

Abstract
MEK1/2 and BRAFV600E inhibitors are used to treat BRAFV600E-positive melanoma, with other cancers under evaluation. Genetic perturbation of copper import or pharmacological reduction of copper with the clinical copper chelator TTM inhibits MEK1/2 kinase activity and reduces BRAFV600E-driven tumorigenesis. In this study, we report that TTM inhibited transformed growth of melanoma cell lines resistant to BRAF or MEK1/2 inhibitors and enhanced the antineoplastic activity of these inhibitors. TTM also provided a survival advantage in a genetically engineered mouse model of melanoma, and when accounting for putative overdosing, trended towards an increase in the survival benefit afforded by BRAF inhibition. This effect was phenocopied by genetically inhibiting copper import in tumors, which was linked to a reduction in MAPK signaling. Thus, TTM reduces copper levels and MAPK signaling, thereby inhibiting BRAFV600E-driven melanoma tumor growth. These observations inform and support clinical evaluation of TTM in melanoma.
Keywords: Copper, cancer, mouse model, melanoma
Introduction
The rate of melanoma has doubled in the last 30 years and will account for an estimated 10,000 deaths this year in the USA (1). While immune checkpoint inhibitors have recently become one of the frontline therapies for advanced melanoma, not all patients respond to these drugs (2). In this regard, oncogenic (V600E) BRAF mutations occur in roughly half of melanomas and to varying degrees in other cancers. The BRAFV600E kinase phosphorylates and activates the MEK1/2 kinases, which in turn phosphorylate and activate the ERK1/2 kinases, chronically stimulating the MAPK pathway (3,4). The mutant-selective, ATP-competitive BRAF inhibitors (BRAFi) vemurafenib and dabrafenib are used to treat late-stage BRAFV600E-positive melanoma and show promise in a variety of other cancers (3,5–7). Resistance to BRAFi nevertheless arises, primarily through reactivation of the MAPK pathway (3,8,9). Combining the allosteric MEK1/2 inhibitors (MEKi) trametinib or cobimetinib with dabrafenib (10,11) or vemurafenib (12), respectively, leads to more durable responses and is now standard-of-care. MEKi are also being tested in a broad spectrum of other cancers (13,14). As such, MEK1/2 are clinically validated drug targets in advanced melanoma and have potential for the treatment of other cancers.
MEK1/2 bind copper (Cu) to promote kinase activity (15,16). Reducing expression of the primary Cu transporter 1 (CTR1) (17) inhibits BRAFV600E signaling and tumorigenesis, an effect rescued by expressing either a Cu-independent MEK1 or activated ERK2. Conversely, mutating sites in MEK1 associated with binding Cu reduces its kinase activity and BRAFV600E signaling and tumorigenesis (16). This novel activity can be pharmacologically targeted with the selective Cu chelator ammonium tetrathiomolybdate (TTM), used to lower Cu levels in patients with Wilson disease (18). Namely, TTM inhibits MEK1/2 kinase activity and growth of BRAFV600E-positive tumors (15,16). This antineoplastic effect is specific to inhibition of MEK1/2, and not other targets, as cells transformed by oncogenic BRAF or engineered to also express activated MEK1 are sensitive to TTM, whereas cells expressing activated ERK2 are resistant (16). Although TTM has not been clinically explored in a cancer credentialed to be BRAFV600E-positive, this drug has been extensively evaluated in cancer trials. By monitoring the serum ceruloplasmin (Cp) activity, a clinical biomarker of Cu (19), cancer patients have been maintained on TTM for as long as 65 months (20). As such, TTM is a well tolerated and affordable drug that can be dosed daily for protracted periods. Hence, this drug has exciting potential for enhancing the antineoplastic activity and/or suppressing resistance to MAPK inhibitors (MAPKi) currently in clinical use.
Materials and Methods
Cell lines
SV40 immortalized Ctr1+/+ and Ctr1−/− mouse embryonic fibroblasts (MEFs) stably expressing MYC-HIS-BRAFV600E were previously described (16) and maintained in DMEM supplemented with 10% FBS. A375 cells (ATCC, catalog # CRL-1619) were maintained in DMEM supplemented with 10% FBS. DM440 and DM443 cells (21) were maintained in IMDM supplemented with 10% FBS. 451Lu parental cells (451Lu-P) and a derivative resistant to BRAF and MEK inhibitors (451Lu-BMR) (22) were a kind gift from Jessie Villanueva (Wistar Institute) and maintained in DMEM supplemented with 5 % FBS with 1 μM trametenib (22). Derived cell lines were generated by stable infection with retroviruses derived from the pBABE vectors described below using established methods (23).
Drugs
Vemurafenib, trametinib (Chemitek), and ammonium tetrathiomolybdate, abbreviated as TTM (Sigma-Aldrich), were dissolved in DMSO for in vitro experiments and 1% DMSO / 1% methylcellulose for in vivo experiments.
Plasmids
pBABEpuro-MYC-HIS-BRAFV600E was previously described (16). pBABEpuro-HA-p61BRAFV600E was created by two-step PCR subcloning of human BRAFV600E cDNA from previously described plasmid pBABEpuro-MYC-HIS-BRAFV600E (16) using primers designed to delete the sequence corresponding to exons 4–8 and to include an N-terminal haemagglutinin (HA) tag. pBABEpuro-HA-COT1, pBABEpuro-HA-CRAF, pBABEpuro-HA-IGF-1R, and pBABEpuro-HA-PDGFRß were created by PCR subcloning the relevant human cDNAs from plasmids pDONR223-MAP3K8, pBABEpuro-CRAF, pBABEbleo-IGF-1R, and pDONR223-PDGFRß (Addgene, plasmids # 23538, 51124, 11212, and 23893, respectively) with primers designed to include an N-terminal HA tag. pBABEpuro-HA-IKKß was created by PCR subcloning human IKKß cDNA from plasmid pDONR-IKKß (Addgene, plasmid # 64609) with primers designed to include an N-terminal HA tag. pBABEpuro-HA-MEK1DD (S218D/S222D), pBABEpuro-HA-MEK1F53S, pBABEpuro-HA-MEK1Q56P, pBABEpuro-HA-MEK1K57N, pBABEpuro-HA-MEK1D67N, pBABEpuro-HA-MEK1L115P, pBABEpuro-HA-MEK1C121S, pBABEpuro-HA-MEK1P124S, pBABEpuro-HA-MEK1Y134C, pBABEpuro-HA-MEK1E203K, and pBABEpuro-HA-MEK1P264S were created by altering the nucleic acid sequence of the wild-type human MAP2K1 cDNA from the pDONR223-MAP2K1 plasmid (Addgene, plasmid # 23406) by site-directed mutagenesis to encode the indicated amino acid changes, followed by PCR subcloning with primers designed to include an N-terminal HA tag. pBABEpuro-HA-MEK2DD (S218D/S222D), pBABEpuro-HA-MEK2E27K, pBABEpuro-HA-MEK2E45K, pBABEpuro-HA-MEK2Q60P, pBABEpuro-HA-MEK2C125S, pBABEpuro-HA-MEK2S154F, and pBABEpuro-HA-MEK2E207K were created by altering the nucleic acid sequence of the wild-type human MAP2K2 cDNA from the pDONR223-MAP2K2 plasmid (Addgene, plasmid # 23555) by site-directed mutagenesis to encode the indicated amino acid changes, followed by PCR subcloning with primers designed to include an N-terminal HA tag. pBABE-HA-NRASQ61L was created by altering the nucleic acid sequence of the wild-type human NRAS cDNA from pBABEpuro-NRAS described previously (24) by site-directed mutagenesis to encode the indicated amino acid change, followed by PCR subcloning with primers designed to include an N-terminal HA tag.
Soft agar assay
2.5 × 104 of the indicated cells were resuspended in 1 ml of 0.3 % bactoagar solution containing vehicle or drug and plated over a 2 ml bottom layer of 0.6% bactoagar solution containing vehicle or drug in a 6-well plate. Cells were fed weekly with 250 μl of media containing vehicle or drug. After 4 weeks, colonies of ≥ 50 cells were counted in a blinded fashion as previously described (16). Drug concentrations for A375 cells were as follows: 25, 50, or 100 nM TTM; 3.125, 6.25, or 12.5 nM vemurafenib; or 75, 150, or 300 pM trametinib. Drug concentrations for DM440 and DM443 cells were as follows: 400 nM TTM; 50 nM vemurafenib; or 600 pM trametinib. Drug concentrations for 451Lu-P and 451Lu-BMR cells were as follows: 1000 nM TTM, 1000 nM, or 100 nM trametinib. Combinations of drugs were used at the indicated concentrations in fixed-ratio doses for purposes of synergy calculations.
Genetically engineered mouse model of melanoma
Mice with BrafCA, Ptenfl, Tyrosinase∷CreERT2tg, or Ctr1fl alleles (17,25–28) were obtained from The Jackson Laboratory (Bar Harbor) or as a kind gift of either David Kirsch or Dennis Thiele (Duke University). To determine the effects of vemurafenib, TTM, or both drugs on survival of mice developing melanoma, BrafCA/CA;Ptenfl/fl mice were bred to Ptenfl/fl;Tyrosinase∷CreERT2tg/+ mice to generate BrafCA/+;Ptenfl/fl; Tyrosinase∷CreERT2tg/+ (BPC) mice. To determine the effects of inactivating Ctr1 in tumors on the survival of mice developing melanoma, BrafCA/CA;Ptenfl/fl;Ctr1fl/+ mice were crossed to Ptenfl/fl;Ctr1fl/+;Tyrosinase∷CreERT2tg/+ mice to generate Ctr1+/+ and Ctr1fl/fl BPC littermates. To induce melanomagenesis, a patch of hair on the right flank of 28-day old mice cleared by shaving and treatment for 10 minutes with VEET depilatory cream (Reckitt Benckiser LLC), after which 2 μl of 5 μM 4-hydroxytamoxifen (abbreviated as 4-HT, Sigma-Aldrich) in DMSO was applied topically once a day for 3 consecutive days, as previously described (26). The flank was shaved as needed to allow visualization of nevi formation. Upon nevi formation, mice were treated daily by oral gavage with the vehicle (250 μl of 1 % DMSO / 1 % methylcellulose), vemurafenib (50 mg/kg), or TTM (200 μl of 10 mg/ml for a fixed dose of 2 mg corresponding to 80 mg/kg in adult mice), or the indicated combinations of drugs at the aforementioned doses. The appearance, behavior, weight, and tumor volume (calculated by use of the modified ellipsoid formula 1/2 (Length × Width × Height) of mice were monitored daily. Drug holidays were provided if the weight of a mouse dropped 15% below maximum, and was maintained until the weight recovered back to maximum weight. Mice were euthanized upon reaching a moribundity endpoint, which was primarily a maximum tumor volume of 1.0 cm3. Mice were not included in analysis if they developed a spontaneous offsite tumor, presumably due to leaky Cre expression (29), or developed tumor ulceration. In the censored analysis, only the mice that either reached tumor volume endpoint or lived at least to the median survival for vehicle-treated mice (44 days) were included. In the repeat analysis of vehicle versus TTM, the preclinical trial was performed exactly as above with two modifications, 1) tumor initiation was performed in heavier 44-day old mice instead of 23-day old mice and 2) TTM was dosed based on animal weight (80 mg/kg) instead of the fixed dose of 2 mg. These studies were conducted in accordance with protocols approved by the Duke University and University of Pennsylvania Institutional Animal Care and Use Committee and performed with support from the Division of Laboratory Animal Resources at Duke University and University Laboratory Animal Resources at the University of Pennsylvania in accordance with all relevant ethical guidelines.
Mouse xenografts
5 × 106 MEFs resuspended in phosphate buffer saline were injected subcutaneously into flanks of CB17.Cg-PrkdcscidLystbg-J/Crl (Fox Chase SCID® beige) mice (Charles River Laboratory) as previously described (16). Drug treatments were as follows: vehicle or 80 mg/kg/day TTM in vehicle every day via oral gavage as described in detail above. The Institutional Animal Care and Use Committee at the University of Pennsylvania approved all studies. The appearance, behavior, weight, and tumor volume of mice were monitored daily. Mice were euthanized upon reaching a moribundity endpoint, which was primarily a maximum tumor volume of 1.0 cm3.
Immunoblotting
Lysates were isolated using standard protocols from A375, DM440, and DM443 cells expressing the indicated transgenes or A375 cell lines grown on poly-hema coated plates (to promote anchorage independent growth) treated with or without either a fixed concentration of TTM (100 nM), vemurafenib (12.5 nM), or trametinib (300 pM) or increasing concentrations of TTM (25 nM, 50 nM, and/or 100 nM), vemurafenib (3.125 nM, 6.25 nM, and/or 12.5 nM), or trametinib (75 pM, 150 pM, and/or 300 pM) for 7 days. Equal quantities were resolved by SDS–PAGE and immunoblotted as previously described (16) with mouse anti-ßACTIN, rabbit anti-phospho(Thr 202/Tyr 204)-ERK1/2, mouse anti-ERK1/2, mouse anti-HA, rabbit anti-phospho(Ser 235/236)-S6, or mouse anti-S6 primary antibody (Cell Signaling Technology, catalog # 3700, 9101, 9107, 4858, and 2317), followed by detection with either a goat anti-mouse IgG or a goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, catalog # 7074 and 7076), and visualized using enhanced chemiluminescence detection reagents (Cell Signaling Technology). The fold change in the ratio of phosphorylated protein to ßACTIN loading control was determined by boxing the appropriate bands of phosphorylated and ßACTIN per representative image with the rectangular selection tool and calculating the total volume of the band in pixels using the Image Lab™ Software (Bio-Rad). The total volume of the phosphorylated protein band in pixels was then normalized to the total volume of the ßACTIN protein band in pixels.
Reverse transcriptase-PCR
RNA was purified from tumor cell lines and reverse transcribed (RT) as previously described (23), afterwhich the indicated cDNAs were amplified with the primers: 5′- CTTTATCCAGCCCTCAC-3′ and 5′- ACCCTAACTGACACACATTCC-3′ that flank the multiple cloning site in pBABEpuro. The loading control GAPDH was amplified with the primers: 5′-GAGAGACCCTCACTGCTG-3′ and 5′-GATGGTACATGACAAGGTGC-3′.
Immunohistochemistry
Tumors were resected from euthanized BPC mice at endpoint, fixed for 24 hours in formalin and embedded in paraffin, after which 5 μm sections were cut with a RM2125 RTS microtome (Leica Biosystems Incorporated) and mounted on slides using standard methodologies. Slides were de-paraffinized, rehydrated, and stained with rabbit anti-phospho(Thr 202/Tyr204)-ERK1/2 or rabbit anti-phospho(Ser240/244)-S6 (Cell Signaling Technology, catalog # 4376 and 5364) by the Duke Pathology Research Histology Laboratory. Photographs were taken of 5 random, high-power fields per slide on a Vanox S microscope (Olympus Corporation of the Americas,) in a blinded fashion. Images were analyzed using Image J software (NIH) to determine the immuno-positive area by color thresholding, as previously described (16).
Serum ceruloplasmin (Cp) activity
Serum was collected from euthanized BPC mice at endpoint and flash frozen in liquid N2. At the time of analysis, serum samples from all cohorts were thawed and analyzed for Cp activity, as previously described (19). Briefly, 3.75 μl of serum was plated in duplicate in a 96-well plate. 37.5 μl of 0.1 M sodium acetate pH 6.0 (Sigma-Aldrich) was added to each well, and the plate was incubated at 37° C for 5 minutes. 15 μl of 2.5 mg/ml o-dianisidine (Sigma-Aldrich) was added to each well, and the plate was incubated at 37° C for 30 minutes. The reaction was quenched by adding 150 μl of 9 M sulfuric acid (Sigma-Aldrich). The absorbance at 540 nm was recorded with a Promax GloMax Multi+Luminometer (Promega Corporation).
ICP-MS analysis
The presence of Mo in the aforementioned serum samples was measured by ICP-MS using standard methodologies. Briefly, serum was digested overnight in 10 volumes of 9:1 9 M HNO3 : 5 M HCl (Sigma-Aldrich) at room temperature in tubes pre-washed with HNO3 and rinsed with ultra-pure water (17.45 MΩ-cm). The next day, samples were boiled for 30 minutes and allowed to cool. A total of 100 μl of each sample was transferred to new, pre-washed screw-top tubes and diluted 1:1 with ultra-pure water. Samples were then submitted to the Analytical Spectroscopy Service Laboratory at North Carolina State University for analysis on an Elan DRCII ICP-MS instrument (Perkin Elmer) for the presence of Mo.
Statistics
Statistical analysis of soft agar colony formation was performed with two-way Analysis of Variance (ANOVA) with a Bonferroni post test to compare transformed growth of each cell line with vector control cells treated with the same drug. The Combination and Bliss Indexes to test for synergy in drug combinations was performed as previously described (30,31). Statistical analysis of initial and maximum weights of BPC mice was performed with one-way ANOVA with a Sidak’s multiple comparisons post test to compare each Adverse Event group with each non-Adverse Event group. Statistical analysis of Kaplan-Meier survival curves was performed using the Mantel-Cox log-rank method to compare each treatment group to vehicle-treated control mice in a pairwise manner or to compare Ctr1fl/fl mice to Ctr1+/+ mice. Transformed Gompertz tumor volumes using the Gompertz function (y = ln[ln(Vmax)-ln(V)]) were plotted against time as previously described (32). The best-fit linear regression of the straight line was performed and has the format y = a + bx in which the specific growth rate α = −b. Tumor growth curves were fit using the exponential growth equation to calculate doubling time. Statistical analysis of serum Mo detected by ICP-MS and quantitative IHC to detect P-ERK1/2 and P-S6 in tumors from drug-treated mice was performed with one-way ANOVA with a Tukey’s multiple comparisons post test to compare each group to each other group. Statistical analysis of quantitative IHC to detect P-ERK1/2 and P-S6 in tumors from Ctr1fl/fl and Ctr1+/+ mice, ceruloplasmin activity in drug-treated mice, and time from 4-HT treatment to pigmentation for Ctr1fl/fl and Ctr1+/+ mice was performed with a two-tailed Student’s t-test. Statistical analysis of tumor volumes of treatment groups in comparison to vehicle group at end point was performed using a one-tailed, unpaired t-test, with a 95% confidence interval for two group datasets or one-way analysis of variance (ANOVA) with a 95%, 99%, or 99.9% confidence interval and Tukey’s multiple comparison post test for ≥ 3 datasets. All statistics were performed using Prism 6 software (GraphPad Software Incorporated) except for calculations of Combination Index, which was calculated using CalcuSyn software (BioSoft), and Bliss Index, which was calculated using Excel 2013 (Microsoft).
Results and discussion
TTM counters MAPK-dependent forms of resistance to MAPKi
To assess whether TTM can counter resistance to current MAPKi, a major challenge in the treatment of melanoma (9), BRAFV600E-mutant A375 melanoma cells were engineered to stably express various mediators of BRAFi resistance, including those identified from clinical samples (33–39), as assessed by RT- PCR and immunoblot (Fig. 1A and Supplementary Fig. S1). Specifically, we tested the p61BRAFV600E splice variant (36), over-expressed CRAF (37), and oncogenic NRASQ61L (35), which all maintain MAPK signaling in the presence of vemurafenib by promoting RAF dimerization. We also tested COT, which phosphorylates and activates MEK1/2 independent of RAF activity (38) and IKKβ, which activates NF-κB signaling to promote survival in the presence of MAPKi independent of the MAPK pathway (33). Finally, we tested IGF-1R (34) and PDGFRβ (35), which promote survival in the presence of MAPKi through activation of the PI3K/AKT pathway. The resultant cell lines were then assayed for anchorage independent growth in triplicate in three independent experiments in the presence of vehicle, TTM, vemurafenib, or trametinib at IC50 concentrations confirmed in vector control cells to both inhibit MAPK signaling, as evidenced by reduced phosphorylated (P-)ERK1/2 and P-S6 levels detected by immunoblot (Fig. 1B and Supplementary Fig. S2), and inhibit colony formation in soft agar (Fig. 1C). As expected, all seven proteins conveyed resistance to vemurafenib, while the MAPK-independent resistance drivers IKKß, IGF-1R, and PDGFRß additionally imparted resistance to TTM and trametinib despite reduced P-ERK1/2 and P-S6 (Fig.1B, 1C, and Supplementary Fig. S3). CRAF imparted some resistance to TTM but not to trametinib, while p61BRAFV600E, COT, and NRASQ61L did not lead to resistance to TTM or trametinib (Fig. 1C). Thus, TTM inhibited the transformed growth of A375 cells expressing most MAPK-dependent drivers of resistance to MAPKi.
Figure 1. TTM counters MAPK-dependent forms of resistance to MAPKi.
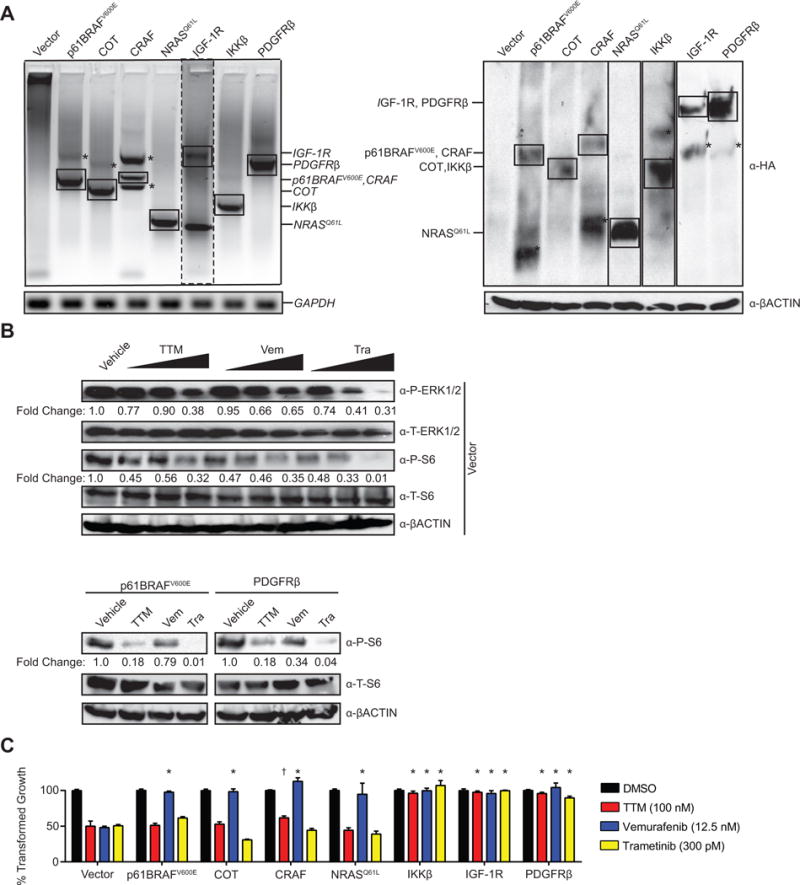
(A) RT-PCR (left) or immunoblot (right) detection of ectopic HA-tagged p61BRAFV600E, COT, CRAF, NRASQ61L, IGF-1R, IKKβ, or PDGFRβ stably expressed in A375 cells. GAPDH and ßACTIN: loading control. *nonspecific amplification or immunoreactivity. (B) Immunoblot detection of phosphorylated (P) or total (T) ERK1/2 and/or S6 in vector control A375 cells treated with vehicle, TTM (25, 50, or 100 nM) vemurafenib; or, vemurafenib (Vem, 3.125, 6.25, or 12.5 nM), or trametinib (Tra, 75, 150, or 300 pM) or A375 cells stably expressing HA-tagged p61BRAFV600E, or HA-tagged PDGFRβ treated with vehicle, TTM (100 nM), vemurafenib (Vem, 12.5 nM), or trametinib (Tra, 300pM) at their IC50 concentrations. ßACTIN: loading control. Bottom: Fold change in phosphorylated to ßACTIN. One of three replicates (see Supplementary Fig. S6, and S7 for other replicates). (C) % transformed growth expressed as the normalized % soft agar colony formation (mean ± s.e.m, triplicate samples, three experiments) of A375 cells stably expressing HA-tagged p61BRAFV600E, COT, CRAF, NRASQ61L, IKKβ, IGF-1R, or PDGFRβ when treated with vehicle (black bar) or the IC50 doses of TTM (red bar), vemurafenib (blue bar), or trametinib (yellow bar). *p<0.01 and **p<0.001 versus vector control cells treated with the same drug. Statistical analysis was performed with two-way Analysis of Variance (ANOVA) with a Bonferroni multiple comparisons post test to compare transformed growth of each cell line with vector control cells treated with the same drug.
TTM counters resistance to MAPKi imparted by MEK1/2 mutants
Activating mutations in MEK1 and MEK2 can also confer resistance to MAPKi and have been detected in tumors of patients developing resistance to BRAFi or the combination of BRAFi and MEKi (9,22,39–47). Given this, A375 cells were engineered to express MEK1 (Fig. 2A and Supplementary Fig. 1) or MEK2 (Fig. 2B and Supplementary Fig. 1) with these and other (48,49) mutations, as assessed by immunoblot, and then treated as above. Most, but not all these mutants imparted some level of resistance to vemurafenib, consistent with the finding that activation of MEK1/2 downstream of oncogenic BRAF confers resistance to BRAFi (9,22,39–47). A subset of MEK1 and MEK2 mutants conferred some level of resistance to trametinib, while none of the mutants imparted resistance to TTM (Fig. 2C and 2D). Thus, TTM counters resistance to MAPKi mediated by MEK1/2 mutants in A375 cells, including mutants that conferred resistance to trametinib.
Figure 2. TTM counters MAPKi resistance imparted by MEK1/2 mutants.
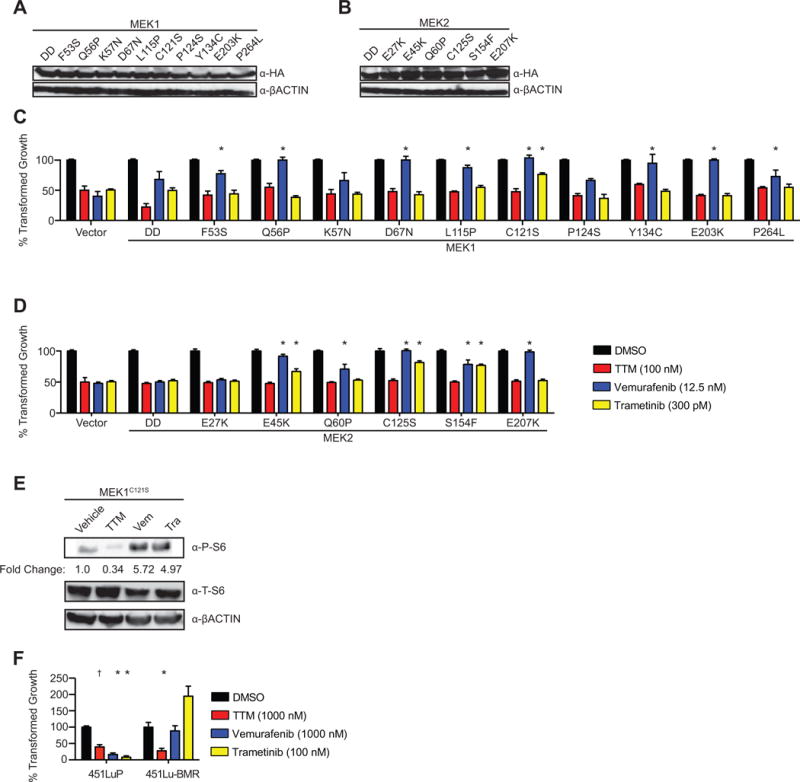
(A, B) Immunoblot detection of ectopic HA-tagged (A) MEK1 mutants DD, F53S, Q56P, K57N, L115P, C121S, P124S, Y134C, E203K, or P264L and (B) MEK2 mutants DD, E27K, E45K, Q60P, C125S, S154F, or E207K stably expressed in A375 cells. ßACTIN: loading control. (C) Immunoblot detection of phosphorylated (P) or total (T) S6 in A375 cells expressing HA-tagged MEK1C121S treated with vehicle, TTM (100 nM), vemurafenib (Vem, 12.5 nM), or trametinib (Tra, 300pM). ßACTIN: loading control. Bottom: Fold change in phosphorylated to ßACTIN. One of three replicates (see Supplementary Fig. S8 for other replicates). (D, E, F) % transformed growth expressed as the normalized % soft agar colony formation (mean ± s.e.m, triplicate samples, three experiments) of (D) A375 cells stably expressing MEK1 mutants DD, F53S, Q56P, K57N, L115P, C121S, P124S, Y134C, E203K or P264L and (E) MEK2 mutants DD, E27K, E45K, Q60P, C125S, S154F, or E207K, when treated with vehicle (black bar) or the IC50 doses of TTM (red bar), vemurafenib (blue bar), or trametinib (yellow bar) or (F) 451Lu-P cells or 451Lu-BMR cells when treated with vehicle (black bar) or TTM (red bar), vemurafenib (blue bar), or trametinib (yellow bar). †p<0.05 and *p<0.01* versus vector control cells treated with the same drug. Statistical analysis was performed with two-way Analysis of Variance (ANOVA) with a Bonferroni multiple comparisons post test to compare transformed growth of each cell line with vector control cells treated with the same drug.
TTM counters MAPKi resistance by suppressing MAPK signaling
To elucidate the mechanism by which TTM suppresses the resistance to MAPKi, the phosphorylation status of S6 was analyzed by immunoblot in A375 cells engineered to be resistant to vemurafenib, vemurafenib and trametinib, or vemurafenib and trametinib and TTM vis-à-vis expression of p61BRAFV600E, MEK1C121S, or PDGFRß, respectively. Consistent with the effects of these drugs on the transformed growth of these cells (Fig. 1C, 2C, and 2D), all three drugs inhibited MAPK signaling, as assessed by immunoblot detection of P-S6 levels, in cells expressing the MAPK-independent resistance driver PDGFRß (Fig. 1B and Supplementary Fig. 3). Further, vemurafenib failed to suppress MAPK signaling in cells expressing p61BRAFV600E or MEK1C121S (Fig. 1B, 2E, Supplementary Fig. 3, and Supplementary Fig. 4). Trametinib inhibited MAPK signaling in cells expressing p61BRAFV600E, but not MEK1C121S (Fig. 1B, 2E, Supplementary Fig. 3, and Supplementary Fig. 4). Finally, TTM reduced the level of P- S6 in these latter two cell lines (Fig. 1B, 2E, Supplementary Fig. 3, and Supplementary Fig. 4). These findings, coupled with the ability of TTM to inhibit MEK1/2 kinase activity (15,16), support the notion that TTM suppresses MAPK-driven resistance to MAPKi by inhibiting the kinase activity of MEK1/2.
TTM counters MAPKi resistance in other BRAF-mutant cell lines
To independently evaluate whether TTM counters resistance to BRAFi and MEKi in other BRAF-mutant cell lines, we note that in addition to A375 cells, knockdown of Ctr1 or treatment with TTM was previously shown to inhibit the transformed growth of other BRAF-mutant human melanoma cell lines such as DM440 and DM443 (16). Thus, these two cell lines were engineered to express no transgene, p61BRAFV600E, MEK1C121S, or PDGFRß, as assessed by RT-PCR or immunoblot (Supplementary Fig. S5A and S5B). These cells were then assayed for growth in soft agar when treated with vehicle, vemurafenib, trametinib, or TTM. As observed with A375 cells, DM440 and DM443 cells expressing p61BRAFV600E were only resistant to vemurafenib, those expressing MEK1C121S were resistant to vemurafenib and in the case of DM443, somewhat resistant to trametinib. Finally, both cell lines expressing PDGFRß were resistant to all three drugs (Supplementary Fig. S5C and S5D). Thus, in a total of three different BRAF-mutant human melanoma cell lines, TTM countered resistance to BRAFi and MEKi.
TTM counters MAPKi resistance driven by prolonged drug treatment
TTM was next assayed for its ability to counter MAPKi resistance in cells with acquired, rather than genetically engineered resistance. Specifically, chronic treatment of the BRAFV600E-mutant melanoma cell line 451Lu (451Lu-P) with increasing concentrations of trametinib was previously shown to give rise to the derivate line 451Lu-BMR, which exhibited a 10- to 100-fold increase in IC50 for both vemurafenib and trametinib (22). Thus, growth in soft agar of both 451Lu-P and 451Lu-BMR cells was measured in the presence of vehicle, vemurafenib, trametinib, or TTM. As expected, vemurafenib and trametinib both reduced the anchorage-independent growth of 451Lu, but not 451Lu-BMR cells. On the other hand, TTM reduced the growth of both cell lines (Fig. 2F). Thus, TTM can also reduce the growth of BRAFV600E-mutant melanoma cells in the context of acquired MAPKi resistance.
TTM enhances the antineoplastic effects of MAPKi
To evaluate the effect of combining TTM with current MAPKi, anchorage independent growth of A375 cells was measured in the presence of either dual or triple combinations of vemurafenib, trametinib, and/or TTM at three different drug concentrations in fixed-ratio combinations. The combination of vemurafenib with trametinib was additive, while combining TTM with either vemurafenib or trametinib was synergistic by both Combination (30) and Bliss (31) Indexes. Combining all three drugs was the most synergistic (Fig. 3A and 3B). To evaluate whether TTM could be combined with current MAPKi to counter resistance to BRAFi or MEKi, growth in soft agar of the aforementioned A375 cells expressing p61BRAFV600E, MEK1C121S, or PDGFRß (Fig. 1A, 2A, and Supplementary Fig. 1) was measured in the presence of either dual or triple combinations of vemurafenib, trametinib, and/or TTM. Addition of TTM to vemurafenib or the combination of vemurafenib and trametinib reduced transformed growth in cells engineered to be resistant to vemurafenib by expression of p61BRAFV600E (Supplementary Fig. S6A). Dual combinations of TTM and vemurafenib, TTM and trametinib, or the combination of all three drugs reduced transformed growth in cells engineered to be resistant to both vemurafenib and trametinib by expression of MEK1C121S (Supplementary Fig. S6B). As expected, cells expressing PDGFRβ were resistant to single, double, or triple combinations of these drugs (Supplementary Fig. S6C). As such, TTM enhanced the antineoplastic activity of BRAFi and MEKi in A375 cells, including in settings of resistance to these inhibitors.
Figure 3. TTM enhances the antineoplastic effects of MAPKi.
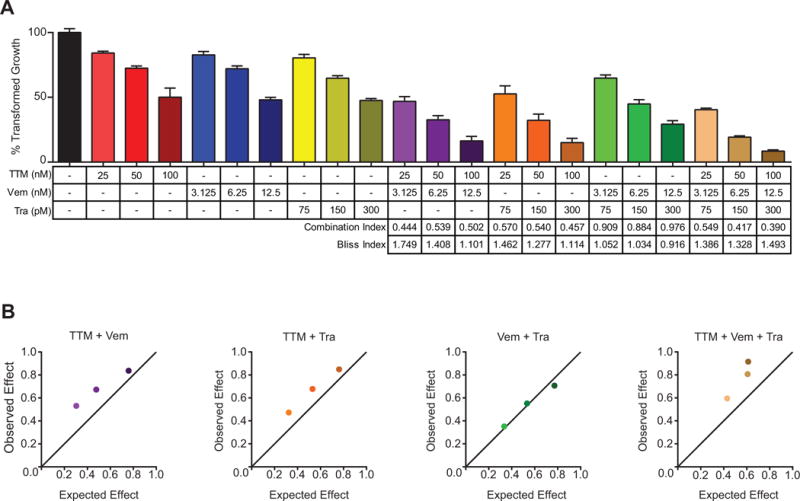
(A) % transformed growth expressed as the normalized % soft agar colony formation (mean ± s.e.m, triplicate samples, three experiments) of A375 cells treated with vehicle (black bar), TTM (red bars), vemurafenib (Vem, blue bars), trametinib (Tra, yellow bars), TTM and vemurafenib (TTM+Vem, purple bars), TTM and trametinib (TTM+Tra, orange bars), vemurafenib and trametinib (Vem+Tra, green bars), or TTM, vemurafenib, and trametinib (TTM+Vem+Tra, brown bars) at the indicated doses alone or in fixed-ratio combinations. Synergistic combinations are indicated by the Combination Index values < 1 and Bliss Index values > 1. (B) Graphical representation of the above Bliss Index for the indicated combinations.
TTM can lead to toxicity preferentially in mice with low weight
To evaluate the therapeutic potential of TTM in vivo, we turned to the genetically engineered BrafCA/+;Ptenfl/fl;Tyr∷CreER-T2tg/+ (BPC) mouse model of melanoma in which topical application of 4-hydroxytamoxifen (4-HT) activates CreER in melanocytes to promote recombination and activation of the BrafCA allele and inactivation of the Ptenfl alleles, thereby initiating melanomagenesis (26). To model human clinical trials, mice were randomly assigned to a treatment group once disease was established, namely lesions detected at the site of induction. Treatments consisted of daily oral dosing with vehicle, 50 mg/kg vemurafenib, a fixed dose of 2 mg of TTM that in adult mice had previously been demonstrated to reduce tumor growth (16), or 2 mg of TTM with 50 mg/kg vemurafenib. Mice were treated until reaching an endpoint, namely maximum tumor volume, moribundity, or excessive weight loss.
Offsite tumors eventually develop in this strain, ostensibly due to leaky Cre expression (29). This necessitated inducing tumorigenesis and beginning treatments in young (6–8 weeks old) mice weighing as little as half the weight of adults previously used to study the antineoplastic effects of TTM. Another complication of this study design was dosing of Cu chelators in Wilson disease or cancer patients is always benchmarked to serum ceruloplasmin (Cp) activity to avoid overdosing, as there can be variability in the response to TTM from patient to patient or even in the same patient over time (50). However, weekly blood collection over the course of a mouse lifetime, beginning as early as 6 weeks of age, is not only impractical, but a risk for an adverse event. These two limitations, very young mice given the fixed dose of TTM used in adult mice and an inability to monitor Cu levels, appeared to result in overdosing of TTM in mice of low weight, resulting in the adverse event of excessive weight loss (reduction of 15% maximum weight). This effect that was even more pronounced when TTM was combined with vemurafenib. Without censoring for these adverse events, the combination of TTM with vemurafenib proved to be worse than vehicle (Supplementary Fig. S7). Consistent with an interpretation of TTM overdosing due to the lower weight of young mice, animals in the TTM or TTM and vemurafenib cohorts that succumbed to adverse events were found to weigh significantly less at the time therapy was initiated (Fig. 4A) or have a lower maximal weight over the course of the experiment (Fig. 4B). These findings highlight the importance of monitoring serum Cp activity when evaluating this drug in human cancer patients, although other interpretations are possible.
Figure 4. TTM can lead to toxicity preferentially in mice with low weight.
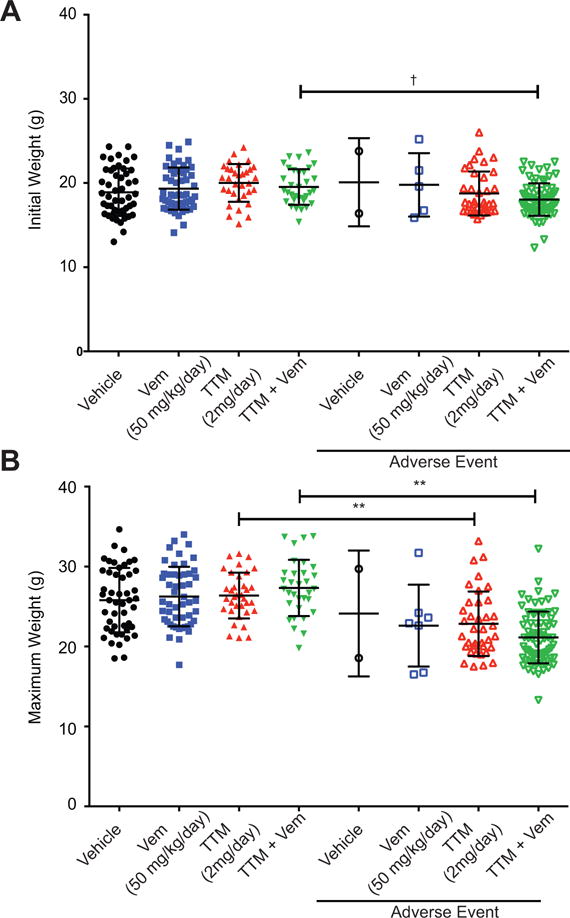
(A, B) Scatter dot plots (mean ± s.e.m) of (A) initial weight at start of treatment or (B) maximum weight obtained during treatment with indicated doses of vehicle (n=50), TTM (n≥71), vemurafenib (Vem, n≥57), or TTM and vemurafenib (TTM+Vem, n≥106) that either did (right) or did not (left) reach and adverse event endpoint. †p<0.05, *p<0.01, and **p<0.001. Statistical analysis was performed with one-way Analysis of Variance (ANOVA) with a Sidak’s multiple comparisons post test to compare each Adverse Event group with each non-Adverse Event group.
TTM extends survival in a genetically engineered mouse model of melanoma
Given the aforementioned association between animal body weight and adverse events, the above preclinical trial was censored to only include mice that tolerated TTM, namely those that either reached tumor volume endpoint or lived at least to the median survival for vehicle-treated mice (44 days) to account for these early adverse events. This analysis revealed that vemurafenib or TTM provided a similar survival advantage over vehicle-treated mice, while the combination of these two drugs trended towards the greatest survival benefit (Fig. 5A). This survival benefit was associated with a reduction in tumor growth (Supplementary Fig. S8A). To experimentally evaluate whether TTM truly has antineoplastic activity when dosing is adjusted to weight, we repeated the preclinical trial by treating mice with either vehicle or TTM at a dose of 80 mg/kg, and in mice in 2 weeks older, when there was no difference between the initial weigh of the mice in either cohort (Fig. 5B). This time, mice treated with TTM did not develop the adverse event of weight loss; mice from both cohorts reached endpoint at the same weight (Fig. 5C). Moreover, while vehicle-treated BPC mice had a median survival of 42.5 days, TTM-treated mice exhibited a statistically significant survival benefit with a median survival of 56 days (Fig. 5D), the same advantage as observed in the censored dataset (Fig. 5A). Thus, with the very important proviso revealed by this study regarding the critical need to benchmark TTM dosing to either serum Cp activity or mouse body weight, this drug has potential to be combined with MAPKi for the treatment of BRAF mutation-positive melanoma.
Figure 5. TTM extends survival in a genetically engineered mouse model of melanoma.
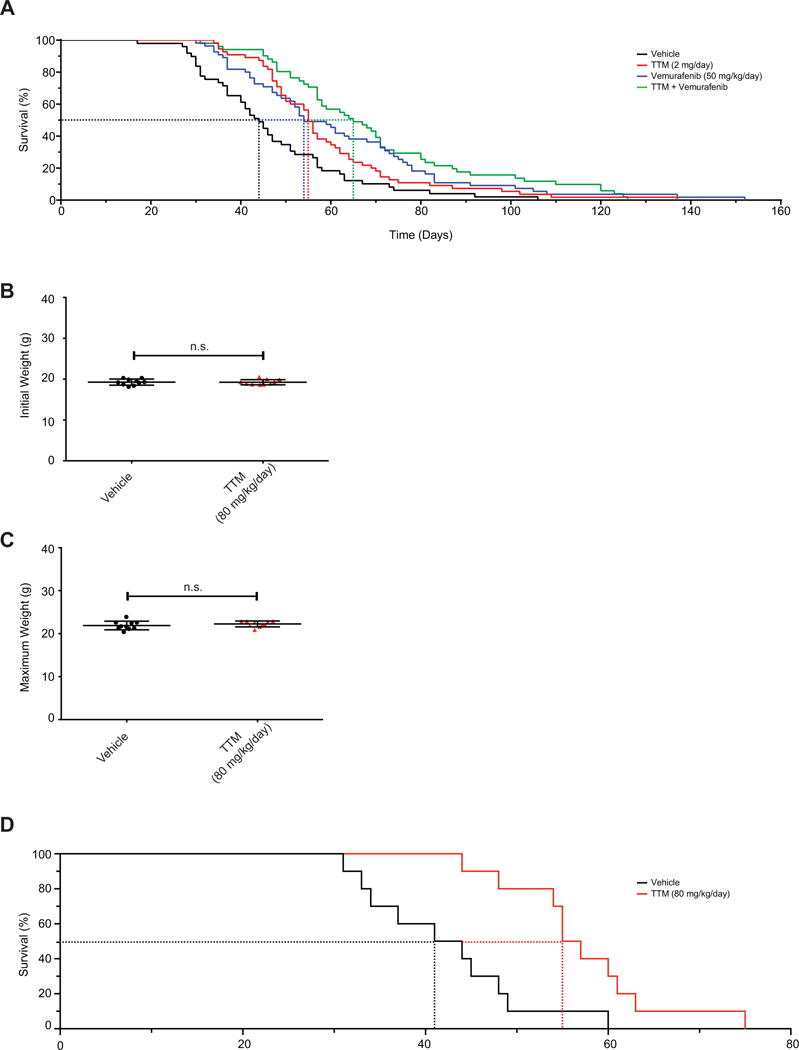
(A) Kaplan-Meier analysis of BPC mice that either reached tumor volume endpoint or lived at least to the median survival for vehicle-treated mice when orally treated daily with vehicle (solid black line, n=49, dotted line: 44 days median survival), TTM (solid red line, n=55, dotted line: 55 days median survival, p=0.004 compared to vehicle), vemurafenib (solid blue line, n=55, dotted line: 54 days median survival, p=0.001 compared to vehicle), or TTM and vemurafenib (solid purple line, n=51, dotted line: 65 days median survival, p=0.0001 compared to vehicle) from the appearance of a pigmented lesion. Statistical analysis was performed with a Mantel-Cox log-rank method to compare each group to vehicle-treated control mice in a pairwise manner. (B, C) Scatter dot plots (mean ± s.e.m) of (B) initial weight at start of treatment or (C) maximum weight obtained during treatment with indicated doses of vehicle (n=10) or TTM (n=10) that either did not reach adverse event endpoint. Statistical analysis was performed with a Student’s t-test. (D) Kaplan-Meier analysis of BPC mice that reached tumor volume endpoint when orally treated daily with vehicle (solid black line, n=10, dotted line: 42.5 days median survival) or TTM (solid red line, n=10, dotted line: 56 days median survival, p=0.0028 compared to vehicle) from the appearance of a pigmented lesion. Statistical analysis was performed with a Mantel-Cox log-rank method to compare each group to vehicle-treated control mice in a pairwise manner.
The antineoplastic effects of TTM in vivo are linked to reduced Cu levels and suppression of MAPK signaling
To explore the mechanism underlying the antineoplastic activity of TTM observed in vivo, we evaluated whether this drug was absorbed, reduced Cu levels, and suppressed the MAPK pathway. Inductively coupled plasma-mass spectrometry (ICP-MS) analysis of serum samples isolated at end point from mice treated with TTM or TTM and vemurafenib (51) confirmed the presence of molybdenum (Mo), the central atom in TTM, indicating that the drug was absorbed (Fig. 6A). Analysis of serum Cp activity at endpoint confirmed a reduction of Cu in mice receiving TTM alone or in combination with vemurafenib (Fig. 6B). Immunohistochemical analysis revealed no obvious reduction in P-ERK1/2 and P-S6 staining in tumors from mice treated with vemurafenib, but a trend towards reduced staining in mice treated with TTM. Treatment with the combination of TTM and vemurafenib led to a significant decrease in P-ERK1/2 and P-S6 staining, indicating synergistic MAPK inhibition from dual Cu chelator and BRAFi treatment in vivo (Fig. 6C and 6D). These data suggest that the survival benefit afforded by TTM and its ability to enhance the antineoplastic activity of vemurafenib is due to a reduction of Cu and MAPK signaling in tumors.
Figure 6. TTM treatment reduces serum Cp activity and tumor P-ERK1/2 and P-S6 immunostaining.
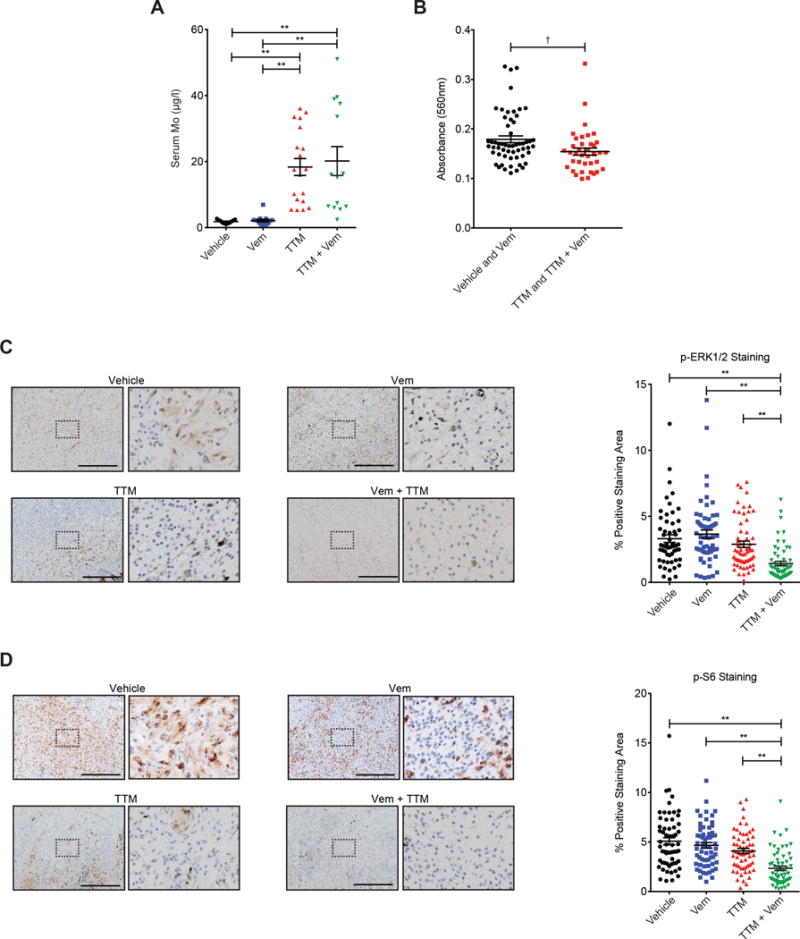
(A, B) Scatter dot plots (mean ± s.e.m) of (A) serum Mo levels (μg/ml, n≥11 mice from each cohort) and (B) serum Cp activity (absorbance at 560 nm, n≥38 in each group) at endpoint from randomly selected mice of the indicated treatment groups from Fig. 5. (C, D) Immunohistochemical detection of P-ERK1/2 and P-S6 in (C) a representative image (scale bar=500 μm) and (D) a scatter dot plot (mean ± s.e.m) of % positive-staining area per field of P-ERK1/2 (n≥11 mice from each cohort, 5 fields per mouse) or P-S6 immunoreactivity (n=12 mice from each cohort, 5 fields per mouse) in tumors at endpoint from randomly selected mice of the indicated treatment cohorts from Fig. 5. †p<0.05 and **p<0.001. Statistical analysis was performed with one-way Analysis of Variance (ANOVA) with (A, C, D) a Tukey’s multiple comparisons post test to compare each group to each other, or (B) a Student’s t-test.
Loss of Ctr1 inhibits MAPK signaling and extends survival in a genetically engineered mouse model of melanoma
To genetically test the hypothesis that the mechanism of action of TTM in vivo is a reduction of Cu levels in tumors, thereby suppressing MAPK signaling and extending survival, a Ctr1fl/fl genotype encoding conditional null alleles of the primary specific transporter of Cu (17) was crossed into the BPC background. Recombination of the Ctr1fl alleles specifically in the tumors (Supplementary Fig. S8B) was associated with a reduction in P-ERK1/2 (Fig. 7A) and P-S6 levels (Fig. 7B). Inactivation of the Ctr1 gene did not affect tumor initiation or alter the number of visible lymph node lesions (Supplementary Fig. S8C). Instead, inactivation of the Ctr1 gene in tumors resulted in a reduction in tumor growth (Supplementary Fig. S8D) that was associated with a survival benefit (Fig. 7C), as observed above in mice treated with TTM (Fig. 5A and 5B). Admittedly, loss of Ctr1 does not completely block Cu import (52) and in agreement, tumors from Ctr1fl/fl BPC mice still exhibit some immunoreactivity to P-ERK1/2 and P-S6 antibodies (Fig. 7A and B). Accordingly, oral treatment with 80 mg/kg/day of TTM resulted in the progressive reduction of tumors in mice injected with either BRAFV600E transformed Ctr1+/+ or Ctr1−/− MEFs (Supplementary Fig. S8E), although this may also suggest involvement of the tumor microenvironment. Collectively however, the signaling and genetic analysis supports a conclusion that the antineoplastic activity of TTM observed in vivo is due in large part to a reduction in Cu levels and suppression of MAPK signaling in the tumor.
Figure 7. Loss of Ctr1 extends survival in a genetically engineered mouse model of melanoma.
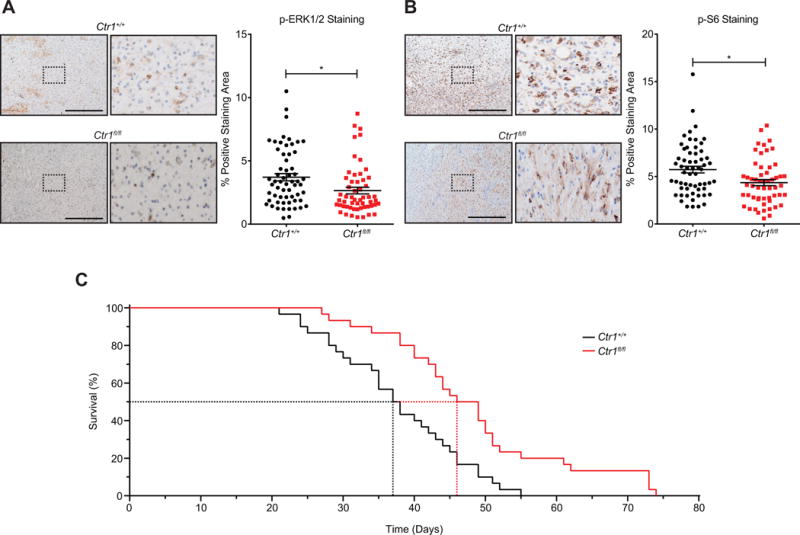
(A) Representative images (scale bar=500 μm) and (B) scatter dot plots (mean ± s.e.m) of % positive-staining area per field for immunohistochemical detection of P-ERK1/2 (n≥11 mice from each cohort, 5 fields per mouse) or P-S6 (n≥11 mice from each cohort, 5 fields per mouse) in tumors at endpoint from randomly selected Ctr1+/+ versus Ctr1fl/fl BPC mice. *p<0.01. Statistical analysis was performed with a Student’s t-test. (C) Kaplan-Meier analysis of Ctr1+/+ (solid black line, n=30, dotted line: 37.5 days median survival) or Ctr1fl/fl (solid red line, n=30, dotted line: 47.5 days median survival, p=0.001 compared to Ctr1+/+) BPC mice. Statistical analysis was performed with a Mantel-Cox log-rank method to compare Ctr1+/+ to Ctr1fl/fl mice.
Summary
We demonstrate that TTM inhibits transformed growth of melanoma cells encoding MAPK-dependent drivers of resistant to BRAFi and/or MEKi, suggesting there is value in repurposing TTM to combat resistance to current MAPKi. TTM also enhanced the ability of MAPKi to inhibit transformed growth of melanoma cells; further suggesting TTM may improve the durability of response to such drugs. In vivo, apparent overdosing of TTM led to toxicities in the genetically engineered BPC mouse model of melanoma, presumably due to an excessive reduction in Cu levels, although other explanations are possible. In this regard, TTM dosing is benchmarked in the clinic to serum Cp activity to avoid excessively low Cu levels. Indeed, with regular monitoring of serum Cp activity, TTM has been chronically dosed for upwards of 65 months in cancer patients (20). Encouragingly, by accounting for this potential overdosing, TTM alone or in combination with vemurafenib led to a survival benefit, perhaps the most important parameter to improve upon, in BPC mice. In fact, benchmarking TTM dosing to weight led to a survival benefit in the absence of weight loss in this mouse model. Mechanistically, the antineoplastic activity of TTM appears to be through inhibiting MEK1/2 in tumors, as this drug or inactivation of the Ctr1 gene led to a reduction in P-ERK1/2 and P-S6 staining in tumors and a survival advantage. Admittedly, neither TTM nor Ctr1 loss were curative, consistent with the clinical findings that MAPKi are oncostatic in melanoma patients (10). Nevertheless, the findings that TTM is well tolerated in cancer patients, counters MAPK-dependent forms of resistance, is additive or synergistic when combined with other MAPKi, and leads to a survival advantage in a genetically engineered mouse model of melanoma support evaluating TTM for the treatment of BRAF mutation-positive melanoma and potentially other cancers.
Supplementary Material
Acknowledgments
We thank David Kirsch and Dennis Thiele for mice strains, Kris Wood for plasmids, Jessie Villanueva for cell lines, and members of the Counter laboratory for helpful discussions.
Financial Support: This work was supported by the NCI (R01CA177587 to C.M. Counter and K01CA178145 to D.C. Brady) and the Lymphoma Foundation (to C.M. Counter).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Atkins MB, Larkin J. Immunotherapy Combined or Sequenced With Targeted Therapy in the Treatment of Solid Tumors: Current Perspectives. J Natl Cancer Inst. 2016;108:djv414. doi: 10.1093/jnci/djv414. [DOI] [PubMed] [Google Scholar]
- 3.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–67. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solit DB, Rosen N. Towards a unified model of RAF inhibitor resistance. Cancer Discov. 2014;4:27–30. doi: 10.1158/2159-8290.CD-13-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 10.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 11.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 13.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–92. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 15.Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32:1284–95. doi: 10.1128/MCB.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–6. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4:235–44. doi: 10.1016/j.cmet.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Brewer GJ, Johnson V, Dick RD, Kluin KJ, Fink JK, Brunberg JA. Treatment of Wilson disease with ammonium tetrathiomolybdate. II. Initial therapy in 33 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 1996;53:1017–25. doi: 10.1001/archneur.1996.00550100103019. [DOI] [PubMed] [Google Scholar]
- 19.Merle U, Eisenbach C, Weiss KH, Tuma S, Stremmel W. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J Hepatol. 2009;51:925–30. doi: 10.1016/j.jhep.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 20.Jain S, Cohen J, Ward MM, Kornhauser N, Chuang E, Cigler T, et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann Oncol. 2013;24:1491–8. doi: 10.1093/annonc/mds654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Augustine CK, Toshimitsu H, Jung SH, Zipfel PA, Yoo JS, Yoshimoto Y, et al. Sorafenib, a multikinase inhibitor, enhances the response of melanoma to regional chemotherapy. Mol Cancer Ther. 2010;9:2090–101. doi: 10.1158/1535-7163.MCT-10-0073. [DOI] [PubMed] [Google Scholar]
- 22.Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen H-Y, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–9. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Hayer KM, Counter CM. A genetically defined normal human somatic cell system to study ras oncogenesis in vivo and in vitro. Methods Enzymol. 2006;407:637–47. doi: 10.1016/S0076-6879(05)07050-3. [DOI] [PubMed] [Google Scholar]
- 24.Lim K-H, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–9. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosenberg M, Muthusamy V, Curley DP, Wang Z, Hobbs C, Nelson B, et al. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis. 2006;44:262–7. doi: 10.1002/dvg.20205. [DOI] [PubMed] [Google Scholar]
- 29.Hooijkaas AI, Gadiot J, van der Valk M, Mooi WJ, Blank CU. Targeting BRAFV600E in an inducible murine model of melanoma. Am J Pathol. 2012;181:785–94. doi: 10.1016/j.ajpath.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 31.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3 doi: 10.1002/prp2.149. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rygaard K, Sprang-Thomsen M. Quantitation and gompertzian analysis of tumor growth. Breast Cancer Res Treat. 1997;46:303–12. doi: 10.1023/a:1005906900231. [DOI] [PubMed] [Google Scholar]
- 33.Wood KC, Konieczkowski DJ, Johannessen CM, Boehm JS, Tamayo P, Botvinnik OB, et al. MicroSCALE screening reveals genetic modifiers of therapeutic response in melanoma. Sci Signal. 2012;5:rs4. doi: 10.1126/scisignal.2002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–74. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 42.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–9. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- 46.Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–20. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 47.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang W, Kessler DS, Erikson RL. Biochemical and biological analysis of Mek1 phosphorylation site mutants. Mol Biol Cell. 1995;6:237–45. doi: 10.1091/mbc.6.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang W, Erikson RL. Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc Natl Acad Sci U S A. 1994;91:8960–3. doi: 10.1073/pnas.91.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521–7. doi: 10.1001/archneur.63.4.521. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki KT, Ogra Y, Ohmichi M. Molybdenum and copper kinetics after tetrathiomolybdate injection in LEC rats: specific role of serum albumin. J Trace Elem Med Biol. 1995;9:170–5. doi: 10.1016/S0946-672X(11)80043-X. [DOI] [PubMed] [Google Scholar]
- 52.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J Biol Chem. 2002;277:40253–9. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.