ii) availability of tissues from prostatectomy specimen.
| Number of patients | 78 | |
| RACE | Caucasian | 38 |
| African American | 17 | |
| Others/unknown | 23 | |
| Mean BMI | 28.47±4.48 | |
| Mean Pre-op PSA | 7.99±6.48 | |
| Gleason | 5–6 | 35 |
| 7 | 33 | |
| 8–9 | 9 | |
| Stage | T1 | 43 |
| T2 | 35 | |
| Positive Margins | 27 | |
| PSA failure | 23 | |
TABLE 3.
Correlation between various parameters in prostatectomy samples from patients with localized prostate cancer.
| Protein 1 | Protein 2 | Spearman Correlation | Adjusted P-Value |
|---|---|---|---|
| PTEN | EIF4E | 0.45 | 0.012 |
| P-Akt | EIF4E | 0.41 | 0.021 |
| P-P70S6K | EIF4E | 0.51 | 0.002 |
| AR | EIF4E | 0.58 | <0.001 |
| Ki67 | P-EIF4E | 0.42 | 0.012 |
| P-mTOR | P-EIF4E | 0.42 | 0.021 |
| P-4EBP1 | P-EIF4E | 0.48 | 0.012 |
eIF4E phosphorylation regulates resistance to mTOR inhibitors in CRPC cells
Next we investigated the effect of eIF4E phosphorylation on cell viability and resistance to mTOR inhibitors. EIF4E was knocked down in PC-346C cells by a siRNA pool (Figure 5A), which inhibited cell viability (67.3% decrease in viability compared to cells treated with control siRNA, p=0.046) (Figure 5B,C). We also investigated the effect of overexpressing eIF4E in C4-2 cells (Figure 5D). Significantly, eIF4E overexpression caused an increase in Akt phosphorylation, and increased viability, both in the presence of vehicle (28% increase, p=0.0307) or RAD001 (77.13% increase, p=0.0159) (Figure 5E). More importantly, eIF4E overexpression had a significant effect on the ability of both bicalutamide and RAD001 to inhibit C4-2 viability. Whereas in cells transfected with an empty vector, bicalutamide induced a 50% inhibition, in those transfected with eIF4E-HA, the same drug induced a 40% inhibition (p<0.001). Similarly, eIF4E overexpression reduced the ability of RAD001 to decrease growth in the same cell line from 96% to 82% (p<0.01). Taken together, these results indicate that eIF4E overexpression induces resistance to RAD001, alone or with bicalutamide, in CRPC cells.
Figure 5. eIF4E phosphorylation levels regulate proliferation in vitro.

(A) Western blots of PC346C transfected with either control siRNA or eIF4E siRNA and treated with the different mTOR inhibitors alone or in combination with Bicalutamide (Caso) for 48 hours. (B and C) MTT viability assay of PC346C transfected with either control siRNA or eIF4E siRNA treated with RAD001, Bicalutamide or the combination. Neither RAD001 nor bicalutamide had significant effect on viability of PC-346C cells transfected with control siRNA (p=0.0755 for vehicle vs bicalutamide; p=0.0680 for vehicle vs RAD001) (Figure 5B); but in eIF4E knockdown cells RAD001 caused 62.1% loss in cell viability (p=0.0014), while in combination with bicalutamide, this effect increased to 80.8% (p=0.00098) (Figure 5C). (D) Western blots of C4-2 cells transfected with either empty vector or pHA-eIF4E and treated with the different mTOR inhibitors alone or in combination with Bicalutamide (Caso) for 48 hours. The eIF4E overexpressing cells demonstrate two bands representing eIF4E – an upper band which demonstrates the expression of the transfected plasmid (which is slightly larger than the endogenous eIF4E due to the HA tag) and a lower band representing the endogenous eIF4E. (E) MTT viability assay of C4-2 cells transfected with either an empty vector or pHA-eIF4E and treated with RAD001, collected up to day 6.
Bicalutamide-induced eIF4E phosphorylation is mediated by an increase in ERK phosphorylation
Previous studies indicated that eIF4E phosphorylation is regulated by mitogen activated protein kinases (MAPK) (32); therefore we investigated the role of this pathway in mediating bicalutamide-induced eIF4E phosphorylation. Bicalutamide increased ERK phosphorylation, but not that of p38MAPK in C4-2 cells (Figure 6A). This effect was independent of RAD001-induced eIF4E phosphorylation, since RAD001 increased AR transcriptional activity on a PSA promoter (Figure 6B), while bicalutamide inhibited PSA (Figure 6A). Treatment with RAD001 increased phosphorylation of both ERK1/2 (in PC-346C cells), and p38MAPK (in C4-2 cells) (Figure 6C), which are known regulators of Mnk1/2 (33). We show both RAD001 and the dual mTOR/PI3K inhibitor BEZ235 upregulate the MAPK/Mnk/eIF4E pathway in C4-2 cells, whereas in PC-346C, only RAD001, but not BEZ235 had a similar effect. However, AR inhibition by castration induced both ERK and p38MAPK phosphorylation. Athymic nu/nu mice were subcutaneously implanted with 22Rv1 cells and either left intact (n=6) or castrated (n=6). Mice were euthanized 28 days after castration or sham operation, and the tumors were excised and processed for Western Blotting (Figure 6D). Results indicated that despite the CRPC status of these cells, castration induced an increase in not only eIF4E(S209) phosphorylation, but also p38MAPK(T180/Y182) and ERK(T202/Y204) phosphorylation.
Figure 6. Activation of eIF4E phosphorylation at Ser209 by RAD001 and bicalutamide is mediated by an increase in ERK and p38MAPK activation.

(A) C4-2 and PC-346C cells were treated for 48 hours with DMSO (vehicle), 10 μM bicalutamide, 1 nM RAD001 or both, then the cells were lysed and evaluated for the various proteins indicated. P-ERK indicates ERK(Y204/T202) and p-p38MAPK indicates p38MAPK(Y180/Y182). T-ERK and t-p38MAPK indicates total (phosphorylated and unphosphorylated) protein. (B) Luciferase assay of C4-2 cells transfected with plasmid expressing hPSA-luc and treated with vehicle, RAD001 (0.1, 1 or 10nM) or BEZ235 (5, 10 or 20nM). AR transcriptional activity was increased by all concentrations of RAD001 while it was decreased by all concentrations of BEZ235. (C) Western blots of C4-2 and PC346C treated with the control vehicle DMSO, the mTOR inhibitor RAD001 or the PI3K/mTOR dual inhibitor BEZ235 for 48 hours and blotted for proteins from the mTOR and MAPK pathways. (D) Western blots of 22Rv1 tumors extracted from athymic nu/nu mice subjected to sham-operation (intact) or castration. Results show an increase in phosphorylation of eIF4E, p38MAPK and ERK in castrated mice compared to intact. (E) Western blot of PC346C cells treated with either the mTOR inhibitor RAD001, ERK1/2 inhibitor PD98059 or the p38MAPK inhibitor SB203580 for 48 hours. (F) MTT viability assay of PC346C cells treated with RAD001, PD98059, SB203580 and the combinations. The combination of ERK and P38MAPK inhibitor sensitized PC346C cells to the mTOR inhibitor RAD001. Results are shown in absorbance fold change to first day of treatment.
To determine whether the increase in ERK or p38 signaling mediated the effect on eIF4E phosphorylation, PC-346C cells, were treated with RAD001, the ERK1/2 inhibitor PD98059 and the p38MAPK inhibitor SB203580 (Figure 6E). RAD001, but not PD980959 or SB203580, increased eIF4E phosphorylation levels. In support of a role for eIF4E phosphorylation in RAD001 resistance, the combination of PD98059 and SB203580 sensitized PC-346C cells to RAD001 (Figure 6F). Taken together, these results demonstrate that high ERK and p38MAPK phosphorylation results in elevated levels of eIF4E phosphorylation which induces resistance to mTOR inhibitors.
Suppression of eIF4E phosphorylation by Mnk inhibitors sensitize CRPC cells to mTOR and AR inhibitors
Next, we investigated the effects of suppressing eIF4E phosphorylation, rather than its expression, on sensitivity of CRPC cells to RAD001. The Mnk1/2 inhibitor CGP57380 prevented eIF4E phosphorylation (Figure 7A,B), but did not affect eIF4E expression (Figure 7B). Whereas RAD001 increased eIF4E phosphorylation, this effect could be overcome by CGP57380 in both PC-346C and in C4-2 cells (Figure 7C). Similar to bicalutamide, increase in eIF4E phosphorylation was observed upon enzalutamide treatment, which was also prevented by CGP57380 (Suppl.Fig. 5A).
Figure 7. Suppression of eIF4E phosphorylation at Ser209 by a Mnk1/2 inhibitor sensitizes CRPC cells to RAD001.

(A) Western blots of PC346C cells treated with the Mnk1/2 inhibitor CGP57380 demonstrating decrease in eIF4E phosphorylation over 4 days. The effect of CGP57380 (5μM) in PC-346C cells is biphasic and caused an initial decrease at 2 hours followed by an increase at 24 hours (less than initial) which was followed by a subsequent decrease after 72 hours. (B) Western blots of C4-2 cells treated for 48 hours with CGP57380, RAD001 or the combination and blotted against phospho and total eIf4E, total Mnk and the loading control tubulin. (C) Immunofluorescence of C4-2 and PC346C cells treated for 48 hours with the Mnk inhibitor CGP57380, the mTOR inhibitor RAD001 and the combination and stained for phospho-eIF4E demonstrating the increase in phosphorylation of eIF4E when treated with RAD001 and the decrease of the same effect in CGP57380 treated cells. The combination treatment also shows inhibition of the increase of phospho-eIF4E induced by RAD001 if CGP57380 is present. (D) MTT viability assay of C4-2, PC346C, 22Rv1 and CWR-R1 cells treated with CGP57380, RAD001 or the combination compared to control for up to 7 days. All cell lines had viability decreased in presence of the Mnk inhibitor CGP57380 (Less effective in R1 cells) and this effect was augmented with the combination. Results are shown in absorbance fold change to first day of treatment and sensitivity to the drug combination was defined as >50% loss of viability. (E) PI single stain flow cytometry of PC346C cells treated with RAD001, CGP57380 or the combination for analysis of cell cycle showing increase in G1 phase arrest in these cells with both drugs separately and even more with the combination. (F) Western blots of different mTOR pathway proteins of C4-2 and PC346C treated for 48 hours with RAD001, CGP57380 or the combination.
Cell viability assays demonstrated viability suppression by CGP57380 in both C4-2 (decrease=82.1%, p<0.0001) and in PC-346C cells (decrease=51.3%, p=0.00002) (Figure 7D). Combination with RAD001 suppressed growth even further (C4-2: 96.2%, p=0.00007 vs CGP57380 alone; PC-346C: 80.5%, p=0.00000118). CGP57380 also sensitized both lines to enzalutamide (Suppl.Fig. 7B,C). Cell cycle analysis by flow cytometry demonstrated G1 arrest with both RAD001 and CGP57380 that was compounded when used in combination (increased G1 48% to 62.4%) (Figure 7E), and induced apoptosis in both C4-2 (increased apoptosis 2.29 to 13.8%) and PC-345C cells (increased apoptosis 3.12 to 8.24%) treated with both CGP57380 and RAD001 (Suppl Figure 5D). Thus inhibition of eIF4E phosphorylation sensitized these cells to a combination of RAD001 with AR antagonists.
Significantly, in C4-2 cells, CGP57380 suppressed Akt phosphorylation at S473 (Figure 7F). Interestingly, Figure 7F shows that p70S6 kinase phosphorylation was suppressed by CGP57380 in PC-346C but not in C4-2 cells. However, this effect is independent of PTEN expression since neither PTEN-null LNCaP nor PTEN expressing CWR-R1 or 22Rv1 cells show a decrease in p70S6K phosphorylation with CGP57380 treatment, nor were it affected by the PI3K inhibitor BKM120 in either C4-2 or PC-346C cells (Suppl.Fig. 6). In addition, 4E-BP1, but not p38MAPK phosphorylation was upregulated by CGP57380 in PC-346C but not in C4-2 cells. Additional combination with bicalutamide did not significantly affect the viability of either C4-2 or PC-346C cells. Overall, these results demonstrate that anti-androgens upregulate eIF4E phosphorylation, which in turn increases proliferation rates and induces resistance of CRPC cells to further treatment with combinations of mTOR and AR inhibitors.
eIF4E phosphorylation promotes cap-independent translation which is insensitive to mTOR inhibitors
Finally, we investigated the role of eIF4E phosphorylation on targets of cap-dependent translation including survivin. In C4-2 cells, RAD001 (which affects cap-dependent translation), but not bicalutamide (which affects AR-mediated transcription), severely decreased survivin levels (Figure 8A) despite no effect by RAD001 on survivin mRNA expression (Suppl.Fig. 7). However, in PC-346C cells, neither RAD001 nor bicalutamide had any effect on survivin, indicating loss of dependence on cap-dependent translation (Figure 8A). In contrast, protein levels of c-myc and cyclinD1, which express IRES in all cells, were not similarly affected, despite suppression of mRNA levels, likely due to a switch to cap-independent translation upon RAD001 treatment. These results indicate a role for high eIF4E phosphorylation in PC-346C cells in resistance to RAD001 by switching to a cap-dependent translation mechanism.
Figure 8. Bicalutamide-induced eIF4E phosphorylation at Ser209 switches translation from a cap-dependent to a cap-independent mechanism resistant to RAD001, whereas the Mnk inhibitor CGP57380 reverses this switch.
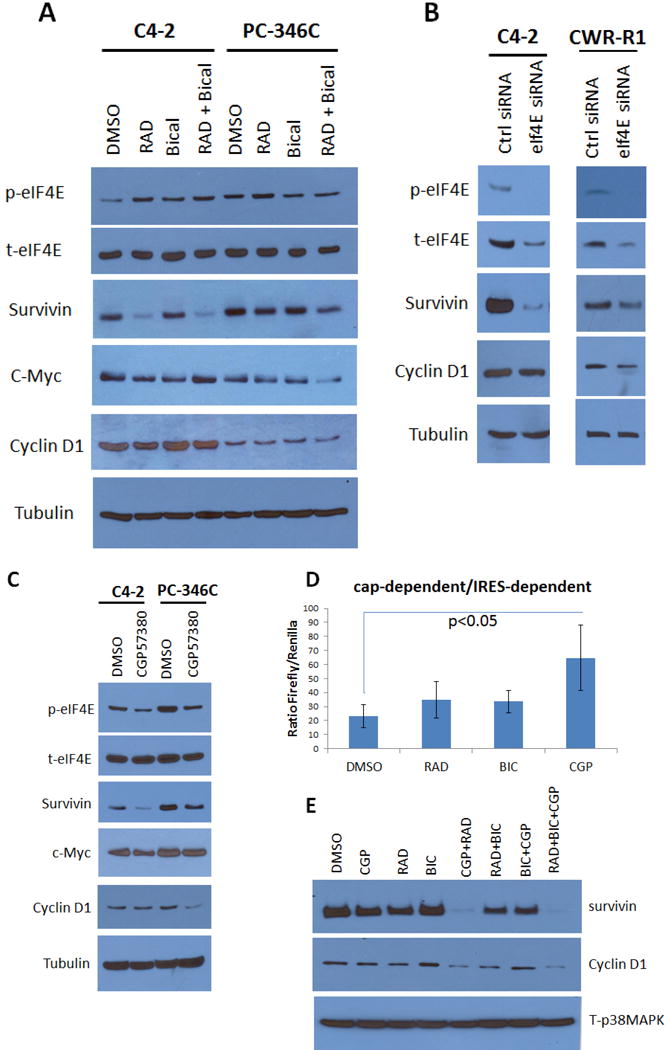
(A) Western blots of C4-2 and PC346C treated with DMSO, RAD001, Bicalutamide and the combination for 48 hours and blotted for eIF4E targets. In C4-2 the treatment with RAD001 or RAD+Bicalutamide had an effect on survivin expression but not in PC346C cells. (B) Western blots of C4-2 and CWR-R1 transfected with either control siRNA or eIF4E siRNA for 48 hours. EIF4E knock down affects surviving expression in both cell lines. (C) Western blots of C4-2 and PC346C treated with either vehicle or the Mnk inhibitor CGP57380 for 48 hours. Again we see eIF4E phosphorylation decrease corresponding with decrease in survivin expression in C4-2 but not in PC346C. (D-E) C4-2 (D) and PC346C (E) were transfected with the pFL-EMCV-IRES-RL construct and treated with RAD001, Bicalutamide, CGP57380 and the possible combinations for 48 hours. Readings represent a mean±S.D. of the ratio of Firefly/Renilla from triplicates.
Hence, we investigated whether reducing eIF4E phosphorylation switched translation back to a cap-dependent mechanism. siRNA knockdown (k/d) of eIF4E reduced eIF4E levels and eliminated eIF4E phosphorylation in both RAD001-sensitive C4-2 and RAD001-insensitive CWR-R1 cells (Figure 8B). eIF4E k/d reduced survivin levels in both C4-2 cells and CWR-R1 cells, and even cyclinD1 levels were significantly reduced in CWR-R1 upon eIF4E k/d. Similarly, eIF4E dephosphorylated by CGP57380 suppressed eIF4E phosphorylation but not expression (Figure 8C); and resulted in a decrease in survivin, c-myc and cyclinD1 levels upon CGP57380 even in mTOR-resistant PC-346C cells.
We then tested whether inhibition of eIF4E phosphorylation sensitized cells to RAD001 by switching back to cap-dependent translation. A dual-luciferase reporter system in which the viral IRES element of encephalomyocarditis virus (EMCV) is positioned between genes encoding Firefly luciferase (F-luc) and Renilla luciferase (R-luc) proteins was used for this purpose (described in (34)) (Figure 8D). In this system, F-luc and R-luc expression results from cap-dependent and cap-independent translation initiation, respectively (34). C4-2 and PC-346C cells were transfected with the dual reporter and treated as shown. The data is expressed as ratio of F-Luc/R-Luc where high values demonstrate cap-dependent translation and low values indicate cap-independent translation. CGP57380, but not RAD001 or bicalutamide, increased the ratio of cap-dependent/cap-independent translation, supporting our hypothesis. In addition, CGP57380 treatment sensitized survivin levels to RAD001 (Figure 8E), thereby indicating that upon suppression of eIF4E phosphorylation, survivin levels were mostly maintained by cap-dependent translation, hence it was susceptible to inhibition by RAD001. Significantly, cyclinD1, which can be regulated by both cap-dependent and –independent translation, was also similarly affected (Figure 8E). This supports the notion that inhibition of eIF4E phosphorylation switches translation back to a cap-dependent mechanism that then sensitizes PCa cells to mTOR inhibitors.
DISCUSSION
A phase II study to test the efficacy and tolerability of RAD001 in combination with bicalutamide in men pretreated with bicalutamide alone reported only 2/36 (5.6%) patients experienced an initial PSA decline >50% (9). In contrast, the same combination in a bicalutamide-naïve cohort was effective (>50% reduction in PSA in 15/24 (62.5%)) (8). Here, we show that the differences in response of the two groups lie in the upregulation of eIF4E phosphorylation induced by prior use of bicalutamide. We demonstrate that AR expression suppresses eIF4E phosphorylation, while inhibition of AR by bicalutamide (or enzalutamide) increases it, which induces resistance to the combination. The efficacy of combinations of AR and mTOR inhibitors lie in coordinated inhibition of both AR-induced transcription and mTOR-induced cap-dependent translation. However, AR inhibition also increases eIF4E phosphorylation, which shifts translation to cap-independent pathways, thereby rendering mTOR inhibitors ineffective. Our data show that pretreatment with bicalutamide increased eIF4E(S209) phosphorylation, thereby inducing cap-independent translation resistant to these combinations. In contrast, in bicalutamide-naïve cells, eIF4E phosphorylation remained low, and the tumor cells responded to the same. This resistance can be overcome by inhibition of eIF4E phosphorylation with Mnk1/2 or ERK1/2 inhibitors.
Various studies indicated efficacy of combinations of a rapamycin analog and an AR antagonist in CRPC models (21, 22, 35). Rapamycin and its analogs increased eIF4E(S209) phosphorylation at S209, despite no direct activation site for rapamycin on this protein (36–38). We now show that the effects of both RAD001 and bicalutamide on eIF4E phosphorylation are mediated by increased activation of the MAPK pathway which regulates Mnk1/2, the kinase that phosphorylates eIF4E at S209. However, it is likely that the mechanisms by which the two drugs activate MAPK are independent of each other and that while RAD001 uses a translation-dependent mechanism, bicalutamide uses a AR-transcription-dependent one (Suppl.Fig. 8).
We determined that higher phospho-eIF4E affected not only the response of CRPC cells to RAD001+bicalutamide, but also to combinations of other mTOR inhibitors (INK128, BEZ235) and AR antagonists (enzalutamide). As further proof that eIF4E phosphorylation is the cause of resistance to the combination of RAD001 and bicalutamide, suppression of eIF4E phosphorylation with the Mnk1/2 inhibitor CGP57380 or ERK1/2 inhibitors sensitized the resistant cells to RAD001. This strategy has previously been shown to be successful in other tumors (39). Previous reports showed that eIf4E activation correlated with reduced patient survival and PCa progression (17, 40). In addition, we now demonstrate that increased eIF4E phosphorylation correlates with proliferation in patient tissues.
Our data indicate that eIF4E phosphorylation regulated proliferation and survival by allowing cap-independent translation of key downstream targets. A case in point is the cell survival protein survivin which has a high turnover rate and is usually translated by a cap-dependent mechanism only. As a result, in C4-2 cells, this protein is easily suppressed by RAD001 which, as a specific inhibitor of mTORC1, selectively inhibits cap-dependent translation. However, in PC-346C cells expressing very high levels of phospho-eIF4E(S209), survivin is not affected by RAD001, indicating the advent of cap-independent translation. Combination with the Mnk-inhibitor CGP57380 reduced eIF4E phosphorylation and sensitized survivin levels to RAD001. Even cyclinD1, which has a known IRES site (41), was suppressed by RAD001 when co-treated with CGP57380. It may be noted that dual Mnk/AR inhibitors were previously shown to be effective in inhibiting growth of 22Rv1 and LNCaP derived CRPC cells (42, 43). Thus, our data indicate that bicalutamide-resistant patients would benefit from pretreatment with Mnk and ERK inhibitors that would sensitize them to the combination of RAD001+bicalutamide. These results should be kept in mind with the advent of multiple clinical trials to test the efficacy of enzalutamide or abiraterone acetate in combination with various mTOR inhibitors (44).
MATERIALS AND METHODS
Patient Characteristics
78 formalin-fixed paraffin-embedded (FFPE) primary prostate tumor and corresponding surrounding non-tumor tissues were available from archives of VA Northern California Health Care System (VANCHCS), Laboratory and Pathology Services. All tissue was collected in accordance with a protocol approved by the VANCHCS Institutional Review Board (IRB) under a waiver of consent as per the regulations of the VANCHCS IRB. Patients included had undergone prostatectomy 2002–2012, and at least 5 year follow-up data was available for analysis. Tumor and non-tumor areas were identified by a pathologist. 60μm core samples were extracted from specified areas of the donor blocks, which were arranged in triplicate in a tissue microarray (TMA) using a Beecher Instruments Manual Tissue Arrayer. Hematoxylin-eosin staining was used as a reference for interpreting additional sections of the TMA stained with various antibodies.
Cell culture and materials
Cell lines
LNCaP and 22Rv1 (ATCC, Manassas, VA), C4-2, C4-2B (MD Anderson, Houston, TX), CWR-R1 (Dr Elizabeth Wilson, University of North Carolina), PC346C (Dr. W.M. van Weerden, Josephine Nefkens Institute, Erasmus MC, Rotterdam, Netherlands) and pRNS1-1 cells (Dr. Johng Rhim, University of the Health Sciences, Bethesda, MD) were cultured in RPMI 1640 medium with 10% fetal bovine serum and 1% antibiotic-antimycotic solutions. All cells except LNCaP and 22Rv1 were authenticated by verification of molecular profile as reported in the original publications. LNCaP and 22Rv1 were purchased less than 12 months prior to report.
Pharmaceuticals
BKM120, BEZ235 and RAD001 were kindly provided by Novartis Pharmaceuticals, Basel, Switzerland. Rapamycin were purchased from LC Laboratories, Massachusetts, USA. PD98059, SB23580 and INK128 were obtained from Selleckchem, Houston, USA. CGP57380 was purchased from Tocris Bioscience, Bristol, UK. Casodex (bicalutamide) was kindly provided by AstraZeneca, Cheshire, UK. Enzalutamide was kindly provided by Medivation, San Francisco, CA, USA.
Antibodies Used
PTEN(9559), p44/42(Erk1/2)(9107), phospho-p44/42(Thr202/Tyr204)(9101), p38MAPK(8690), phospho-p38MAPK(Thr180/Tyr182)(4511), eIF4E(9742), phospho-eIF4E(Ser209)(9741), Mnk(2195), phospho-Mnk(Thr197/202)(2111), phospho-4EBP1(Ser65)(9451), AKT(4685), phospho-Akt(Ser473)(9271), mTOR(2983), phospho-mTOR(Ser2448)(2971), phospho-p70S6K(Thr421/Ser424)(9205), AR(3202), α-Tubulin(2125), c-Myc(9402), Survivin(2808), Cyclin D1(2978), BCL-2(2872), p-eIF4G(2441) and EGFR(2232) were from Cell Signaling Technology (Beverly, MA), 4EBP1(6936), AR(N-20)(816), β-Actin(130656) were from Santa Cruz Biotechnology (Santa Cruz, CA). GAPDH(MAB374) was from EMD Millipore, Darmstadt, Germany. phospho-eIF4E(Ser209)(ab76256) for immunohistochemistry and immunofluorescence were from Abcam, Cambridge, UK. Ki67(7240) antibody was from DAKO via Agilent Technologies, Santa Clara, CA.
Plasmids and siRNA
Cells were transiently transfected using Lipofectamine2000 (Invitrogen, Grand Island, NY) according to manufacturer’s specifications. The following plasmids were used in the transfections: pCDNA3 (Invitrogen, Carlsbad, CA), pRL-HCV-FL (kindly provided by Dr. Xinbin Chen, UC Davis School of Veterinary Medicine), pcDNA3-FLAG PTEN (Addgene), pHA-eIF4E (Addgene), eIF4E siRNA (sense: GGAUGGUAUUGAGCCUAUGTT, antisense: CAUAGGCUCAAUACCAUCCTT), and control siRNA (Cell Signaling Technology, Beverly, MA).
Mouse studies
Mice (4–5-week-old, nu/nu athymic male) were obtained from Harlan Sprague-Dawley, Inc. and implanted subcutaneously (s.c.) with sustained release testosterone pellets (12.5mg, 90-day release; Innovative Research of America). All animal experiments were carried out in accordance with a protocol approved by the UC Davis Institutional Animal Care and Use Committee (IACUC). Suspensions of CWR22 (kindly provided by Dr. Clifford Tepper, Department of Biochemistry, UC Davis) or 22Rv1 cells in 50% Matrigel solubilized basement membrane (BD Biosciences), and tumors were established by s.c. injections of 2.5×106 cells/site into both flanks. Tumor-bearing mice were left intact or castrated by standard procedures (45). When palpable tumors were observed, animals were randomly assigned to one of the following groups (n=6 per group) and were treated with (a) vehicle (peanut oil only), (b) bicalutamide, delivered by oral gavage at a dose of 50mg/Kg, 100μL per dose, five-times per week, dissolved in ethanol, and delivered as a suspension in peanut oil, (c) rapamycin at a dose of 4mg/Kg 5-times a week by oral gavage, delivered as a suspension in peanut oil and (d) bicalutamide+rapamycin. At the end, mice were euthanized; and tumors collected for paraffin embedding. Blood from the euthanized animals was fractionated and serum collected for analysis.
Statistical Analysis
Analyses were conducted using the statistical software environment R, version 3.2.1 (R Core Team, 2015). Mouse Studies: Marker levels were compared between groups using Kruskal-Wallis tests to test globally for any differences among groups, followed by pairwise comparisons using Wilcoxon rank sum tests in the case of a significant Kruskal-Wallis test. Correlations between markers were estimated using Spearman correlation coefficient. Based on resampling, with 6 mice/group, the power of the Wilcoxon rank sum test to detect a difference in expression of cytoplasmic phospho-eIF4E between groups is approximately 99%, using the observed data distributions as the true data distributions for the purposes of sampling. Tumor Microarrays: Correlations between the expressions of two proteins were estimated using Spearman rank correlation. Protein expression was compared between matched cancer and normal tissue using Wilcoxon signed rank tests. All tests are non-parametric. Based on resampling, with 78 paired cancer/non-cancer samples the power of the Wilcoxon signed-rank test to detect a difference in expression of eIF4E between cancer and non-cancer samples is approximately 71%, after adjustment for multiple testing and using the observed data distributions as the true data distributions for the purposes of sampling.
Other Methods
Western blotting, MTT viability assay, flow cytometry, immunohistochemistry and immunofluorescence were performed as described elsewhere (46). Dual-Glo luciferase assay was conducted as described in (34). PSA ELISA was described in (47). All studies were repeated in triplicate biological replicates, and were repeated at least two times with consistent results. Data is presented as mean±S.D. of three replicates. Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Supplementary Material
Acknowledgments
We thank Novartis Pharmaceuticals for the gift of RAD001. We also thank Ms. Stephanie Soares, Department of Urology, University of California Davis, School of Medicine, for the construction of the tissue microarrays used in this study, and Yu Wang, Department of Urology, for assistance with mice experiments. Human PSA-luciferase construct (hPSA-luc) was kindly provided by Dr. XuBao Shi, University of California Davis, Department of Urology. CWR-R1 cells were provided by Dr Elizabeth Wilson (University of North Carolina), pRNS-1-1 cells were from Dr. Johng Rhim, University of the Health Sciences, Bethesda, MD, while PC-346C cells were from Dr. W.M. van Weerden, Josephine Nefkens Institute, Erasmus MC, Rotterdam, Netherlands. We also thank Dr. Xinbin Chen (UC Davis School of Veterinary Medicine) for the pRL-HCV-FL plasmid, and Maitreyee K. Jathal and Thomas M. Steele (UC Davis, Department of Urology) for samples of 22Rv1 xenograft tumors.
FUNDING:
This work was supported by a Biomedical Laboratory Research & Development (BLRD) Merit Award (I01BX000400, PMG) from the Department of Veterans Affairs, and by Awards R01CA133209 (PMG) and R01CA185509 (PMG) from the National Institutes of Health.
Footnotes
DECLARATION OF INTEREST
The authors declare no conflict of interest. The work reported here does not represent the views or opinions of the Department of Veteran Affairs or the United States Government.
References
- 1.MacVicar GR, Hussain MH. Emerging therapies in metastatic castration-sensitive and castration-resistant prostate cancer. Curr Opin Oncol. 2013;25(3):252–60. doi: 10.1097/CCO.0b013e32835ff161. [DOI] [PubMed] [Google Scholar]
- 2.Wu Y, Chhipa RR, Cheng J, Zhang H, Mohler JL, Ip C. Androgen receptor-mTOR crosstalk is regulated by testosterone availability: implication for prostate cancer cell survival. Anticancer Res. 2010;30(10):3895–901. [PMC free article] [PubMed] [Google Scholar]
- 3.Fang Z, Zhang T, Dizeyi N, Chen S, Wang H, Swanson KD, et al. Androgen Receptor Enhances p27 Degradation in Prostate Cancer Cells through Rapid and Selective TORC2 Activation. J Biol Chem. 2012;287(3):2090–8. doi: 10.1074/jbc.M111.323303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghosh PM, Malik SN, Bedolla RG, Wang Y, Mikhailova M, Prihoda TJ, et al. Signal transduction pathways in androgen-dependent and -independent prostate cancer cell proliferation. Endocr Relat Cancer. 2005;12(1):119–34. doi: 10.1677/erc.1.00835. [DOI] [PubMed] [Google Scholar]
- 5.Chamie K, Ghosh PM, Koppie TM, Romero V, Troppmann C, deVere White RW. The effect of sirolimus on prostate-specific antigen (PSA) levels in male renal transplant recipients without prostate cancer. Am J Transplant. 2008;8(12):2668–73. doi: 10.1111/j.1600-6143.2008.02430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amato RJ, Jac J, Mohammad T, Saxena S. Pilot study of rapamycin in patients with hormone-refractory prostate cancer. Clin Genitourin Cancer. 2008;6(2):97–102. doi: 10.3816/CGC.2008.n.015. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Mikhailova M, Bose S, Pan CX, deVere White RW, Ghosh PM. Regulation of androgen receptor transcriptional activity by rapamycin in prostate cancer cell proliferation and survival. Oncogene. 2008;27(56):7106–17. doi: 10.1038/onc.2008.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow H, Ghosh PM, deVere White R, Evans CP, Dall’Era MA, Yap SA, et al. A phase 2 clinical trial of everolimus plus bicalutamide for castration-resistant prostate cancer. Cancer. 2016 doi: 10.1002/cncr.29927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakabayashi M, Werner L, Courtney KD, Buckle G, Oh WK, Bubley GJ, et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int. 2012;110(11):1729–35. doi: 10.1111/j.1464-410X.2012.11456.x. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 12.Sonenberg N. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem Cell Biol. 2008;86(2):178–83. doi: 10.1139/O08-034. [DOI] [PubMed] [Google Scholar]
- 13.Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21(24):3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ueda T, Sasaki M, Elia AJ, Chio II, Hamada K, Fukunaga R, et al. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A. 2010;107(32):13984–90. doi: 10.1073/pnas.1008136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24(15):6539–49. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lachance PE, Miron M, Raught B, Sonenberg N, Lasko P. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol Cell Biol. 2002;22(6):1656–63. doi: 10.1128/MCB.22.6.1656-1663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A. 2010;107(32):14134–9. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goetz C, Everson RG, Zhang LC, Gromeier M. MAPK signal-integrating kinase controls cap-independent translation and cell type-specific cytotoxicity of an oncolytic poliovirus. Mol Ther. 2010;18(11):1937–46. doi: 10.1038/mt.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianchini A, Loiarro M, Bielli P, Busa R, Paronetto MP, Loreni F, et al. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis. 2008;29(12):2279–88. doi: 10.1093/carcin/bgn221. [DOI] [PubMed] [Google Scholar]
- 21.Schayowitz A, Sabnis G, Goloubeva O, Njar VC, Brodie AM. Prolonging hormone sensitivity in prostate cancer xenografts through dual inhibition of AR and mTOR. Br J Cancer. 2010;103(7):1001–7. doi: 10.1038/sj.bjc.6605882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Zhu J, Efferson CL, Ware C, Tammam J, Angagaw M, et al. Inhibition of tumor growth progression by antiandrogens and mTOR inhibitor in a Pten-deficient mouse model of prostate cancer. Cancer Res. 2009;69(18):7466–72. doi: 10.1158/0008-5472.CAN-08-4385. [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O’Neill A, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20(2):173–86. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Siddiqui S, Bose S, Mooso B, Asuncion A, Bedolla RG, et al. Nrdp1-mediated regulation of ErbB3 expression by the androgen receptor in androgen-dependent but not castrate-resistant prostate cancer cells. Cancer Res. 2010;70(14):5994–6003. doi: 10.1158/0008-5472.CAN-09-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mooso B, Madhav A, Johnson S, Roy M, Moore ME, Moy C, et al. Androgen Receptor regulation of Vitamin D receptor in response of castration-resistant prostate cancer cells to 1alpha-Hydroxyvitamin D5 - a calcitriol analog. Genes Cancer. 2010;1(9):927–940. doi: 10.1177/1947601910385450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi XB, Xue L, Tepper CG, Gandour-Edwards R, Ghosh P, Kung HJ, et al. The oncogenic potential of a prostate cancer-derived androgen receptor mutant. Prostate. 2007;67(6):591–602. doi: 10.1002/pros.20544. [DOI] [PubMed] [Google Scholar]
- 27.Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res. 1999;253(1):100–9. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez Canedo C, Demeulder B, Ginion A, Bayascas JR, Balligand JL, Alessi DR, et al. Activation of the cardiac mTOR/p70(S6K) pathway by leucine requires PDK1 and correlates with PRAS40 phosphorylation. Am J Physiol Endocrinol Metab. 2010;298(4):E761–9. doi: 10.1152/ajpendo.00421.2009. [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, et al. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276(34):31858–62. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- 30.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15(21):2852–64. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shveygert M, Kaiser C, Bradrick SS, Gromeier M. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol Cell Biol. 2010;30(21):5160–7. doi: 10.1128/MCB.00448-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16(8):1909–20. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, Leger-Abraham M. Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci U S A. 2015;112(30):E4036–45. doi: 10.1073/pnas.1512118112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Squillace RM, Miller D, Wardwell SD, Wang F, Clackson T, Rivera VM. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. Int J Oncol. 2012;41(2):425–32. doi: 10.3892/ijo.2012.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stead RL, Proud CG. Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a) FEBS Lett. 2013;587(16):2623–8. doi: 10.1016/j.febslet.2013.06.045. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007;27(21):7405–13. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65(16):7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 39.Wen Q, Wang W, Luo J, Chu S, Chen L, Xu L, et al. CGP57380 enhances efficacy of RAD001 in non-small cell lung cancer through abrogating mTOR inhibition-induced phosphorylation of eIF4E and activating mitochondrial apoptotic pathway. Oncotarget. 2016 doi: 10.18632/oncotarget.8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res. 2009;69(9):3866–73. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem. 2005;280(12):10964–73. doi: 10.1074/jbc.M407874200. [DOI] [PubMed] [Google Scholar]
- 42.Ramamurthy VP, Ramalingam S, Gediya L, Kwegyir-Afful AK, Njar VC. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget. 2015;6(5):3195–210. doi: 10.18632/oncotarget.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mbatia HW, Ramalingam S, Ramamurthy VP, Martin MS, Kwegyir-Afful AK, Njar VC. Novel C-4 heteroaryl 13-cis-retinamide Mnk/AR degrading agents inhibit cell proliferation and migration and induce apoptosis in human breast and prostate cancer cells and suppress growth of MDA-MB-231 human breast and CWR22Rv1 human prostate tumor xenografts in mice. J Med Chem. 2015;58(4):1900–14. doi: 10.1021/jm501792c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16(3):378–86. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Idris AI. Ovariectomy/orchidectomy in rodents. Methods Mol Biol. 2012;816:545–51. doi: 10.1007/978-1-61779-415-5_34. [DOI] [PubMed] [Google Scholar]
- 46.Mooso BA, Vinall RL, Tepper CG, Savoy RM, Cheung JP, Singh S, et al. Enhancing the effectiveness of androgen deprivation in prostate cancer by inducing Filamin A nuclear localization. Endocr Relat Cancer. 2012;19(6):759–77. doi: 10.1530/ERC-12-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikhailova M, Wang Y, Bedolla R, Lu XH, Kreisberg JI, Ghosh PM. AKT regulates androgen receptor-dependent growth and PSA expression in prostate cancer. Adv Exp Med Biol. 2008;617:397–405. doi: 10.1007/978-0-387-69080-3_38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.