

Abstract
Myoferlin, a member of ferlin family of proteins, was first discovered as a candidate gene for muscular dystrophy and cardiomyopathy. Recently myoferlin was shown to be also expressed in endothelial and cancer cells where it was shown to modulate VEGFR-2 and EGFR signaling by enhancing their stability and recycling. Based on these reports, we hypothesized that myoferlin might be regulating IL-6 signaling by modulating IL-6R stabilization and recycling. However, in our immunoprecipitation (IP) experiments, we did not observe myoferlin binding with IL-6R. Instead, we made a novel discovery that in resting cells myoferlin was bound to EHD2 protein and when cells were treated with IL-6, myoferlin dissociated from EHD2 and binds to activated STAT3. Interestingly, myoferlin depletion did not affect STAT3 phosphorylation, but completely blocked STAT3 translocation to nucleus. In addition, inhibition of STAT3 phosphorylation by phosphorylation defective STAT3 mutants or JAK inhibitor blocked STAT3 binding to myoferlin and nuclear translocation. Myoferlin knockdown significantly decreased IL-6-mediated tumor cell migration, tumorsphere formation and ALDH positive cancer stem cell population, in vitro. Furthermore, myoferlin knockdown significantly decreased IL-6-meditated tumor growth and tumor metastasis. Based on these results, we have proposed a novel model for the role of myoferlin in chaperoning phosphorylated STAT3 to the nucleus.
Keywords: Myoferlin, IL-6, STAT3, nuclear translocation, cancer stem cell, HNSCC
Introduction
IL-6, a pleiotropic cytokine, is involved in a number of cellular processes including, cell proliferation, cell motility, cell survival and immune response.1–4 IL-6 is one of the main chemokines present in serum samples of cancer patients and elevated IL-6 levels have also been shown to be an independent predictor of poor survival, tumor recurrence and tumor metastasis in a number of malignancies including breast, prostate and head and neck cancers.5–8 Recently, we have shown that IL-6 promotes tumor metastasis by inducing epithelial-mesenchymal transition (EMT).2 IL-6 mediates its biological function predominantly through binding to IL-6 receptor-α (IL-6Rα) on the cell surface.9 IL-6 binding to its receptor in turn induces conformational changes leading to the formation of IL-6/IL-6Rα/gp130 hexameric complex (a gp130 homodimer plus IL-6/IL-6Rα hetrodimers).10–12 This complex then recruits Janus (JAK) kinases and phosphorylates them.13 Activated JAKs, phosphorylate cytosolic STAT3 which then translocates to the nucleus to function as a transcription factor.14 This nuclear translocation of activated STAT3 is critical for the IL-6/STAT3 signaling. But, protein shuttling across nuclear membrane is tightly regulated15 and very little is known about the molecular events that facilitate the nuclear translocation of STAT3. In this study, we observed that phosphorylation of STAT3 by IL-6 leads to its binding with myoferlin and co-migration to the nucleus. Although myoferlin has been shown to regulate cell membrane fusion, repair and recycling,16, 17 there are no studies that has investigated the role of myoferlin in IL-6/STAT3 signaling particularly its role in chaperoning STAT3 to the nucleus.
Myoferlin, a member of the ferlin family of proteins, was originally discovered as a muscle specific protein.18 The ferlin family of proteins share similar domain architecture that includes a carboxy-terminal transmembrane domain and multiple amino-terminal C2 domains which promote their binding to negatively charged phospholipids.19–21 The ferlin family of proteins (dysferlin, otoferlin and myoferlin) has been extensively studied in muscle cell function and it has been shown to play an important role in plasma membrane integrity, myoblast fusion and vesicle trafficking.18, 22–24 Mutations of dysferlin are linked to limb girdle muscular dystrophy type B and Myoshi myopathy because skeletal muscle fiber sarcolemma in these patients fail to repair damaged muscle cells,21 whereas mutations of otoferlin leads to non-syndromic deafness because synaptic vesicles fail to fuse at the plasma membrane.25 Myoferlin is a 230-kDa protein that is highly expressed in myoblasts18, 26 and myoferlin depletion studies in mice showed strong muscular dystrophy due to the failure of myoblasts to fuse and form large multinucleate myotubes.23 Recently, a number of different groups have shown that myoferlin is also expressed in endothelial and cancer cells.17, 27–31 Bernatchez et al. have shown that myoferlin modulates VEGF/VEGFR-2 signaling in vascular endothelial cells by stabilizing VEGFR-2.17 Myoferlin depletion in endothelial cells also significantly decreased angiogenic tyrosine kinase receptor (Tie-2) expression.32 In cancer cells, myoferlin was shown to regulate EGF/EGFR signaling by stabilizing EGFR.27
In this study, we observed that in the resting cancer cells myoferlin is bound to EHD2 protein and when the cancer cells are treated with IL-6, myoferlin dissociates from EHD2, binds to activated STAT3 and chaperone it to nucleus to mediate its transcriptional role. Myoferlin depletion in the cancer cells significantly decreased tumor cell migration and tumorsphere formation, in vitro and tumor progression, in vivo. Taken together, our results from this study provide a novel oncogenic role for myoferlin in modulating IL-6/STAT3 signaling that plays an important function in a number of cellular processes including cell proliferation, cell migration and cancer stem cell phenotype.2–4
Results
Myoferlin expression is significantly upregulated in tumor samples from head and neck squamous cell carcinoma (HNSCC) patients and HNSCC cell lines
We have recently shown by immunohistochemistry (IHC) that myoferlin expression is markedly upregulated in HNSCC and it directly correlates with poor overall survival.33 In this study, we measured myoferlin expression levels in 30 frozen samples from HNSCC patients (20 primary tumors and 10 adjacent normal controls) by real-time PCR. Our results show that myoferlin expression was significantly higher in HNSCC tumor samples (Mann-Whitney test; p value 0.0008) as compared to adjacent normal tissue (Fig. 1a). We next examined whether myoferlin expression is also higher in head and neck tumor cell lines as compared to normal epithelial cells. Indeed, both myoferlin mRNA and protein levels were significantly higher in head and neck cancer cell lines as compared to normal epithelial cells (Fig. 1b–c).
Figure 1. Myoferlin levels are upregulated in primary tumor samples from head and neck cancer patients and HNSCC cell lines.
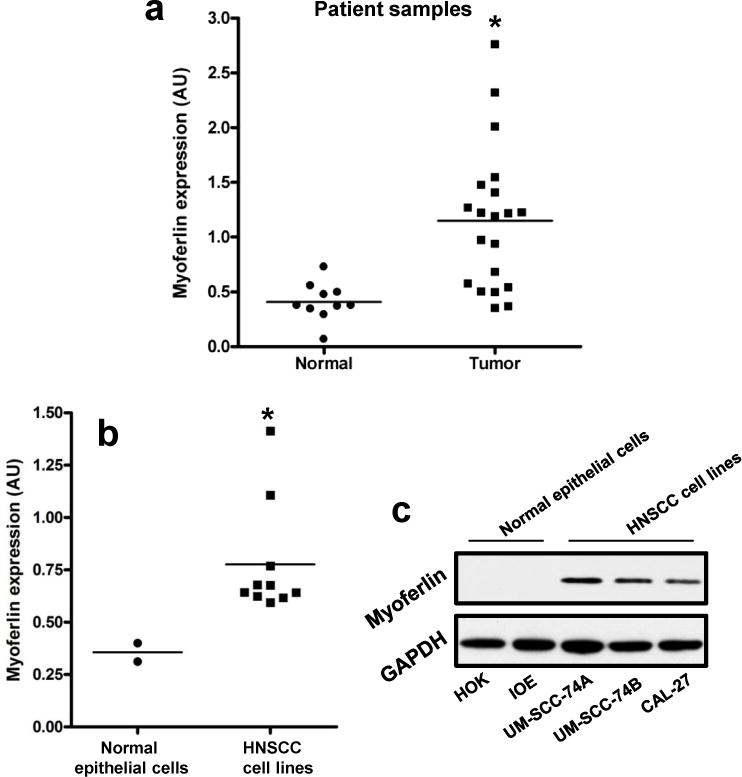
(a) Myoferlin expression in primary tumor samples (n=20) and in adjacent normal mucosa (n =10) of HNSCC patients was analyzed by RT-PCR. (b–c) Myoferlin expression in 10 HNSCC cell lines (UM-SCC-74A, UM-SCC-74B, CAL-27, UM-SCC-25, UM-SCC-36, FADU, UM-SCC-38, UM-SCC-5, UM-SCC-10A and UM-SCC-11A) and 2 normal epithelial cells (Human oral keratinocytes [HOK] and immortalized oral epithelial cells [IOE]) were analyzed by RT-PCR (b) and Western blotting (c). mRNA expression for myoferlin was normalized to OAZ1 using the 2−ΔΔCt method. Equal protein loading was verified by stripping the blots and reprobing with GAPDH antibody.
IL-6 induces STAT3-myoferlin binding and co-migration to nucleus
We and others have previously shown that IL-6 activates a number of different pathways including JAK/STAT3 pathway (Fig. 2a).2, 4 Recently myoferlin was shown to bind to VEGFR-2 and promote its stability and recycling.17 We initially examined if myoferlin might be modulating IL-6 signaling by regulating IL-6 receptor (IL-6R) stability and recycling. However, our immunoprecipitation (IP) and HA pulldown experiments suggest that myoferlin does not bind to IL-6R (data not shown). Instead, we observed that in resting cells myoferlin is bound to EHD2 protein and upon stimulation with IL-6, myoferlin dissociates from EHD2 and binds to activated STAT3 (Fig. 2b and 2d). STAT3 is cytoplasmic protein that is predominantly activated at the cell surface and then migrates to nucleus to function as a transcription factor.34 We next examined if myoferlin binding to phosphorylated STAT3 affected its translocation to nucleus. IL-6 treatment of CAL27 cells induced a time-dependent co-migration of myoferlin and STAT3 to nucleus (Fig. 3a–b). We next examined if this IL-6-induced co-migration of myoferlin and STAT3 is also observed in other HNSCC cell lines. We repeated these experiments using another HNSCC cell line (UM-SCC-74B) and we observed a similar co-migration of myoferlin and STAT3 to nucleus (Fig. 3c and Supplemental Fig. 1a). Recent studies have shown that myoferlin expression is also elevated in breast cancer cells and it promotes cancer cell motility, invasion and EMT.27, 35 We next examined if myoferlin and STAT3 co-migrated to nucleus in response to IL-6 treatment in breast cancer cells. Indeed, IL-6 treatment of breast cancer cells (MDA-MB-231) induced co-migration of myoferlin and STAT3 to nucleus (Fig. 3d and Supplemental Fig. 1b). We next knocked down myoferlin in tumor cells by siRNA and examined if myoferlin regulates IL-6-induced STAT3 phosphorylation and nuclear translocation. Our results show that myoferlin knockdown does not affect IL-6-mediated STAT3 phosphorylation (Fig. 3e) but myoferlin knockdown significantly blocked STAT3 translocation to nucleus (Fig. 3f and Supplemental Fig. 2).
Figure 2. IL-6 induces STAT3 phosphorylation and binding to myoferlin.
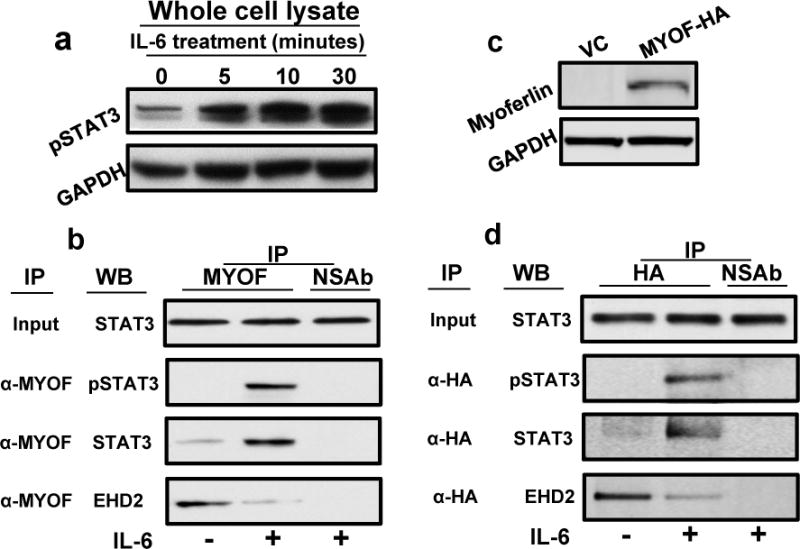
(a) CAL27 cells were treated with IL-6 for different time intervals and STAT3 activation was examined by Western blotting. (b) CAL27 cells were treated with IL-6 for 30 minutes and whole cell lysate was prepared and myoferlin immunoprecipitated (IP). Non-specific IgG was used as control. Proteins bound to myoferlin were resolved by SDS-PAGE and presence of pSTAT3 (Y705), STAT3 and EHD2 in IP was analyzed by Western blotting. Whole cell lysate was used as input control. (c–d) HA-tagged myoferlin was overexpressed in immortalized oral epithelial cells (IOE) that normally express very little or no myoferlin. (c) Myoferlin expression in IOE cells was verified by Western blotting. (d) Proteins bound to myoferlin were pull-down by anti-HA antibody and presence of pSTAT3 (Y705), STAT3 and EHD2 was analyzed by Western blotting.
Figure 3. Myoferlin knockdown does not inhibit STAT3 phosphorylation but blocks STAT3 translocation to nucleus.
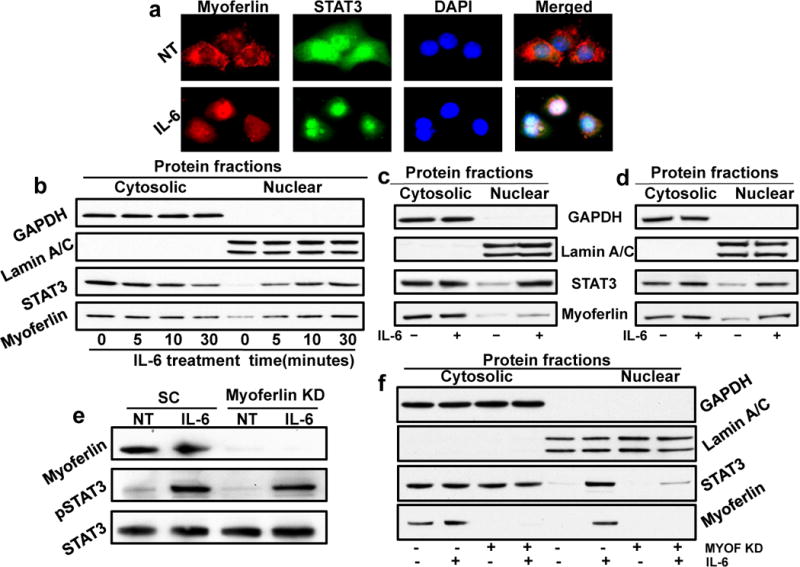
(a) CAL27 cells were cultured in Labtech chambers and treated with IL-6 for 30 minutes. Cells were then stained for myoferlin (red), STAT3 (green) and nucleus (DAPI, blue) and analyzed by fluorescence microscope at 600X. (b) CAL27 cells were treated with IL-6 for different time intervals. Nuclear and cytosolic fractions were isolated from these cells and Western blotted for GAPDH, Lamin A/C, STAT3 and myoferlin. (c–d) UM-SCC-74B (c) and MDA-MB-231 (d) cells were treated with IL-6 for 30 min. Nuclear and cytosolic fractions were isolated from these cells and Western blotted for GAPDH, Lamin A/C, STAT3 and myoferlin. (e-f) Myoferlin was knocked down by siRNA in CAL27 cells (MYOF KD). Cells transduced with scrambled siRNA (SC) were used as control. Seventy two hours post transfection; cells were treated with IL-6 for 30 minutes. (e) Whole cell lysates were Western blotted for myoferlin, STAT3 and pSTAT3. (f) Nuclear and cytosolic fractions were Western blotted for GAPDH, Lamin A/C, STAT3 and myoferlin.
STAT3 phosphorylation is required for STAT3 binding to myoferlin and translocation to nucleus
We next examined if STAT3 phosphorylation is required for myoferlin binding and translocation to nucleus. To answer this question, we employed two different approaches. In the first set of experiments, we used a JAK inhibitor (JAK inhibitor-1) to block STAT3 phosphorylation and examined myoferlin binding to STAT3 and translocation to nucleus. Our result show that JAK inh-1 markedly reduced IL-6-mediated STAT3 phosphorylation (Fig. 4a), myoferlin binding to STAT3 (Fig. 4b) and nuclear translocation of STAT3 and myoferlin (Fig. 4c). In the second set of experiments, we overexpressed STAT3 phosphorylation defective mutant (STAT3YF, kindly provided by Dr. Tom Smithgal, University of Pittsburgh) with a Tyr-705 to Phe-705 substitution.36 Tumor cells overexpressing STAT3YF mutant showed marked decrease in IL-6-mediated STAT3 phosphorylation (Fig. 4d), myoferlin binding to STAT3 (Fig. 4e) and nuclear translocation of STAT3 and myoferlin (Fig. 4f). These results therefore suggest that STAT3 phosphorylation at Tyr-705 is critical for myoferlin binding and nuclear translocation.
Figure 4. STAT3 phosphorylation is required for STAT3 binding to myoferlin and translocation to nucleus.
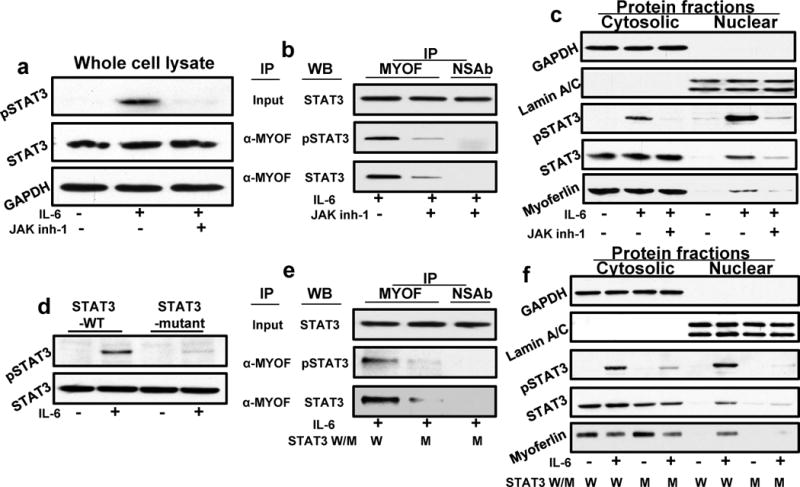
(a–c) CAL27 cells were treated with IL-6 for 30 minutes in the presence or absence of JAK inhibitor (JAK inh-1). (a) Whole cell lysates were Western blotted for pSTAT3, STAT3 and GAPDH. (b) CAL27 cells were treated with IL-6 for 30 minutes; whole cell lysate was prepared and myoferlin immunoprecipitated (IP). Non-specific IgG was used as control. Whole cell lysate was used as input control. Proteins bound to myoferlin were resolved by SDS-PAGE and presence of pSTAT3 (Y705) and STAT3 in IP samples was analyzed by Western blotting. (c) Nuclear and cytosolic fractions were Western blotted for GAPDH, Lamin A/C and pSTAT3, STAT3 and myoferlin. (d-e) CAL27 cells were transfected with STAT3 phosphorylation (Y705) defective mutant (STAT3YF). (d) Cells were treated with IL-6 for 30 minutes and whole cell lysates were Western blotted for pSTAT3 and STAT3. (e) Cells were treated with IL-6 for 30 minutes and whole cell lysate was prepared and myoferlin immunoprecipitated (IP). Non-specific IgG was used as control. Whole cell lysate was used as input control. Proteins bound to myoferlin were resolved by SDS-PAGE and presence of pSTAT3 (Y705) and STAT3 in IP was analyzed by Western blotting. (f) Nuclear and cytosolic fractions were Western blotted for GAPDH, Lamin A/C and pSTAT3, STAT3 and myoferlin.
Myoferlin knockdown significantly decreases tumor cell migration, tumorsphere formation and ALDH-positive tumor cells
IL-6/STAT3 signaling has been shown to promote tumor cell motility and stem cell phenotype.2, 37 We next examined if myoferlin knock down affects IL-6-mediated tumor cell migration and cancer stem cell phenotype. To stably knockdown myoferlin, we used two different shRNAs [purchased from Sigma (shRNA1) and Santa Cruz (shRNA2)]. Cells transduced with scrambled shRNA were used as control (Fig. 5a). Our results show that myoferlin knockdown using both shRNAs significantly decreased IL-6-mediated tumor cell motility (Fig. 5b). Myoferlin knockdown also significantly decreased ALDH positive cells in IL-6 overexpressing CAL27 cells (Fig. 5c) and tumorsphere formation in CAL27-IL-6 cells (Fig. 5d–e). Similarly, myoferlin knockdown in UM-SCC-74A cells (HNSCC cell line that naturally express high levels of IL-6) significantly decreased tumor cell migration and tumorsphere formation (data not shown). In addition, myoferlin knockdown significantly decreased the expression of IL-6/STAT3 target genes (snail and nanog) in UM-SCC-74A cells (Fig. 5f).
Figure 5. Myoferlin knockdown decreases cell migration, ALDH positive cells and tumorsphere formation.
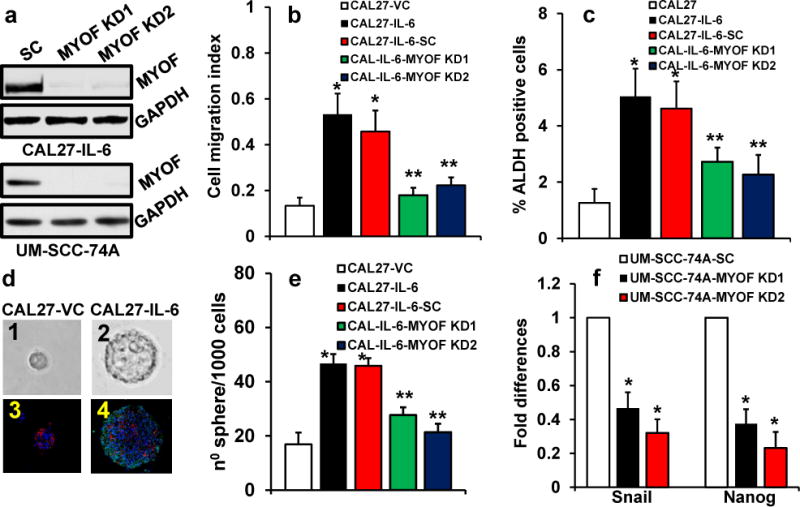
(a–f) Myoferlin expression in CAL27 overexpressing IL-6 (CAL27-IL-6) or UM-SCC-74A cells was stably knockdown by two different shRNAs. Control cells were transduced with scramble shRNA control (SC). (a) Myoferlin knockdown in CAL27-IL-6 and UM-SCC-74A cells was verified by Western blotting. (b) Cell migration was examined by xCELLigence system and expressed as cell migration index. (c) CAL27 cells expressing vector alone (CAL27-VC) or CAL27 overexpressing IL-6 (CAL27-IL-6) or myoferlin knockdown cells (CAL27-IL-6-MYOF KD1 or KD2) or CAL27-IL-6 cells transduced with scramble shRNA control (CAL27-IL-6-SC) were stained for ALDH activity and analyzed by flow cytometry. (d–e) Cells were cultured on ultralow binding plates in serum free medium. After 10 days, number of tumorsphere (>50 μM) were counted. (d1–2) Representative pictures of tumorsphere from CAL27-VC and CAL27-IL-6 groups, respectively. (d3–4) Tumorsphere were gently added to Matrigel and allowed to polymerize (solidify). Matrigel containing tumorsphere were then paraffin embedded and sectioned for immunohistochemistry (IHC). Tumorspheres were stained for E-cadherin (red), pSTAT3 (green) and nucleus (DAPI, blue) and then analyzed by fluorescence microscope at 400X. (e) Tumorsphere formation efficiency in CAL27 cells. (f) The expression of IL-6/STAT3 target genes snail and nanog were analyzed by real-time PCR in UM-SCC-74A (a cell line that naturally express high levels of IL-6) after myoferlin knockdown.
Myoferlin knockdown significantly decreases tumor growth and tumor metastasis, in vivo
We had previously shown that IL-6-STAT3 pathway is critical for tumor metastasis.2 To further examine the role of myoferlin in IL-6-STAT3 signaling pathway and tumor metastasis, we stably knocked down myoferlin in CAL27-IL-6 or UM-SCC-74A (a highly metastatic HNSCC cell line that naturally expresses high levels of IL-6)38 cells by 2 shRNAs. Our in vitro biochemical and functional experiments suggest that both these shRNAs were effective in blocking myoferlin expression and function. In our in vivo experiments, we used cells that were transduced by myoferlin shRNA purchased from Sigma-Aldrich. Cells transduced with scrambled shRNA were used as control. IL-6 overexpression in CAL27 cells significantly increased tumor growth as compared to CAL27 cells transduced with vector alone (CAL27-VC) (Fig. 6a). In addition, CAL27-IL-6 tumors showed markedly higher expression of nanog (Supplementary Fig. 3a). Myoferlin knockdown in CAL27-IL-6 cells (CAL27-IL-6-MYOF KD) significantly decreased tumor growth and nanog expression as compared to CAL27-IL-6 (Fig. 6a–b and Supplementary Fig. 3a). Similarly, myoferlin knockdown in UM-SCC-74A cells (UM-SCC-74A-MYOF KD) significantly decreased tumor growth and nanog expression (Fig. 6 c–d and Supplementary Fig. 3b).
Figure 6. Myoferlin knockdown significantly decreases tumor growth and tumor metastasis.
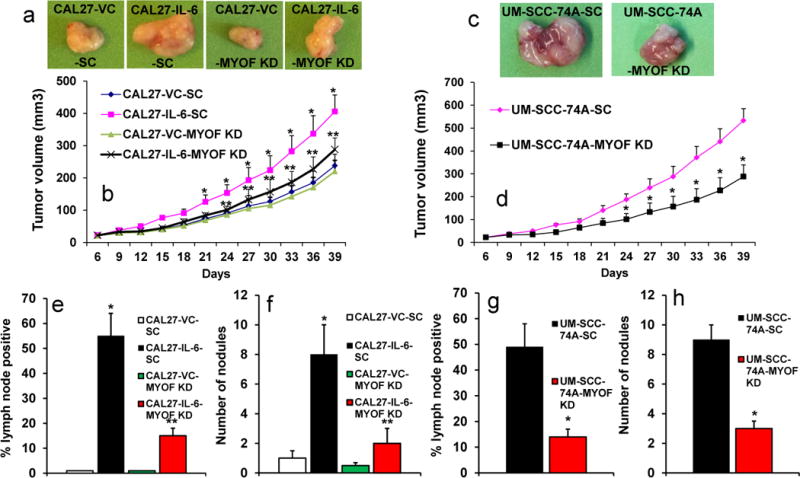
(a–d) Tumor cells (CAL27 or UM-SCC-74A) were implanted in the flanks of SCID mice and tumor growth and tumor metastasis to draining lymph nodes and lungs was analyzed. (a) Representative pictures of tumors from CAL27 cells overexpressing IL-6 or vector alone (VC) and transduced with scramble shRNA control (CAL27-VC-SC or CAL27-IL-6-SC) or transduced with myoferlin shRNA (CAL27-VC-MYOF KD or CAL27-IL-6-MYOF KD) groups. (b) Tumor growth curves for CAL27-VC-SC, CAL27-IL-6-SC, CAL27-VC-MYOF KD and CAL27-IL-6-MYOF KD tumors. (c) Representative pictures of tumors from UM-SCC-74A-SC and UM-SCC-74A-MYOF KD groups, respectively. (d) Tumor growth curves for UM-SCC-74A-SC and UM-SCC-74A-MYOF KD tumors. (e & g) Tumor metastasis to draining lymph nodes for CAL27 (e) and UM-SCC-74A tumors (g). (f & h) Tumor metastasis to lungs for CAL27 (f) and UM-SCC-74A tumors (h).
Lymph nodes from animals carrying CAL27-VC tumors were negative for metastatic disease whereas 55% of lymph nodes from animals carrying CAL27-IL-6 tumors were positive for metastatic disease (Fig. 6e). Myoferlin knockdown in CAL27-IL-6 cells (CAL27-IL-6-MYOF KD) significantly decreased in lymph node metastatic disease (Fig. 6e). In addition, lungs from animals carrying CAL27-IL-6-MYOF KD tumors showed significant decrease in metastatic nodes (Fig. 6f). Similarly, myoferlin knockdown in UM-SCC-74A cells significantly deceased lymph node (Fig. 6g) and lung metastasis (Fig. 6h).
Proposed mechanism of myoferlin role in IL-6/STAT3 signaling
Based on the results from this study, we propose a novel mechanism by which myoferlin modulates IL-6/STAT3 signaling (Fig. 7). In the resting state, myoferlin is bound to EHD2 protein (Fig. 7a). IL-6 binding to IL-6R induces STAT3 phosphorylation (Fig. 7b) and also leads to myoferlin dissociation from EHD2 and binding to phosphorylated STAT3 and chaperoning pSTAT3 to nucleus (Fig. 7c). In the nucleus, STAT3 functions as a transcription factor and regulates a number of IL-6/STAT3 downstream genes including snail and nanog.
Figure 7. Myoferlin chaperone model.
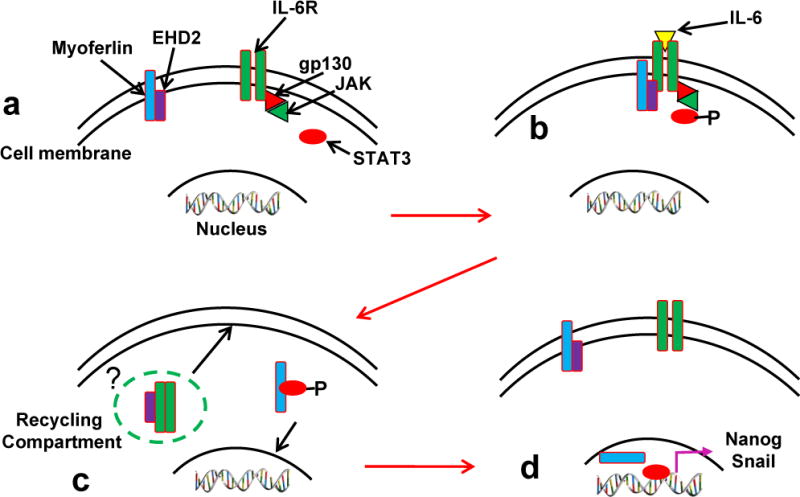
(a) In the resting cells, myoferlin is bound to EHD2 protein at the plasma membrane. (b) IL-6 binding to its receptor recruits STAT3, myoferlin and EHD2 to the IL-6R-gp130-JAK complex at the plasma membrane and phosphorylates myoferlin and STAT3. (c) Phosphorylation of myoferlin dissociates it from EHD2 and binds to pSTAT3. (d) Myoferlin chaperones STAT3 to nucleus while EHD2 shuttles IL-6R back to plasma membrane.
Discussion
In this study, we demonstrate that a muscle specific protein “myoferlin” which is absent or expressed at very low levels in normal mucosa is markedly upregulated in head and neck cancer cells. Recent studies have shown a similar elevated expression of myoferlin in breast, lung and pancreatic cancer cells.27, 29–31 All the previously published studies in muscle, endothelial cells and cancer cells have thus far examined the role of myoferlin in plasma membrane function particularly membrane repair, vesicle trafficking and receptor stability.16, 17, 22, 23, 27 Based on these studies, we initially hypothesized that myoferlin might be regulating IL-6 signaling by modulating IL-6R recycling and stability. However, we did not observe myoferlin binding to IL-6R and its recycling to plasma membrane. Instead, we observed that myoferlin binds to activated STAT3 and co-migrates to nucleus. Myoferlin has been previously shown to be present in the nucleus of muscle cells.18 Recently, we examined sub-cellular expression of myoferlin in tumor samples from HNSCC patients. We observed that nuclear myoferlin expression is highly predictive of poor overall survival (p<0.0001) and patients whose tumors were nuclear myoferlin positive had 5.4 times the hazard of death than patients whose tumors had myoferlin present in cytosol or at plasma membrane (95%, CI: 3.4–8.8).33 There is no published study so far that has examined the functional role of myoferlin in the nucleus. Therefore, in this study, we examined if myoferlin modulates IL-6/STAT3 signaling by binding to phosphorylated STAT3 and chaperoning it to the nucleus.
In our immunoprecipitation and HA pulldown experiments, we observed that myoferlin is bound to EHD2 protein in resting cells and IL-6 treatment of these cells induced myoferlin dissociation from EHD2 and binding to phosphorylated STAT3. EHD2 protein is predominantly studied for its role in endocytic recycling and has been shown to bind to myoferlin in muscle cells.24 It is possible that EHD2 protein might also function as a natural inhibitor of myoferlin by binding to it and blocking its IL-6/STAT3 signaling functions. Recent studies have shown that EHD2 could function as a tumor suppresser protein.39–42 EHD2 overexpression was inversely associated with overall survival and EHD2 knockdown significantly enhanced breast cancer and esophageal squamous cell carcinoma (ESCC) cell migration, cell invasion and enhanced epithelial to mesenchymal transition (EMT).39, 40 In this study, we observed that dissociation of EHD2 from myoferlin in activated tumor cells (treated with IL-6) led to enhanced tumor cell motility and cancer stem cell phenotype. Similarly, myoferlin knockdown in cancer cells significantly inhibited tumor cell motility, tumor invasion and promoted mesenchymal to epithelial transition (MET).35, 43 These results support our inference that EHD2 might mediate its tumor suppressor function by inhibiting myoferlin function and IL-6/STAT3 signaling. However, further studies are required to validate the role of EHD2 in negatively regulating myoferlin function.
There have been contradicting reports regarding the requirement of tyrosine phosphorylation in STAT3 protein for its translocation to the nucleus.44–46 Liu et al have recently shown that STAT3 nuclear import is independent of tyrosine phosphorylation.44 However, a number of other studies have shown that tyrosine phosphorylation (particularly mediated by growth factors; e.g. IL-6, VEGF) is required for STAT3 translocation to the nucleus.45, 46 Our results suggest that in IL-6 mediated STAT3 nuclear translocation, tyrosine phosphorylation is essential for its binding to myoferlin and subsequent translocation to nucleus. However, in myoferlin deficient cells, STAT3 tyrosine phosphorylation alone was not enough to mediate its translocation to nucleus. It is possible that at basal level, STAT3 nuclear translocation might not be dependent on tyrosine phosphorylation and is predominantly mediated by binding to importin-a3.44 But in the growth factor (e.g. IL-6) mediated nuclear translocation, STAT3 phosphorylation markedly upregulates its nuclear translocation by enhancing STAT3 binding to a chaperone protein “myoferlin”. Similar chaperones mediated enhanced nuclear trafficking of glucocorticoid receptors have been reported.47
Recently, we have shown that high expression of IL-6 is directly associated with nuclear localization of myoferlin in head and neck tumors.33 In addition, nuclear myoferlin expression was predictive of tumor recurrence, perineural invasion, extracapsular spread (ECS) and distal metastasis. Similarly, a number of gene microarray studies have reported an association between myoferlin expression and metastatic cancer.48–50 However, molecular mechanism(s) that underline myoferlin-mediated aggressive and metastatic phenotype in cancer cells is not very well understood. Our results from this study provide a novel insight into the role of myoferlin in regulating IL-6/STAT3 signaling cascade that has been shown to promote tumor growth, chemoresistance and tumor metastasis,2, 51, 52 Results from this study also provide a scientific rationale to design small molecule inhibitors that could disrupt myoferlin/STAT3 protein-protein interactions thereby providing a novel therapeutic option for the treatment of head and neck cancer patients.
Materials and Methods
Patient tumor samples, reagents and Cell lines
Tumor and normal tissue samples used in this study were collected from HNSCC patients undergoing treatment at the James Cancer Hospital after obtaining informed consent. The use of patient samples was approved by the Ohio State University institutional review board. CAL27 and MDA-MB-231 were obtained from ATCC (Manassas, VA). UM-SCC-74A and UM-SCC-74B cell lines were obtained from the laboratory of Dr. Thomas E. Carey at the University of Michigan.53 The identity of all the tumor cell lines was confirmed by STR genotyping (Identifiler Kit, Applied Biosystems, Carlsband, CA). Immortalized (HPV E6/E7) oral epithelial cells (IOE) were kindly provided by Drs. William Foulkes and Ala-Eddin Al Moustafa (McGill University, Montreal, Quebec, Canada).54 Normal human oral keratinocytes (HOK) were obtained from ScienCell (Carlsbad, CA). All tumor cell lines were cultured in DMEM supplemented with 10% fetal bovine serum containing 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA) and 1% Non-essential amino acids. IOE and HOK were grown in keratinocyte growth medium (Invitrogen, Carlsbad, CA). HA-tagged myoferlin plasmid (pCDNA3.1-Myoferlin HA, Principal Investigator; Dr. William Sessa)17 was obtained from Addgene (Cambridge, MA). Primary antibodies against STAT3 (# 9132), STAT3 (pY705, # 9131), Lamin (# 2032), Tubulin (#2148) and E-Cadherin (# 3195) were obtained from Cell Signaling (Danvers, MA); EHD2 (# ab23935) was from Abcam (Cambridge, MA); Myoferlin (# HPA014245) was from Sigma (St. Louis, MO) and GAPDH (# MAB374) from Millipore (Billerica, MA). Recombinant IL-6 was purchased from PeproTech (Rocky Hill, NJ). JAK inhibitor 1 was obtained from EMD Millipore (Billerica, MA) and ALDH staining kit was obtained from Stemcell Technologies, (Vancouver, Canada).
Quantitative PCR
RNA from the tumor samples (n=20), normal controls (n=10), HNSCC cell lines (n=10) and normal epithelial cells (n=2) was extracted using TRIzol reagent (Invitrogen). Myoferlin, snail or nanog RNA was transcribed into cDNA and amplified with TaqMan primer/probes (myoferlin; Hs00203853-m1, nanog; Hs02387400-g1 and snail; Hs00195591-m1). mRNA expression for myoferlin, snail or nanog was normalized to OAZ1 using the 2−ΔΔCt method.55
Myoferlin overexpression
HA-tagged myoferlin was transfected in immortalized oral epithelial cells (IOE) using lipofectamine 3000 (Invitrogen).2 Myoferlin overexpression was verified by Western blotting.
Myoferlin knockdown with siRNA or shRNA
CAL27 and UM-SCC-74A cells were transduced with Dharmacon siGENOME SMART pool siRNA for Myoferlin. In brief, tumor cells were cultured in 60 mm dishes and transfected with 100 nM of siRNA SMART pool. Cells transfected with scrambled siRNA were used as control. After 16–18 hours of incubation, cells were washed with PBS and further incubated in fresh medium. Seventy two hours post transfection, cells were used for experiments. For stable knockdown, we used two shRNAs; MISSION® Myoferlin shRNA Lentiviral transduction particles from Sigma-Aldrich and Myoferlin shRNA (m) Lentiviral particles from Santa Cruz Biotechnology. MISSION® pLKO.1-puro carrying scrambled shRNA was used as a control. The lentiviral constructs were transduced in the cells in 96 well plate using polybrene to the final concentration of 4μg/ml. The transfected cells were selected with puromycin treatment and myoferlin knockdown was confirmed by Western blotting.
Immunoprecipitation
Tumor cells were cultured in 6 cm dishes and treated with IL-6. After 30 minutes, cells were washed with PBS and whole cell lysate prepared. The whole cell lysate was then incubated with 50μl of protein A/G agarose beads (Thermo Scientific-Pierce, Rockford, IL) for 30 min at 4°C. After pre-clearing step, cell lysate was incubated with anti-myoferlin antibody overnight at 4°C on rocker. Next, protein A/G Agarose beads slurry was added to the cell lysate and further incubated for 4 hours at 4°C. After incubation, agarose beads were collected by centrifuging the slurry. Proteins bound to agarose beads were separated by adding 2x sample buffer and boiling it at 95°C for 5 min.
Cytoplasmic and nuclear protein extraction
Nuclear and cytoplasmic proteins from tumor cells were separated using the NE-PER Nuclear & Cytoplasmic extraction kit (Pierce, Rockford, IL). Cells were treated with IL-6 for different time points and then harvested for cytosolic and nuclear protein extraction. Cell pellet was treated with cell membrane lysis reagents from the extraction kit to release the cytoplasmic contents. The cytoplasmic proteins were collected by centrifugation leaving the intact nuclei in the pellet. The nuclear pellet was washed with PBS to reduce carryover of the cytoplasmic proteins to the nuclear protein fraction. Nuclear lysis buffer was then added to the pellet to lyse the nuclei and release the nuclear proteins. Reducing agent and loading buffer were then added to the cytoplasmic and nuclear extracts and analyzed by Western blotting.
Western Blot Analysis
Cell lysates were separated using NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred onto PVDF membranes. Nonspecific bindings were blocked by incubating membranes with 5% Milk or 3% BSA in TBST for 1 hour at room temperature. Membranes were then incubated with the respective primary antibody in TBST + 5% Milk or 3% BSA at 4°C overnight. The blots were washed and then incubated with sheep anti-mouse (1:3,000) or with donkey anti-rabbit (1:4,000) IgG-labelled with horseradish peroxidase for 1 hour at room temperature and specific protein bands were detected using ECL-plus kit (Thermo Scientific-Pierce, Rockford, IL). Equal protein loading was verified by stripping the blots and then re-probing it for GAPDH.
Immunofluorescent staining
Tumor cells were cultured in Labtech chambers and treated with recombinant IL-6 (50 ng/ml). At the end of incubation, cells were fixed with 4% paraformaldehyde and permeablized by treatment with 100% methanol for 10 minutes at −20°C. Next, slides were washed with PBS, blocked with normal goat IgG for 1 hour and incubated overnight at 4°C with rabbit anti-myoferlin and mouse anti-STAT3 antibodies. After three washes, chamber slides were then incubated with secondary antibodies (Alexa Fluor 488 labeled-goat anti-mouse-IgG or goat Alexa Fluor 594 labelled-anti-rabbit-IgG). Chamber slides were then mounted with ProLong gold with DAPI (Invitrogen). The immunofluorescent staining images were captured using Nikon Eclipse 80i microscope and overlaid using NIS-Elements-Basic software (Nikon, Melville, NY).
Tumor cell motility assay
We used Xcelligence system (ACEA Biosciences, San Diego, CA) to measure tumor cell motility.2 In brief, 160 μl of DMEM media with 10% FBS was carefully added to the lower chambers. Next, upper chamber was then carefully assembled with lower chamber and 50 μl of serum free media added to the top wells. After 1 hour, cell suspension (50,000 cells in 100 μl) was added to each well. Cell migration from upper chamber to lower chamber was monitored for 24 hrs.
ALDH staining
Tumor cells were cultured in 6 cm plates, trypsinized and adjusted to 1 × 106 cells/ml. Cell were then subdivided into control and test groups and stained for ALDH (Stemcell Technologies, Vancouver, Canada). After staining, cells were analyzed by flow cytometry.
Tumorsphere formation
Tumor cells were cultured on ultralow binding plates (Corning, Lowell, MA) for 7 days as previously described.56 At the end of incubation, number of tumorsphere >50μm were counted. Tumorspheres were gently spun down, mixed with Matrigel and allowed to solidify at 37°C. Matrigel plugs were then fixed, paraffin embedded and processed for immunohistochemistry. Tumorsphere sections were deparaffinized and standard heat induced antigen retrieval method was used as described before.57 Slides were then washed with TBST, blocked with normal goat serum for 1 hour, and then incubated with rabbit anti-E-cadherin and mouse anti-nanog antibodies. After overnight incubation, slides were washed with PBS and further incubated with secondary antibodies (Alexa Fluor 488 labeled-goat anti-mouse-IgG and Alexa Fluor 594 labeled-goat anti-rabbit-IgG-). Slides were then mounted with ProLong gold with DAPI (Invitrogen). The immunofluorescent staining images were captured using Nikon Eclipse 80i microscope and overlaid using NIS-Elements-Basic software.
Tumor growth and metastasis model
Animal studies were approved by The Ohio State University IACUC Animal ethic committee. Myoferlin was stably knocked down in CAL27-IL-6 (CAL27 overexpressing IL-6) and UM-SCC-74A cells (naturally express high IL-6). Tumor cells (1×106) were implanted subcutaneously in the flanks of SCID mice (n=5 for each group).38 Tumor volume was measured twice a week starting day 6 till the end of the study. After 39 days, animals were euthanized and tumors, regional lymph nodes, and lungs were harvested. To label and identify flank draining lymph nodes, 25 μl of 5% dye (Evans Blue, Sigma) was injected in the foot pads of animals 30 minutes before euthanizing.58 Primary tumors, lymph nodes and lungs were fixed with paraformaldehyde and processed for immunohistochemistry as described in tumorsphere formation.
Statistical analysis
We used a conservative Bonferroni correction method for the sample size calculations. Results are presented as mean ± s.e.m. Mann-Whitney test was used to examine the significance of myoferlin expression in patient samples. For rest of the in vitro and in vivo experiments, we used two-way analysis of variance and Student’s t test to analyze the data. The difference between different groups with p value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported in part by funding from NIH/NCI (CA178649 to PK) and The Ohio State University Comprehensive Cancer Center (PK and TNT).
Footnotes
Disclosure of Potential Conflict of Interest: All the authors have no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8(Suppl 2):S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011 doi: 10.1158/1541-7786.MCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lederle W, Depner S, Schnur S, Obermueller E, Catone N, Just A, et al. IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer. 2011;128:2803–2814. doi: 10.1002/ijc.25621. [DOI] [PubMed] [Google Scholar]
- 4.Lesina M, Kurkowski Magdalena U, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, et al. Serum Interleukin 6 as a Prognostic Factor in Patients with Prostate Cancer. Clinical Cancer Research. 2000;6:2702–2706. [PubMed] [Google Scholar]
- 6.Dethlefsen C, Højfeldt G, Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Research and Treatment. 2013;138:657–664. doi: 10.1007/s10549-013-2488-z. [DOI] [PubMed] [Google Scholar]
- 7.Duffy SA, Taylor JM, Terrell JE, Islam M, Li Y, Fowler KE, et al. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer. 2008;113:750–757. doi: 10.1002/cncr.23615. [DOI] [PubMed] [Google Scholar]
- 8.Riedel F, Zaiss I, Herzog D, Gotte K, Naim R, Hormann K. Serum levels of interleukin-6 in patients with primary head and neck squamous cell carcinoma. Anticancer research. 2005;25:2761–2765. [PubMed] [Google Scholar]
- 9.Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, et al. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825–828. doi: 10.1126/science.3136546. [DOI] [PubMed] [Google Scholar]
- 10.Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–1157. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 11.Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–581. doi: 10.1016/0092-8674(89)90438-8. [DOI] [PubMed] [Google Scholar]
- 12.Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, et al. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–1810. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong Z, Wen Z, Darnell J. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 15.Wente SR, Rout MP. The Nuclear Pore Complex and Nuclear Transport. Cold Spring Harbor Perspectives in Biology. 2010;2:a000562. doi: 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernatchez PN, Sharma A, Kodaman P, Sessa WC. Myoferlin is critical for endocytosis in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C484–492. doi: 10.1152/ajpcell.00498.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernatchez PN, Acevedo L, Fernandez-Hernando C, Murata T, Chalouni C, Kim J, et al. Myoferlin Regulates Vascular Endothelial Growth Factor Receptor-2 Stability and Function. Journal of Biological Chemistry. 2007;282:30745–30753. doi: 10.1074/jbc.M704798200. [DOI] [PubMed] [Google Scholar]
- 18.Davis DB, Delmonte AJ, Ly CT, McNally EM. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum Mol Genet. 2000;9:217–226. doi: 10.1093/hmg/9.2.217. [DOI] [PubMed] [Google Scholar]
- 19.Jiménez JL, Bashir R. In silico functional and structural characterisation of ferlin proteins by mapping disease-causing mutations and evolutionary information onto three-dimensional models of their C2 domains. Journal of the Neurological Sciences. 2007;260:114–123. doi: 10.1016/j.jns.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 20.Perin MS, Friedt VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 22.Demonbreun AR, Lapidos KA, Heretis K, Levin S, Dale R, Pytel P, et al. Myoferlin regulation by NFAT in muscle injury, regeneration and repair. Journal of Cell Science. 2010;123:2413–2422. doi: 10.1242/jcs.065375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, et al. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doherty KR, Demonbreun AR, Wallace GQ, Cave A, Posey AD, Heretis K, et al. The Endocytic Recycling Protein EHD2 Interacts with Myoferlin to Regulate Myoblast Fusion. Journal of Biological Chemistry. 2008;283:20252–20260. doi: 10.1074/jbc.M802306200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roux I, Safieddine S, Nouvian R, Grati Mh, Simmler M-C, Bahloul A, et al. Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 26.Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. Journal of Biological Chemistry. 2002;277:22883–22888. doi: 10.1074/jbc.M201858200. [DOI] [PubMed] [Google Scholar]
- 27.Turtoi A, Blomme A, Bellahcene A, Gilles C, Hennequiere V, Peixoto P, et al. Myoferlin Is a Key Regulator of EGFR Activity in Breast Cancer. Cancer Res. 2013;73:5438–5448. doi: 10.1158/0008-5472.CAN-13-1142. [DOI] [PubMed] [Google Scholar]
- 28.Li R, Ackerman WEt, Mihai C, Volakis LI, Ghadiali S, Kniss DA. Myoferlin depletion in breast cancer cells promotes mesenchymal to epithelial shape change and stalls invasion. PLoS One. 2012;7:e39766. doi: 10.1371/journal.pone.0039766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turtoi A, Musmeci D, Wang Y, Dumont B, Somja J, Bevilacqua G, et al. Identification of Novel Accessible Proteins Bearing Diagnostic and Therapeutic Potential in Human Pancreatic Ductal Adenocarcinoma. Journal of Proteome Research. 2011;10:4302–4313. doi: 10.1021/pr200527z. [DOI] [PubMed] [Google Scholar]
- 30.Difilippantonio S, Chen Y, Pietas A, Schlüns K, Pacyna-Gengelbach M, Deutschmann N, et al. Gene expression profiles in human non-small and small-cell lung cancers. European Journal of Cancer. 2003;39:1936–1947. doi: 10.1016/s0959-8049(03)00419-2. [DOI] [PubMed] [Google Scholar]
- 31.Abba MC, Hu Y, Sun H, Drake JA, Gaddis S, Baggerly K, et al. Gene expression signature of estrogen receptor α status in breast cancer. BMC Genomics. 2005;6:37–37. doi: 10.1186/1471-2164-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu C, Sharma A, Trane A, Utokaparch S, Leung C, Bernatchez P. Myoferlin gene silencing decreases Tie-2 expression in vitro and angiogenesis in vivo. Vascular Pharmacology. 2011;55:26–33. doi: 10.1016/j.vph.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Kumar B, Brown NB, Swanson BJ, Schmitt AC, Old O, Ozer E, et al. High expression of myoferlin is associated with Poor Outcome in Oropharyngeal Squamous Cell Carcinoma Patients and is Inversely Associated with HPV-Status. Oncotarget. 2016 doi: 10.18632/oncotarget.7625. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian SS, Lamb P, Seidel HM, Stein RB, Rosen J. Rapid activation of the STAT3 transcription factor by granulocyte colony-stimulating factor. Blood. 1994;84:1760–1764. [PubMed] [Google Scholar]
- 35.Li R, Ackerman WE, Mihai C, Volakis LI, Ghadiali S, Kniss DA. Myoferlin Depletion in Breast Cancer Cells Promotes Mesenchymal to Epithelial Shape Change and Stalls Invasion. PLoS ONE. 2012;7:e39766. doi: 10.1371/journal.pone.0039766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src Family Kinase Hck Requires a Functional SH3 Domain. Journal of Biological Chemistry. 2002;277:45680–45687. doi: 10.1074/jbc.M204255200. [DOI] [PubMed] [Google Scholar]
- 37.Villalva C, Martin-Lanneree S, Cortes U, Dkhissi F, Wager M, Le Corf A, et al. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer. 2010;128:826–838. doi: 10.1002/ijc.25416. [DOI] [PubMed] [Google Scholar]
- 38.Kumar P, Ning Y, Polverini PJ. Endothelial cells expressing Bcl-2 promotes tumor metastasis by enhancing tumor angiogenesis, blood vessel leakiness and tumor invasion. Lab Invest. 2008;88:740–749. doi: 10.1038/labinvest.2008.46. [DOI] [PubMed] [Google Scholar]
- 39.Li M, Yang X, Zhang J, Shi H, Hang Q, Huang X, et al. Effects of EHD2 interference on migration of esophageal squamous cell carcinoma. Medical Oncology (Northwood, London, England) 2013;30:396. doi: 10.1007/s12032-012-0396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Ren H, Yao L, Chen X, He A. Role of EHD2 in migration and invasion of human breast cancer cells. Tumor Biol. 2015;36:3717–3726. doi: 10.1007/s13277-014-3011-9. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Liu X, Sun Y, Wu D, Qiu A, Cheng H, et al. Decreased expression and prognostic role of EHD2 in human breast carcinoma: correlation with E-cadherin. J Mol Hist. 2015;46:221–231. doi: 10.1007/s10735-015-9614-7. [DOI] [PubMed] [Google Scholar]
- 42.Lv J, Fan N, Wang Y, Wang X, Gao C. Identification of Differentially Expressed Proteins of Normal and Cancerous Human Colorectal Tissues by Liquid Chromatograph-Mass Spectrometer Based on iTRAQ Approach. Cancer investigation. 2015:1–9. doi: 10.3109/07357907.2015.1055757. [DOI] [PubMed] [Google Scholar]
- 43.Eisenberg MC, Kim Y, Li R, Ackerman WE, Kniss DA, Friedman A. Mechanistic modeling of the effects of myoferlin on tumor cell invasion. Proceedings of the National Academy of Sciences. 2011;108:20078–20083. doi: 10.1073/pnas.1116327108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8150–8155. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yahata Y, Shirakata Y, Tokumaru S, Yamasaki K, Sayama K, Hanakawa Y, et al. Nuclear Translocation of Phosphorylated STAT3 Is Essential for Vascular Endothelial Growth Factor-induced Human Dermal Microvascular Endothelial Cell Migration and Tube Formation. Journal of Biological Chemistry. 2003;278:40026–40031. doi: 10.1074/jbc.M301866200. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y-p, Tan Y-n, Wang Z-l, Zeng L, Lu Z-x, Li L-l, et al. Phosphorylation and nuclear translocation of STAT3 regulated by the Epstein-Barr virus latent membrane protein 1 in nasopharyngeal carcinoma. International Journal of Molecular Medicine. 2008;21:153–162. [PubMed] [Google Scholar]
- 47.DeFranco DB. Role of molecular chaperones in subnuclear trafficking of glucocorticoid receptors. Kidney Int. 2000;57:1241–1249. doi: 10.1046/j.1523-1755.2000.00957.x. [DOI] [PubMed] [Google Scholar]
- 48.Amatschek S, Koenig U, Auer H, Steinlein P, Pacher M, Gruenfelder A, et al. Tissue-Wide Expression Profiling Using cDNA Subtraction and Microarrays to Identify Tumor-Specific Genes. Cancer Research. 2004;64:844–856. doi: 10.1158/0008-5472.can-03-2361. [DOI] [PubMed] [Google Scholar]
- 49.Adam PJ, Boyd R, Tyson KL, Fletcher GC, Stamps A, Hudson L, et al. Comprehensive Proteomic Analysis of Breast Cancer Cell Membranes Reveals Unique Proteins with Potential Roles in Clinical Cancer. Journal of Biological Chemistry. 2003;278:6482–6489. doi: 10.1074/jbc.M210184200. [DOI] [PubMed] [Google Scholar]
- 50.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 51.Bourguignon LY, Earle C, Wong G, Spevak CC, Krueger K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene. 2012;31:149–160. doi: 10.1038/onc.2011.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21:7611–7618. doi: 10.1038/sj.onc.1206004. [DOI] [PubMed] [Google Scholar]
- 53.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head & Neck. 2010;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al Moustafa A-E, Foulkes WD, Benlimame N, Wong A, Yen L, Bergeron J, et al. E6//E7 proteins of HPV type 16 and ErbB-2 cooperate to induce neoplastic transformation of primary normal oral epithelial cells. Oncogene. 2004;23:350–358. doi: 10.1038/sj.onc.1207148. [DOI] [PubMed] [Google Scholar]
- 55.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-[Delta][Delta]CT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 56.Kumar B, Yadav A, Lang JC, Teknos TN, Kumar P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes & Cancer. 2015;6:169–181. doi: 10.18632/genesandcancer.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar P, Benedict R, Urzua F, Fischbach C, Mooney D, Polverini P. Combination treatment significantly enhances the efficacy of antitumor therapy by preferentially targeting angiogenesis. Lab Invest. 2005;85:756–767. doi: 10.1038/labinvest.3700272. [DOI] [PubMed] [Google Scholar]
- 58.Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332:170–174. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.