

Abstract
Thoracic NETs [bronchopulmonary NETs (BPNETs) and thymic NETs (TNET)] share a common anatomic primary location, likely a common cell of origin, the “Kulchitsky cell” and presumably, a common etiopathogenesis. Although they are similarly grouped into well-differentiated [typical carcinoids (TC) and atypical carcinoids (AC)] and poorly differentiated neoplasms and both express somatostatin receptors, they exhibit a wide variation in clinical behavior. TNETs are more aggressive, are frequently metastatic, and have a lower 5-year survival rate (~50% vs. ~80%) than BPNETs. They are typically symptomatic, most often secreting ACTH (40% of tumors) but both tumor groups share secretion of common biomarkers including chromogranin A and 5-HIAA. Consistently effective and accurate circulating biomarkers are, however, currently unavailable. Surgery is the primary therapeutic tool for both BPNET and TNETs but there remains little consensus about later interventions e.g., targeted therapy, or how these can be monitored. Genetic analyses have identified different topographies (e.g., significant alterations in chromatin and epigenetic remodeling in BPNETs versus frequent chromosomal abnormalities in TNETs) but there is an absence of clinically actionable mutations in both tumor groups. Liquid biopsies, tools that can measure neoplastic signatures in peripheral blood, can potentially be leveraged to detect disease early i.e., recurrence, predict tumors that may respond to specific therapies and serve as real-time monitors for treatment responses. Recent studies have identified that mRNA transcript analysis in blood effectively identifies both BPNET and TNETs. The clinical utility of this gene expression assay includes use as a diagnostic, confirmation of completeness of surgical resection and use as a molecular management tool to monitor efficacy of PRRT and other therapeutic strategies.
Keywords: Biomarker, bronchopulmonary, carcinoid, NETest, neuroendocrine
Introduction
Thoracic NETs [bronchopulmonary and thymic NETs (TNET)] share a common anatomic primary location but exhibit substantially different spectra of clinical behavior. Although initially considered to share similar embryological origins (foregut), there is a limited understanding of their specific neuroendocrine cells of origin which range from neuroendocrine “K” or Kulchitsky cells to Clara cells. Overall, the conglomeration of such lesions as thoracic NETs is an archaic viewpoint based upon anatomical contiguities rather than cellular and molecular delineation. The different tumors grouped under this colliquation comprise well-differentiated NETs (typical and atypical “carcinoids” and poorly differentiated tumors (small cell and large cell neuroendocrine cancers). Other neuroendocrine lesions that occur in the thoracic cavity include paragangliomas and pheochromocytomas of the sympathetic chain as well neuroendocrine metastases from the gastroenteropancreatic system.
Overall, thymic and bronchopulmonary NETs (BPNETs) vary widely in their symptomatology, histological characteristics, clinical behavior, natural histories, and etiopathology. It is logical, at this time, to recognize them as very different entities linked only by their common thoracic cavity location rather than group them by anatomic location.
At a genetic level, although these tumors exhibit an overall low mutation rate, BPNETs are consistently associated with alterations at the multiple endocrine neoplasia (MEN)-1 locus (including loss of expression and mutations). Conversely, TNETs exhibit a retention of genetic material at this site. In addition, although 5–10% of MEN-1 patients develop BPNET or TNETs, these neoplasms demonstrate somewhat different etiopathological pathways (BP: women/non-smoking compared to men/smoking in TNETs). Although TNETs frequently recapitulate histopathological similarities with BPNETs, the former typically follow a more aggressive behavior than their BPNET counterparts. These may, to some extent, reflect the later stage at which TNETs are diagnosed. Irrespective, both tumor groups express somatostatin receptors and share secretion of common biomarkers including chromogranin A and 5-HIAA. While somatostatin receptor imaging (SRI) may have a role in diagnosis, staging and evaluating targeted therapy, circulating biomarkers are non-specific and do not significantly aid clinical decision-making. While surgery is the primary therapeutic tool for both BPNET and TNETs, there is little consensus about later interventions and how these can be monitored. As such, the recent focus on liquid biopsies, tools that can measure neoplastic signatures in peripheral blood, has led to an interest in these instruments in this neoplasia. The rationale is that such approaches may detect disease early i.e., recurrence, and serve as a real-time monitor for treatment responses. This manuscript evaluates the natural history, pathobiology, and genetic abnormalities underpinning both BPNET and TNETs. Given the absence of actionable mutations in these tumors, the principal focus is the role of one such signature, a circulating mRNA tool, the NETest. The clinical utility of this test is evaluated in the context of BPNET and TNET treatment paradigms.
Evolution of taxonomy
BPNETs
The initial descriptions of BPNETs date from the 19th century and substantially predate that of TNETs which were first described in 1972 by Rosai and Higa (1) (Figure 1). Almost a century prior to this Muller, in 1882, had described a bronchial adenoma at autopsy, followed by Heine [1927] and Reiser [1928]. In 1930, Kramer presciently noted that such “growths” may have malignant potential and shortly thereafter, Geipel [1931] described the histological basis of bronchial Basalzellkrebs (2) (“bronchial adenomata”). In 1937, Hamperl determined that bronchial adenoma comprised two pathological variants, a cylindroid group and “carcinoids”, comprising neuroendocrine or “Kulchitsky” cells. Thereafter, pathologists began to consider pulmonary tumors to reflect neuroendocrine cell-derived (now known as “typical” carcinoids) neoplasia as well as lesions where neuroendocrine cells were a component of the tumor e.g., oat cell carcinomas (current terminology, “small cell lung cancer”). In 1968, Bensch et al. recognized that oat cell carcinoma and carcinoid tumors were histogenetically related (3). Four years thereafter, in 1972, Arrigoni and colleagues from the Mayo Clinic noted that although “carcinoids” generally have a good prognosis, some metastasized and progressed to systemic spread with eventual death (4). This led to the description of so called “atypical” carcinoids. Ten years later, Mills and coworkers described a group of atypical carcinoids (AC), which more closely resembled small-cell lung cancer (5). DeLellis et al. in 1984 (6) noted that bronchopulmonary carcinoids closely resembled the “benign” carcinoid lesions of the small intestine initially described by Oberndorfer in 1907 (7). These observations have formed the basis for the concept of a spectrum of bronchopulmonary lesions. There is now a common acceptance that typical (well-differentiated) carcinoid are represent one end of the neoplastic spectrum and the typical small-cell (oat-cell) carcinoma (small cell lung cancer) the other end (Figure 2).
Figure 1.

The cellular origin (neuroendocrine “Kulchitsky” cells) of bronchopulmonary NETs was first defined by H. Hamperl (top left) in 1937. He classified the tumors as “carcinoids”, which were initially titled “typical” (histology: center top). P. Bernatz (top right) subsequently identified that some tumors exhibited malignant features and termed these “atypical” in contradistinction to the more “benign” “typical” carcinoid. The former are typically more malignant and often metastatic [background: 68Ga-PET CT identifying a lymph node metastasis (orange) from a BPNET]. Bernatz also described the four stage classification system for thymic tumors (pathological specimen: center bottom), including NETs [1961]. J. Rosai (bottom right) and Higa both identified that some thymic tumors exhibited neuroendocrine features and could be classified as NETs [1972]. They were the first to identify a relationship with MEN-1. A. Masaoka (bottom left) devised a staging system for thymic tumors [1981] which remains the current basis for assessing prognosis.
Figure 2.

Timeline of the individuals responsible for identification of the cells, tumor types and different staging/classification systems for bronchopulmonary and thymic neuroendocrine neoplasia. Red, significant events for neuroendocrine tumor disease; yellow, significant events for BPNETs; blue, significant events for thymic NETs.
TNET
TNETs were recognized in 1972 by Rosai and Higa (1) who considered them to be “a mediastinal endocrine neoplasm, of probably thymic origin, related to carcinoid tumor”. They subsequently adopted the term a “thymic carcinoid” to differentiate them from thymomas. The latter had been initially histologically classified in the 1960s, as either non-invasive or invasive (8), before subsequent reconsideration as four histologic subtypes by Bernatz et al. (9). Rosai and Higa’s observations allowed for a more specific appreciation of thymic neoplasia and was the first to acknowledge carcinoid tumors of the thymus and histologically separate them from thymomas. They postulated that earlier reports of thymomas associated with endocrine symptomatology most likely represented unrecognized primary carcinoid tumors of the thymus (10). In addition, they identified a putative relationship to MEN-1 (11). Overall, the tumors are uncommon and since 1972, only ~400 have been described (12).
Previously, most thymic carcinoid investigation has been focused on developing staging systems to better characterize tumor growth. Bergh proposed the first and used a three-tier system which classified thymomas based upon the presence of symptoms, tumor extent and histology (13). Wilkins and Castleman in 1979 updated this by including mediastinal pleura or pericardial invasion in stage II (14). They also identified tumor invasiveness as a negative prognostic feature. Masaoka et al. in 1981 reiterated this observation demonstrating that survival was directly linked to invasion and introduced a four stage system that could be used to stratify therapy (15). The principal difference to the Wilkins classification was in regard to stage II. The Masaoka system was revised by Koga in 1994, whereby stage IIb was included in a new stage I (16). Such an approach has recently been clinically validated [i.e., an invasive (stage III/IV) versus non-invasive (stage I/II) classification system] (17). In 2011, the International Thymic Malignancy Interest Group (ITMIG) adopted the Masaoka-Koga system (18). Irrespective of history or the limitations of each of these studies (all undertaken in mediastinal tumors as a group), TNETs are more often thought of as being related to bronchopulmonary neuroendocrine neoplasia. As such, tumors are often collated together with BPNETs. While there appear to be a number of overlapping areas between the two groups, TNETs exhibit specific characteristics that set them apart from BPNETs. Indeed, the consideration that two different tumors in separate organs (lung/thymus) are considered together based upon their similar location in one body cavity (thorax) is naive. It is well accepted that small bowel and pancreatic NETS are completely different pathological entities despite being in the abdominal cavity (19).
Epidemiology and natural history
BPNETs are more common than TNETs with the incidences respectively being 1.35 and 0.02 per 100,000 populations per year (20) (Table 1). The median ages at presentation with bronchopulmonary and TNETs are 64 and 59 years, respectively (20).
Table 1. Comparator evaluation of bronchopulmonary and thymic neuroendocrine neoplasia.
| Topic | Feature | Bronchopulmonary | Thymic |
|---|---|---|---|
| Epidemiology | Incidence (per/100,000) | 1.35 | 0.02 |
| Prevalence | 20% of lung neoplasms | 2–3% of thymic neoplasms | |
| Clinical | Presentation | 40% incidental | Symptomatic (~50%) |
| Age | 5–6th decade | 5th decade | |
| Gender | M = F | M > F | |
| Metastatic | 25% TC, ≥50% AC | Frequent (>70%) | |
| Histology | Histology | NET (>90%) (grade I, II) | NEC (>70%) |
| Imaging | SRI | Positive >80% | Positive >80% |
| 18FDG-PET | Positive (33% TC, 94% AC) | Positive* | |
| Biomarkers | Monoanalytes (CgA, NSE, ACTH) | Inadequate (<40% for CgA, <25% NSE) | Specific monoanalytes (ACTH ~40%) |
| Prognosis | Survival (5-year) | 50–95% | 30–70% |
| Genetic topography | Mutational burden | Low | Low |
| Nicotine mutation signature | No | Unknown** | |
| Germline MEN-1 | 5–13% | ~25% | |
| Sporadic MEN-1 | Frequent (~50%) | No*** | |
| Chromatin/epigenetic remodeling | Frequent (~50%) | Unknown | |
| Chromosomal abnormalities | Low <35% | Frequent: 31–88% | |
| Potential drug targets (copy number gains) | EGFR (gefitinib), MET (crizotinib), PDGFRβ (imatinib), HER4 (lapatinib), AKT1/FRAP1 (everolimus) | None | |
| Potential Informative cell biology/pathway expression | VHL, P21WAF1CIP1, stathmin, ER# | PAK3, β-catenin, NOTCH2 | |
| Potential prognostic biomarkers | CD44$, OTP$$, RET$$, E-cadherin/β-catenin&, DSG3&& | None |
*, no studies have been undertaken in TNETs (21); **, no exome sequencing available; ***, MEN-1 retains heterozygosity; #, TC =55%; AC =86%; $, increased in poor prognosis; $$, decreased in poor prognosis; &, decreased in TC; &&, DSG3 (desmoglein 3) decreased in AC. NEC, neuroendocrine carcinoma; NET, neuroendocrine tumor; SRI, somatostatin receptor imaging.
BPNETs
Overall, BPNETs constitute ~30% of all NETs and make up ~20% of all lung cancers. They are increasing in incidence and prevalence (22), the reasons for which are unknown but may include histopathological reclassification (22). Over the last 30 years, the age-adjusted incidence rates of BPNETs have more than doubled for all races and both genders (22). They are also currently poorly characterized particularly at an etiopathological and molecular biological level.
BPNETs are usually judged to be sporadic, although approximately 10% have some feature suggesting hereditary origin. These features are either tumor multiplicity (5%) or an association with MEN1 (5%). Up to 5% of BPNETs occur with MEN-1; up to 13% of MEN-1 patients develop BPNETs (23). Tumors exhibit variable malignancy which may not be easy to delineate by standard histopathology (24).
Typical BPNETs are usually classified as low-grade, well-differentiated pulmonary neuroendocrine neoplasms. They usually present in the fifth to sixth decade of life in association with symptoms of cough, wheezing, hemoptysis, and sometimes recurrent post-obstructive pneumonia. BPNETs are often in a central pulmonary location and rarely associated with the classic carcinoid syndrome (often related to metastatic disease). Occasionally (1%) tumors, secrete adrenocorticotropic hormone (ACTH), resulting in Cushing syndrome.
Atypical BPNETs typically present with nonspecific signs and symptoms, including coughing and wheezing. These misdiagnosed asthma-like symptoms contribute to the delay in diagnosis. Most tumors are asymptomatic from the neuroendocrine perspective and a substantial number (~40%) are identified serendipitously on routine chest radiology (25). Some may produce bioactive products including ACTH (~2%), growth hormone-releasing hormone (GHRH) and the carcinoid syndrome (metastatic disease). Typically, less than 5% exhibit hormonally-related symptoms.
Typical carcinoids (TCs) exhibit a good prognosis with a 5-year survival of approximately 90%. However, distant metastases may occur many years even after radical (apparently) complete resection of the primary. Standard clinical guidelines recommend a 15-year follow-up (26). ACs are associated with poor prognosis and a 5-year survival of 50–80%. Both large cell neuroendocrine carcinomas (LCNECs) and SCLCs exhibit a significantly poorer prognosis of ~30% and 5%, respectively (27).
TNETs
TNETs are an uncommon neoplasia, accounting for ~2% of all mediastinal tumors and ~5% of thymic lesions (12). They constitute ~0.4% of all carcinoid tumors (20) and <5% of all anterior mediastinal neoplasms with an age-adjusted incidence rate of 0.018 per 100,000 (USA) and a male predominance (77%) with a peak incidence in the fifth decade (12,28). In contrast, BPNETs exhibit an equal gender distribution (22). Although most TNETs are sporadic ~25% of TNETs arise in patients with MEN-1 and ~8% of MEN-1 patients develop TNETs (29). However, a specific thymic MEN1 genotype has not been identified.
Thymic carcinoid tumors are similar to lesions arising within the bronchi, ranging in differentiation and behavior from TC to AC to small cell carcinoma. Tumors may remain asymptomatic even with progressive disease; TNETs are therefore often diagnosed late with advanced disease at presentation and tend to be large masses at presentation ranging in size from 6–20 cm. Approximately 50% of thymic carcinoids are functionally active (30) and ~40% have ACTH-induced Cushing syndrome. Such lesions are usually smaller on presentation since they present early with clinical manifestations of corticosteroid excess (27). Non-functioning thymic carcinoids may be seen in association with MEN-1. The carcinoid syndrome is uncommon although ~30% have advanced-stage disease at presentation. Overall, TNETs are more aggressive than BPNETs and associated with a less favorable prognosis. Prognosis is poor because of the late presentation with distant metastases and a high incidence of local recurrence despite radical resection. Five-year survival varies from 30–70% (12).
Key clinical unmet needs (Table 2)
Table 2. Unmet needs in bronchopulmonary and thymic neuroendocrine neoplasia.
| Blood biomarker for early detection |
| Specific molecular markers (blood and tissue) predictive of invasion and metastasis |
| Blood biomarkers for detection of minimal residual disease |
| Blood biomarkers to monitor disease progression |
| Blood biomarkers to monitor treatment response |
| Development of lung-specific imaging agents |
| Imaging strategies to accurately assess minimal residual disease |
| Identification of actionable targets (blood and tissue) for therapy |
BPNETs
These include an accurate determination of malignancy, identification of residual disease, the ability to accurately monitor disease progression and to evaluate treatment response. Current tissue-based approaches are not accurate. As surgery is the critical survival determinant for these tumors, the determination of minimum residual disease is a key limitation to early therapeutic intervention. Similarly given the limitations of imaging the need for a biomarker to monitor therapeutic efficacy is a critical requirement (24).
TNETs
The most significant issue is the difficulty in the prediction of prognosis. Furthermore, apart from surgery, there is no consensus regarding an optimal therapeutic strategy. In both tumor types the lack of clarity regarding the type of surgery, treatment protocols and the need for a sensitive, specific and unique circulating biomarker to measure efficacy remain unmet needs.
Cells of origin
Both BPNET and TNETs are considered to develop from elements of the diffuse neuroendocrine cell system generically referred to as “Kulchitsky” cells. BPNETs arise from K cells of the bronchopulmonary epithelium, previously known as Clara cells (22). The latter eponym is no longer used given the evidence that Clara was known to have acquired the tissue from murdered victims of the Nazi regime (31). The K cells are dispersed within the epithelium of the tracheobronchial system and function as regulatory chemoreceptors for sensing of oxygen and carbon dioxide levels (22). K cells are dispersed with in the normal thymus and constitute a minor (~1%) cell population with an unknown function.
Etiopathology
The etiology is largely unclear for both tumor types and the environmental risk factors associated with either unknown. Interestingly, nearly all cases of TNETs associated with MEN-1 are male smokers (32). A role for nicotine in the genesis BPNETs has been largely excluded (22,33).
Histology and staging
BPNETs
Although they share structural, morphological, immunohistochemical, and ultrastructural features, they are considered as four subgroups: TC, AC, large-cell neuro-endocrine carcinoma (LCNEC), and small-cell lung carcinoma (SCLC). Each exhibits considerably different biological and clinical characteristics (Figure 3) (34). A separate rare condition of neuroendocrine cell-derived hyperplasia [diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH)] has been described, but is poorly understood. It may be analogous to ECL cell hyperplasia of the stomach and represent a response to a biological agent. Thus, elevated gastrin levels in the stomach consequent upon high pH may be comparable to a local paracrine agent in the lung generated by hypoxia (35). The TC group generally behaves in an indolent fashion whereas the atypical tumors can range from indolent to highly aggressive. TCs are broadly regarded as benign, but may present with metastatic spread and behave like ACs, displaying a poorer prognosis. Such lesions require aggressive treatment. There is significant inter-observer variation (Kappa =0.32) in lung carcinoid histopathologic classification and is of concern regarding ACs (36). Both SCLC and LCNEC progress rapidly, metastasize early and exhibit a poor prognosis. The slow-growing TC exhibit a good prognosis (5-year survival ~88%), whereas for AC this is 50% and for the highly malignant LCNEC 15–57% and SCLC (<5%) (22).
Figure 3.

Histological and genetic classification systems for bronchopulmonary (red) and thymic (yellow) neuroendocrine neoplasia. Both are classified into 4 groups—typical and atypical carcinoids—and large cell neuroendocrine cancers (LCNEC) and small cell neoplasia. In the lung, carcinoids are typically well-differentiated and are characterized as neuroendocrine tumors (NET). In the thymus, they are also well-differentiated but classified as neuroendocrine cancer (NEC) given their greater propensity to exhibit aggressive disease. At a molecular level, each well-differentiated group, NET or NEC, exhibit very different genetic alterations. Lung NETs are associated with sporadic loss of MEN-1 and genes involved in chromatin remodeling. This genotype does not appear to occur in the thymus. Chromosomal abnormalities are infrequent in both groups, but specific, potentially “druggable” gains of function are only evident for BPNETs. Biologically informative pathways also differ; alterations in hypoxia signaling and cell cycle inhibitors characterize BPNETs, while WNT-signaling and invasion/migration (RAC1/PAK3) characterize thymic NETs. There are currently no prognostic markers for the latter, but BPNET prognosis has been linked to a series of markers, especially CD44 and OTP. ?, no data; green arrow, upregulated; red arrow, down-regulated.
TNETs
TNETs are included in the thymic carcinoma group and, like BPNETs, are classified into two sub-groups: well-differentiated neuroendocrine carcinomas (typical and atypical) and poorly differentiated NECs (small cell and large cell) (37). Thymic carcinoids closely resemble atypical lung carcinoids in their histologic appearance and behavior (10). Histologically, most are identical to well-differentiated neuroendocrine carcinoma. These are malignant tumors and often develop distant metastases, sometimes after long intervals. However, the behavior of this tumor is unpredictable, and there is no satisfactory classification system to accurately predict its progression. World Health Organization (WHO) histological classification divides thymic epithelial tumors in thymomas and thymic carcinomas, the latter also include TNETs. This group (thymic carcinomas) has a significantly lower survival than thymomas. TNETs (even grade 1) are regarded as a malignant lesion prone to metastasize to mediastinal lymph nodes and to distant sites even after total excision. Neither the T and N classification, nor histologic grade is effective in accurately predicting outcome. More aggressive management, including routine adjuvant therapy and re-excision of the subsequent recurrent tumor, are considered necessary to optimize survival. The Masaoka stage of the tumor and effective surgical resection are the only two significant prognostic factors for overall survival (OS) (12); histologic subtype appears to have no bearing on outcome.
The molecular genetic topography
BPNETs
The genetic landscape of BPNETs identifies a very low mutation rate and a non-smoking mutation pattern (i.e., no loss of p53, few G → T transversions) consistent with the absence of a nicotine-driven etiopathogenesis (33). Comparative genomic hybridization (CGH) studies have identified loss of heterozygosity (LOH) at 11q13, the site of the MEN-1 locus, as a common event in BPNETs (38). Chromosomal instability is infrequent and targetable mutations are rarely identifiable. Copy number gains occur at 14q32 (the DLK1-DOI locus, a miRNA-rich region) and ~20–25% of BPNETs exhibit gains in EGFR, MET, PDGFRβ, HER4, AKT1 and FRAP1 (39). The latter are potential targets for mTOR inhibitors. Copy number losses occur in ~20% of tumors in hypoxia-associated VHL and the cell cycle inhibitor, P21WAF1/CIP1 (39). CD44 (cell adhesion and migration), orthopedia homeobox (OTP) gene (neuroendocrine cell development) and the RET proto-oncogene (cadherin) are all increased in BPNETs (40) and may have prognostic utility. Poor prognostic indicators appear to be CD44 elevation and decrease of OTP and RET (41). Stathmin, a marker of proliferation and PI3K signaling in pancreatic NETs (42), is decreased (43). Loss of expression of desmoglein 3, a cell adhesion glycoprotein, was associated with poor prognosis particularly for ACs (44). Loss of other cell adhesion complexes e.g., E-cadherin/β-catenin complex identifies TCs with poor prognosis (45). Both TCs and ACs highly express estrogen receptors (46) but very rarely, nicotinic β2 receptors (46). In contrast, α7 which is associated with an aggressive nicotinic phenotype is variably expressed (47). MEN-1 mutation occurs in 18% and LOH at 11q13 in 36% of sporadic BPNETs (48). Deletion of 11q is the most frequently observed alteration in BPNETs; it is less frequently lost in typical than ACs and may represent a marker of progression (48).
MEN-1 is the most common germline mutation (~5%) while sporadic alterations have been identified in MEN-1, PSIP1 and ARID1 (33). Overall, methylation and histone modifications i.e., chromatin remodeling, are the most prominent genetic alterations occurring in 20–40% of BPNETs. None of these alterations, however, are currently clinically actionable. In contrast to SCLCs and LCNECs, TP53 and RB1 mutations are rare events, suggesting that BP “carcinoids” are not early progenitor lesions of these highly aggressive tumors and that both arise through independent cellular mechanisms. These data also suggest that inactivation of chromatin-remodeling genes may be sufficient to drive transformation in pulmonary (typical and atypical) carcinoids (33).
TNETs
The genetic landscape of TNETs is similar to that of BPNETs in that thymic tumors have very few chromosomal abnormalities (<1 imbalance/tumor which occur in less than a third of patients) (49). Since exome-level analyses have not to date been undertaken, the mutation pattern is unknown but it is doubtful to be nicotine-related except, perhaps, in males with MEN-1. It seems evident, however, that a different cytogenetic mechanism likely occurs in the development of TNETs compared to BPNETs. CGH studies have demonstrated gross chromosomal imbalances in 31–88% of samples (49,50) and, in contradistinction to BPNETs, heterozygosity at the MEN-1 locus, 11q13 is retained (50). At a transcriptional level, TNETs over-express PAK3 (p21-activated kinase 3) and its upstream regulator RAC1. Both these genes are involved in the regulation of cell migration and invasion (51). ACTH-secreting tumors also express elevated β-catenin, pro-opiomelanocortin (POMC) and carboxypeptidase E (CPE) and exhibit decreased NOTCH2 levels (WNT-signaling) (52).
TNETs exhibit an increased mitotic activity and chromosomal aberrations ranging from low-grade TC to AC to high grade LCNEC to small cell cancer (49). These lesions, thymic carcinoids (typical and atypical) and NECs (small cell and large cell), are significantly different molecular biological entities as in BPNETs (49). It is intriguing that while the genetic profile of thymic carcinoids are discrete from that of pulmonary carcinoids (53,54), high grade thymic and BPNETs appear to be similar. This suggests that distinctive differentiation/proliferation pathways may be operative in the development of well-differentiated BPNET and TNETs (typical and atypical). In contrast, higher grade tumors (small cell/large cell) likely undergo common mutation/transformation pathways which is not organ-dependent i.e., mutation from a common epithelial progenitor cell irrespective of BP or thymic origin.
Multiple endocrine neoplasia type 1 (MEN-1)
MEN-1 is an autosomal dominant tumor syndrome arising from an inactivating mutation on a tumor suppressor gene located on chromosome 11 (11q13). The prevalence of BPNET and TNETs in MEN-1 patients is 2–5%. BPNET and TNETs occur in 3–8% of patients with MEN-1. BPs occur more frequently (63%), in women (80% are TC) and are not associated with an increased mortality (23). TNETs almost exclusively occur in men (90%) and 87% are AC associated with an aggressive disease course (55). In an Australian (Tasman kindred), the prevalence of BPNETs was 5%, the majority of whom were women (56). In the Dutch MEN1 database, the prevalence of BPNETs was 13% of whom 66% were female (23). In a recent Mayo Clinic report, 67% were men (55). In the latter study, BPNETs were most commonly diagnosed during routine screening and were associated with an indolent course. BPNETs in MEN-1 have the added feature of being a disease with female preponderance (5:1 ratio); the explanation for this remains unclear. TNETs are predominantly seen in men and are associated with a more aggressive behavior (55). There is a significant male preponderance (10:1) in TNETs with MEN-1. The basis for the marked gender distribution difference is unknown. Several studies have failed to find loss of heterozygosity at 11q13 in TNETs of the MEN-1 syndrome, despite being malignant. Inactivation of the normal MEN-1 copy without mutation or promoter hypermethylation is a possible mechanism requiring confirmation (55). Involvement of the MEN-1 gene has not been tested directly in sporadic TNETs. Irrespective of these differences, TNETs in MEN-1 are histologically indistinguishable from their pulmonary counterparts (29).
The current clinical recommendations for assessing and following-up MEN-1 patients include periodic screening for both BP and thymic disease every 1–2 years using CT/MRI (57). It is likely that the development of a sensitive blood-based biomarker may provide an alternative and beneficial monitoring strategy to life-long radiation-related imaging assessment (Table 2).
Biomarkers
The biochemical profile of BPNET and TNETs are similar. Thus, single secretory products (monoanalytes) including chromogranin A (CgA), neuron specific enolase (NSE) and 5-hydroxy-indoleacetic acid (5-HIAA) can be measured. Their performance metrics for diagnosis and treatment monitoring, however, are poor to ineffective in terms of clinical utility (58). For example, an elevated 24-hour urinary 5-HIAA level may be of use in the diagnosis of a BP or TNET (59). BPNET and TNETs, however, often lack aromatic amino acid decarboxylase, necessary to convert tryptophan to serotonin, and then to urinary 5-HIAA. This as well as the well-described limitations posed by interfering food agents e.g., tryptophan-rich foods and medications e.g., proton-pump inhibitors, render single analyte measurements of limited clinical utility. Specific secretory (monoanalyte) biomarkers e.g., 24 hr urine free cortisol and ACTH (if Cushing’s syndrome is present), and GHRH or IGF-1 (if acromegaly is present) may be useful in selected individuals. However, given the very low incidence of ACTH secreting tumors (<5%), their utility is limited.
In contrast to secretory biomarkers, tissue biomarkers including somatostatin receptor (SSTR) expression have been identified. SSTR are highly expressed on both BPNET and TNET tumor tissue and have been utilized both as a diagnostic (SRI e.g., 68Ga-SSA-PET/CT) or as a biomarker for targeted therapy e.g., peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE (60,61). Tissue expression scores of this receptor have recently been proposed (62), and there is consideration of the measurement of circulating somatostatin receptor mRNA levels (see section below—section “Circulating mRNA”).
For BPNETs, SRI is used both for confirming the diagnosis as well as in staging protocols (24). SRI with conventional 111In-pentetreotide scintigraphy may visualize up to 80% of primary tumors but is most sensitive for metastatic disease (24). Three studies have examined the utility of PRRT (62-64). In the first, in nine bronchial carcinoids, treatment responses were identified in six, disease stabilization was noted in two, and one patient exhibited progressive disease (63). In a more recent, larger study (n=34 bronchial NETs), the disease control rate was 80% (64). A recent retrospective analysis of 114 advanced bronchial NETs treated with 90Yttrium-, 90Y+177Lutetium-, or 177Lu-based PRRT protocols, indicated a median OS of 58.8 months, with median progression-free survival (PFS) of 28 months. Patients treated with 177Lu-DOTATATE exhibited the longest 5-year OS (61.4%). Objective responses were associated with longer OS and PFS (65). No factor was identified that could accurately measure efficacy of therapy in BPNETs or predict response to PRRT.
The diagnosis of TNETs is facilitated by SRI and is particularly effective in identifying tumors in MEN-1 (66). The limitations in detecting recurrence are based upon the difficulty in image identification of small lesions (29,67). Only two instances of thymic carcinoids treated with 177Lu DOTATATE PRRT have been published (63). Only one responded with the second exhibiting progressive disease.
These studies identify the limitations of currently available circulating biomarkers in both diagnosis of the disease and in the prediction of effective therapy. They also identify the need for a circulating biomarker to monitor therapies and predict response to a therapy e.g., PRRT.
Novel biomarkers
Novel biomarkers include but are not limited to circulating genetic information e.g., circulating tumor DNA or RNA or tumor cells, or the identification of actionable mutation events e.g., BRAF mutations. These all fall within the rubric of a “liquid biopsy”. Currently there is a significant interest in the identification and application of such “liquid biopsies” in the field of oncology (68,69). As such, the use of liquid biopsy allows for patient stratification (e.g., as a companion diagnostic), for screening, for monitoring treatment responses e.g., to EFGR inhibitors in lung cancer (70) and for detection of minimal residual disease after surgery/recurrence e.g., in colon cancer (71). All of these areas can be characterized as unmet needs in BPNET/TNET management.
Circulating tumor DNA with or without actionable mutations
Measurements of circulating tumor DNA have not been undertaken in either BPNETs or TNETs. In addition, neither BPNET nor TNETs are associated with targetable mutations except in a minority of cases. At present, therefore, insufficient specific mutations and treatment-targetable mutations have been identified in these diseases. As such, circulating DNA is therefore currently not a viable option for the development of a biomarker for either BPNET or TNETs.
Circulating tumor cells
This strategy is generally not considered as reliable, sensitive or specific for the detection and diagnosis of neuroendocrine tumors and levels do not closely correlate with tumor grade in NETs (72). Nevertheless, one study has evaluated CTCs in BPNETs (73). This group identified that the target protein expression used to capture the cells, EpCAM, was variably expressed in BPNETs. Furthermore, based on the inconsistent results, the authors concluded that CTCs would not be useful in this tumor group (73). Nothing is known regarding CTCs in TNETs, but it may present a similar unsatisfactory outcome to that identified in BPNETs.
Circulating miRNA
miRNA signatures have been evaluated in small bowel NETs (74,75) and are detectable in the circulation (76). Similar peripheral blood studies have not been undertaken in either BPNET or TNETs. One study identified miRNAs in different types of lung cancers and exhibited some utility in identifying BPNETs (77). A second study identified that miRNAs (a different cluster) could differentiate BPNETs from SCLC/LNECs (78). Both studies underscore the problems with this biomarker in that different miRNA profiles were generated in each study. There is a well-described wide age- and population-dependent variability in miRNA-based biomarker profiling (79). Currently, it is considered that miRNA profiles remain experimental and require validation in tissue.
Circulating mRNA
Two different groups have evaluated circulating mRNA in BPNETs. One group was able to detect somatostatin receptor expression (SSTR2a and 5) in peripheral blood using real-time quantitative PCR (80). Expression levels were consistent with the extent of the disease measured by SRI (OctreoscanTM ~75%).
More recently, the complex nature of cancer and its responses to therapy, has become better appreciated (81,82). It is now evident that tumor behavior is optimally characterized by evaluating the canonical pathways or “hallmarks” that comprise tumor biology. A blood multianalyte measurement has been recently developed (NETest) that identifies circulating mRNA as a diagnostic test and whose “omic” configuration can be used as a surrogate marker of neuroendocrine tumor biological behavior (83,84). The NETest encompasses the expression of 51 genes specifically linked to neuroendocrine tumor pathobiology (83). Thus, the “hallmarks” of neuroendocrine tumor behavior can be defined by measured genes that include “omic” measures of proliferation (Ki67), growth factor signaling (RAS/RAF) and metabolism (oxidative phosphorylation, hypoxic signaling) (85). Gene expression measurements correlate directly with tissue levels (84) and provide precise information about the tumor, its pathophysiology and its pathobiological state from stability to progression (75). Gene expression is measured by qPCR, normalized expression is assessed by four different prediction algorithms (SVM, LDA, KNN and Bayes) and the output is scaled to a disease/tumor activity (0–100%) score (84). The score has demonstrated utility (90–95% accuracy) as a diagnostic and has been demonstrated to be substantially more accurate than single analyte measurement for detecting neuroendocrine tumors (86). It is appropriately decreased following various surgical procedures (R0 vs. RI/R2 resection) and can identify residual/recurrent disease (87). The score can also effectively monitor treatment protocols (60,88) and predict disease progression (89).
Blood transcript analysis has been evaluated in both BPNET and TNETs. In 36 BPNETs (14 typical, 22, atypical), >94% expressed elevated levels of circulating NET genes (NETest positive) (Figure 4). The individual gene expression levels could accurately differentiate progressive from stable disease (90). In a separate prospective study with PRRT, NETest was positive in all BPNETs prior to treatment (60). PRRT therapy was effective 62% and gene expression levels correctly identified post-therapy disease status in all. Moreover, measurement of individual gene expression levels (growth factor signaling and metabolism) pre-therapy—accurately predicted treatment response (100%). This represents the first instance of a multianalyte LDT functioning as a companion diagnostic. Previously, such tests have measured a single marker/mutation e.g., BRAF (91).
Figure 4.

Diagnostic utility of a blood-based circulating neuroendocrine tumor gene assay—the 51 marker gene NETest—in bronchopulmonary NETs. Transcripts are elevated in 94% in both typical and atypical tumors. Chromogranin A (CgA) in contrast is only positive (elevated beyond normal levels) in 40%. Typical carcinoids are positive in (36%) and atypical tumors positive (46%).
In BPNETs a circulating mRNA multi-transcript fingerprint correlates with standard morphologic and functional imaging. The blood values can be used to predict and define treatment response or outcome to therapy in BPNETs. In addition, when surgical resections are undertaken significant decreases in the NETest in BPNETs 4–6 weeks after surgery define the extent and success of the resection (92). These observations mirror previous reports in GEP-NETs where extent and completeness of surgery is directly related to the alterations in peripheral blood circulatory levels (87). Of particular note is the observation that elevated post-surgical levels predict the presence of minimal residual disease and subsequent tumor recurrence. Whether this occurs with BPNETs is as yet unknown but it is likely that a similar outcome will be identified.
The NETest is also positive in TNETs. Three G2 tumors, all non-secretors (i.e., no production of serotonin, ACTH etc.) with low CgA levels, expressed detectable gene expression (M Tesselaar, unpublished observations). All three were positive on OctreoScanTM. Although the clinical utility remains to be determined in larger studies it seems likely that the blood fingerprint would function similarly to what has been established in BPNETs, i.e., identify progressive tumors, confirm complete resection after surgery, and predict treatment response.
Coda
Consistently effective and accurate circulating biomarkers for BPNET and TNETs are currently unavailable. Recent studies indicate that mRNA transcript analysis in blood effectively identifies both BPNET and TNETs. The clinical utility of mRNA measurement in BPNET and TNETs includes use as a diagnostic, confirmation of completeness of surgical resection and to monitor efficacy of PRRT and other therapeutic strategies. One might reflect on the nature of destiny and fate in contemplating the primary observations of Siegfried Oberndorfer who initially described carcinoid tumors in 1907 in Dresden. Almost forty years [1944] later he perished in his city of exile, Istanbul, of a thymic carcinoid; only identified histologically in 2005 (Figure 5).
Figure 5.
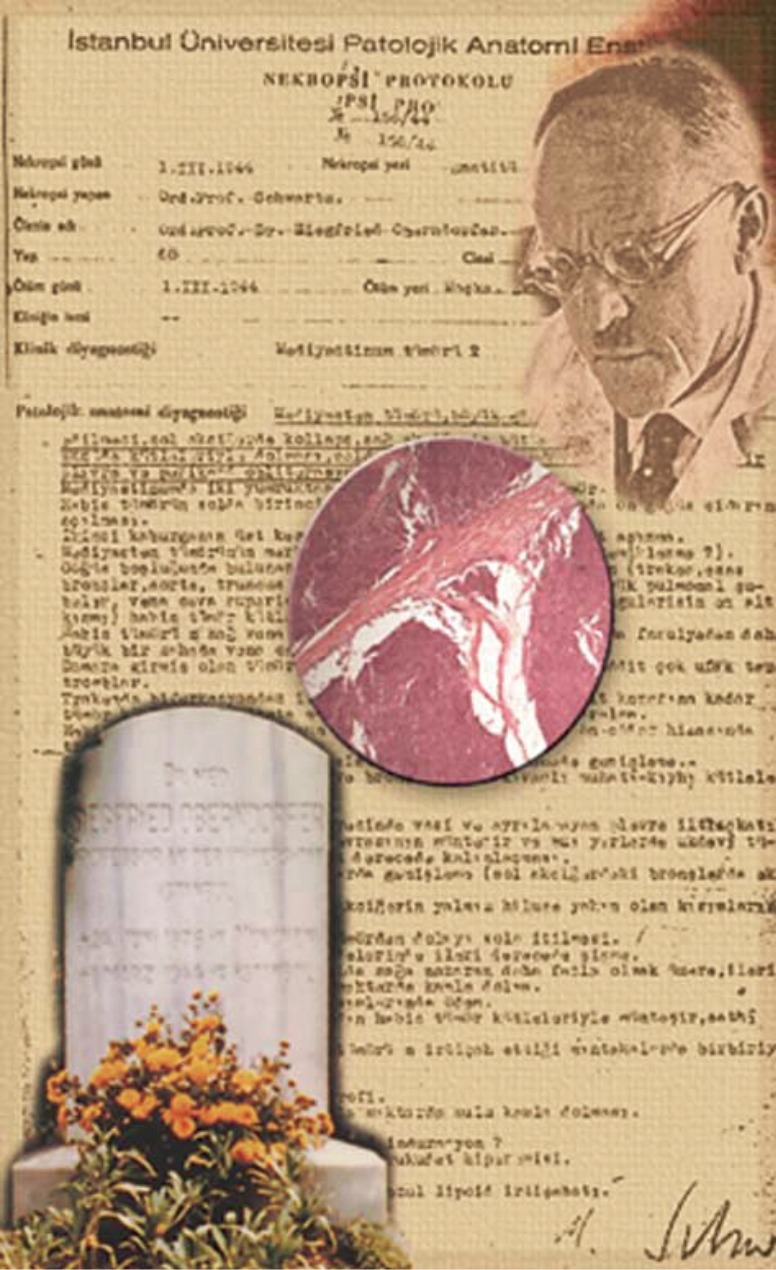
S. Oberndorfer was the first to describe the novel entity “carcinoids” in 1907 in Dresden. He was exiled for ethnic reasons by the Third Reich to Turkey [1933] and died in Istanbul [1944] where he is interred in the Sisli Cemetery (bottom left). The autopsy report (background) undertaken by his fellow refugee (Dr. Schwartz) indicated the cause of death to be a neoplastic mediastinal mass. Subsequent re-examination of histological sections (I. Modlin and G. Kloppel) in 2005 identified the mass to be a thymic neuroendocrine tumor. [Courtesy Modlin I, et al. Arch Surg 2007;142:187-97].
Acknowledgements
Funded by Clifton Life Sciences.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Rosai J, Higa E. Mediastinal endocrine neoplasm, of probable thymic origin, related to carcinoid tumor. Clinicopathologic study of 8 cases. Cancer 1972;29:1061-74. [DOI] [PubMed] [Google Scholar]
- 2.Geipel P. Concerning Our Knowledge of Benign Bronchial Tumors. Frankfurter Zeitschrift fur Pathologie 1931;42:516. [Google Scholar]
- 3.Bensch KG, Corrin B, Pariente R, et al. Oat-cell carcinoma of the lung. Its origin and relationship to bronchial carcinoid. Cancer 1968;22:1163-72. [DOI] [PubMed] [Google Scholar]
- 4.Arrigoni MG, Woolner LB, Bernatz PE. Atypical carcinoid tumors of the lung. J Thorac Cardiovasc Surg 1972;64:413-21. [PubMed] [Google Scholar]
- 5.Mills SE, Cooper PH, Walker AN, et al. Atypical carcinoid tumor of the lung. A clinicopathologic study of 17 cases. Am J Surg Pathol 1982;6:643-54. 10.1097/00000478-198210000-00006 [DOI] [PubMed] [Google Scholar]
- 6.DeLellis RA, Dayal Y, Wolfe HJ. Carcinoid tumors. Changing concepts and new perspectives. Am J Surg Pathol 1984;8:295-300. 10.1097/00000478-198404000-00007 [DOI] [PubMed] [Google Scholar]
- 7.Oberndorfer S. Karzinoide Tumoren des Dünndarmes. Frankfurter Zeitschrift für Pathologie 1907;1:426-9. [Google Scholar]
- 8.Lattes R. Thymoma and other tumors of the thymus. Cancer 1962;15:1224-60. [DOI] [Google Scholar]
- 9.Bernatz PE, Harrison EG, Clagett OT. Thymoma: a clinicopathologic study. J Thorac Cardiovasc Surg 1961;42:424-44. [PubMed] [Google Scholar]
- 10.Lausi PO, Refai M, Filosso PL, et al. Thymic neuroendocrine tumors. Thorac Surg Clin 2014;24:327-32. 10.1016/j.thorsurg.2014.05.007 [DOI] [PubMed] [Google Scholar]
- 11.Rosai J, Higa E, Davie J. Mediastinal endocrine neoplasm in patients with multiple endocrine adenomatosis. A previously unrecognized association. Cancer 1972;29:1075-83. [DOI] [PubMed] [Google Scholar]
- 12.Filosso PL, Yao X, Ahmad U, et al. Outcome of primary neuroendocrine tumors of the thymus: a joint analysis of the International Thymic Malignancy Interest Group and the European Society of Thoracic Surgeons databases. J Thorac Cardiovasc Surg 2015;149:103-9.e2. 10.1016/j.jtcvs.2014.08.061 [DOI] [PubMed] [Google Scholar]
- 13.Bergh NP, Gatzinsky P, Larsson S, et al. Tumors of the thymus and thymic region: I. Clinicopathological studies on thymomas. Ann Thorac Surg 1978;25:91-8. 10.1016/S0003-4975(10)63498-4 [DOI] [PubMed] [Google Scholar]
- 14.Wilkins EW, Jr, Castleman B. Thymoma: a continuing survey at the Massachusetts General Hospital. Ann Thorac Surg 1979;28:252-6. 10.1016/S0003-4975(10)63114-1 [DOI] [PubMed] [Google Scholar]
- 15.Masaoka A, Monden Y, Nakahara K, et al. Follow-up study of thymomas with special reference to their clinical stages. Cancer 1981;48:2485-92. [DOI] [PubMed] [Google Scholar]
- 16.Koga K, Matsuno Y, Noguchi M, et al. A review of 79 thymomas: modification of staging system and reappraisal of conventional division into invasive and non-invasive thymoma. Pathol Int 1994;44:359-67. 10.1111/j.1440-1827.1994.tb02936.x [DOI] [PubMed] [Google Scholar]
- 17.Kondo K, Monden Y. Therapy for thymic epithelial tumors: a clinical study of 1,320 patients from Japan. Ann Thorac Surg 2003;76:878-84; discussion 884-5. 10.1016/S0003-4975(03)00555-1 [DOI] [PubMed] [Google Scholar]
- 18.Filosso PL, Ruffini E, Lausi PO, et al. Historical perspectives: The evolution of the thymic epithelial tumors staging system. Lung Cancer 2014;83:126-32. 10.1016/j.lungcan.2013.09.013 [DOI] [PubMed] [Google Scholar]
- 19.Modlin I, Kidd M. Conversationes de Inebriati. Athenaeum 2016:120-7. [Google Scholar]
- 20.Yao JC, Hassan M, Phan A, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26:3063-72. 10.1200/JCO.2007.15.4377 [DOI] [PubMed] [Google Scholar]
- 21.De Luca S, Fonti R, Palmieri G, et al. Combined imaging with 18F-FDG-PET/CT and 111In-labeled octreotide SPECT for evaluation of thymic epithelial tumors. Clin Nucl Med 2013;38:354-8. 10.1097/RLU.0b013e318286bd84 [DOI] [PubMed] [Google Scholar]
- 22.Gustafsson BI, Kidd M, Chan A, et al. Bronchopulmonary neuroendocrine tumors. Cancer 2008;113:5-21. 10.1002/cncr.23542 [DOI] [PubMed] [Google Scholar]
- 23.de Laat JM, Pieterman CR, van den Broek MF, et al. Natural course and survival of neuroendocrine tumors of thymus and lung in MEN1 patients. J Clin Endocrinol Metab 2014;99:3325-33. 10.1210/jc.2014-1560 [DOI] [PubMed] [Google Scholar]
- 24.Caplin ME, Baudin E, Ferolla P, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol 2015;26:1604-20. 10.1093/annonc/mdv041 [DOI] [PubMed] [Google Scholar]
- 25.Jeung MY, Gasser B, Gangi A, et al. Bronchial carcinoid tumors of the thorax: spectrum of radiologic findings. Radiographics 2002;22:351-65. 10.1148/radiographics.22.2.g02mr01351 [DOI] [PubMed] [Google Scholar]
- 26.Oberg K, Hellman P, Ferolla P, et al. Neuroendocrine bronchial and thymic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23:vii120-3. [DOI] [PubMed] [Google Scholar]
- 27.Phan AT, Oberg K, Choi J, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus). Pancreas 2010;39:784-98. 10.1097/MPA.0b013e3181ec1380 [DOI] [PubMed] [Google Scholar]
- 28.Gaur P, Leary C, Yao JC. Thymic neuroendocrine tumors: a SEER database analysis of 160 patients. Ann Surg 2010;251:1117-21. 10.1097/SLA.0b013e3181dd4ec4 [DOI] [PubMed] [Google Scholar]
- 29.Gibril F, Chen YJ, Schrump DS, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2003;88:1066-81. 10.1210/jc.2002-021314 [DOI] [PubMed] [Google Scholar]
- 30.Rosado de Christenson ML, Abbott GF, Kirejczyk WM, et al. Thoracic carcinoids: radiologic-pathologic correlation. Radiographics 1999;19:707-36. 10.1148/radiographics.19.3.g99ma11707 [DOI] [PubMed] [Google Scholar]
- 31.Winkelmann A, Noack T. The Clara cell: a "Third Reich eponym"? Eur Respir J 2010;36:722-7. 10.1183/09031936.00146609 [DOI] [PubMed] [Google Scholar]
- 32.Goudet P, Bonithon-Kopp C, Murat A, et al. Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d'etude des Tumeurs Endocrines. Eur J Endocrinol 2011;165:97-105. 10.1530/EJE-10-0950 [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Cuesta L, Peifer M, Lu X, et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun 2014;5:3518. 10.1038/ncomms4518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Travis WD, Gal AA, Colby TV, et al. Reproducibility of neuroendocrine lung tumor classification. Hum Pathol 1998;29:272-9. 10.1016/S0046-8177(98)90047-8 [DOI] [PubMed] [Google Scholar]
- 35.Modlin I, Kidd M. Reflections of the Gnostic Society. Mastrobuoni Verlag GmBh: Bordeaux Savant Publications, 2015. [Google Scholar]
- 36.Swarts DR, van Suylen RJ, den Bakker MA, et al. Interobserver variability for the WHO classification of pulmonary carcinoids. Am J Surg Pathol 2014;38:1429-36. 10.1097/PAS.0000000000000300 [DOI] [PubMed] [Google Scholar]
- 37.Travis WD, Brambilla E, Muller-Hermlink H, et al. editors. World Health Organization classification of tumours. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon: IARC Press, 2004. [Google Scholar]
- 38.Cakir M, Grossman A. The molecular pathogenesis and management of bronchial carcinoids. Expert Opin Ther Targets 2011;15:457-91. 10.1517/14728222.2011.555403 [DOI] [PubMed] [Google Scholar]
- 39.Voortman J, Lee JH, Killian JK, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci U S A 2010;107:13040-5. 10.1073/pnas.1008132107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swarts DR, Henfling ME, Van Neste L, et al. CD44 and OTP are strong prognostic markers for pulmonary carcinoids. Clin Cancer Res 2013;19:2197-207. 10.1158/1078-0432.CCR-12-3078 [DOI] [PubMed] [Google Scholar]
- 41.Swarts DR, Van Neste L, Henfling ME, et al. An exploration of pathways involved in lung carcinoid progression using gene expression profiling. Carcinogenesis 2013;34:2726-37. 10.1093/carcin/bgt271 [DOI] [PubMed] [Google Scholar]
- 42.Schimmack S, Taylor A, Lawrence B, et al. Stathmin in pancreatic neuroendocrine neoplasms: a marker of proliferation and PI3K signaling. Tumour Biol 2015;36:399-408. 10.1007/s13277-014-2629-y [DOI] [PubMed] [Google Scholar]
- 43.Tanca A, Addis MF, Pagnozzi D, et al. Proteomic analysis of formalin-fixed, paraffin-embedded lung neuroendocrine tumor samples from hospital archives. J Proteomics 2011;74:359-70. 10.1016/j.jprot.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 44.Fukuoka J, Dracheva T, Shih JH, et al. Desmoglein 3 as a prognostic factor in lung cancer. Hum Pathol 2007;38:276-83. 10.1016/j.humpath.2006.08.006 [DOI] [PubMed] [Google Scholar]
- 45.Galvan JA, Astudillo A, Vallina A, et al. Prognostic and diagnostic value of epithelial to mesenchymal transition markers in pulmonary neuroendocrine tumors. BMC Cancer 2014;14:855. 10.1186/1471-2407-14-855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sica G, Wagner PL, Altorki N, et al. Immunohistochemical expression of estrogen and progesterone receptors in primary pulmonary neuroendocrine tumors. Arch Pathol Lab Med 2008;132:1889-95. [DOI] [PubMed] [Google Scholar]
- 47.Sartelet H, Maouche K, Totobenazara JL, et al. Expression of nicotinic receptors in normal and tumoral pulmonary neuroendocrine cells (PNEC). Pathol Res Pract 2008;204:891-8. 10.1016/j.prp.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 48.Swarts DR, Ramaekers FC, Speel EJ. Molecular and cellular biology of neuroendocrine lung tumors: evidence for separate biological entities. Biochim Biophys Acta 2012;1826:255-71. [DOI] [PubMed] [Google Scholar]
- 49.Strobel P, Zettl A, Shilo K, et al. Tumor genetics and survival of thymic neuroendocrine neoplasms: a multi-institutional clinicopathologic study. Genes Chromosomes Cancer 2014;53:738-49. 10.1002/gcc.22183 [DOI] [PubMed] [Google Scholar]
- 50.Pan CC, Jong YJ, Chen YJ. Comparative genomic hybridization analysis of thymic neuroendocrine tumors. Mod Pathol 2005;18:358-64. 10.1038/modpathol.3800246 [DOI] [PubMed] [Google Scholar]
- 51.Liu RX, Wang WQ, Ye L, et al. p21-activated kinase 3 is overexpressed in thymic neuroendocrine tumors (carcinoids) with ectopic ACTH syndrome and participates in cell migration. Endocrine 2010;38:38-47. 10.1007/s12020-010-9324-6 [DOI] [PubMed] [Google Scholar]
- 52.Bi YF, Liu RX, Ye L, et al. Gene expression profiles of thymic neuroendocrine tumors (carcinoids) with ectopic ACTH syndrome reveal novel molecular mechanism. Endocr Relat Cancer 2009;16:1273-82. 10.1677/ERC-08-0325 [DOI] [PubMed] [Google Scholar]
- 53.Ullmann R, Petzmann S, Klemen H, et al. The position of pulmonary carcinoids within the spectrum of neuroendocrine tumors of the lung and other tissues. Genes Chromosomes Cancer 2002;34:78-85. 10.1002/gcc.10049 [DOI] [PubMed] [Google Scholar]
- 54.Zhao J, de Krijger RR, Meier D, et al. Genomic alterations in well-differentiated gastrointestinal and bronchial neuroendocrine tumors (carcinoids): marked differences indicating diversity in molecular pathogenesis. Am J Pathol 2000;157:1431-8. 10.1016/S0002-9440(10)64780-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh Ospina N, Thompson GB, C. Nichols F 3rd, et al. Thymic and Bronchial Carcinoid Tumors in Multiple Endocrine Neoplasia Type 1: The Mayo Clinic Experience from 1977 to 2013. Horm Cancer 2015;6:247-53. 10.1007/s12672-015-0228-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sachithanandan N, Harle RA, Burgess JR. Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer 2005;103:509-15. 10.1002/cncr.20825 [DOI] [PubMed] [Google Scholar]
- 57.Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990-3011. 10.1210/jc.2012-1230 [DOI] [PubMed] [Google Scholar]
- 58.Modlin I, Kidd M, Taylor A, et al. Neuroendocrine Tumor Biomarkers: Current Status and Perspectives. Neuroendocrinology 2014;100:265-77. 10.1159/000368363 [DOI] [PubMed] [Google Scholar]
- 59.Feldman JM. Urinary serotonin in the diagnosis of carcinoid tumors. Clin Chem 1986;32:840-4. [PubMed] [Google Scholar]
- 60.Bodei L, Kidd M, Modlin IM, et al. Measurement of circulating transcripts and gene cluster analysis predicts and defines therapeutic efficacy of peptide receptor radionuclide therapy (PRRT) in neuroendocrine tumors. Eur J Nucl Med Mol Imaging 2016;43:839-51. 10.1007/s00259-015-3250-z [DOI] [PubMed] [Google Scholar]
- 61.Bodei L, Kidd M, Modlin IM, et al. Gene transcript analysis blood values correlate with (68)Ga-DOTA-somatostatin analog (SSA) PET/CT imaging in neuroendocrine tumors and can define disease status. Eur J Nucl Med Mol Imaging 2015;42:1341-52. 10.1007/s00259-015-3075-9 [DOI] [PubMed] [Google Scholar]
- 62.Specht E, Kaemmerer D, Sanger J, et al. Comparison of immunoreactive score, HER2/neu score and H score for the immunohistochemical evaluation of somatostatin receptors in bronchopulmonary neuroendocrine neoplasms. Histopathology 2015;67:368-77. 10.1111/his.12662 [DOI] [PubMed] [Google Scholar]
- 63.van Essen M, Krenning EP, Bakker WH, et al. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with foregut carcinoid tumours of bronchial, gastric and thymic origin. Eur J Nucl Med Mol Imaging 2007;34:1219-27. 10.1007/s00259-006-0355-4 [DOI] [PubMed] [Google Scholar]
- 64.Ianniello A, Sansovini M, Severi S, et al. Peptide receptor radionuclide therapy with (177)Lu-DOTATATE in advanced bronchial carcinoids: prognostic role of thyroid transcription factor 1 and (18)F-FDG PET. Eur J Nucl Med Mol Imaging 2016;43:1040-6. 10.1007/s00259-015-3262-8 [DOI] [PubMed] [Google Scholar]
- 65.Mariniello A, Bodei L, Tinelli C, et al. Long-term results of PRRT in advanced bronchopulmonary carcinoid. Eur J Nucl Med Mol Imaging 2016;43:441-52. 10.1007/s00259-015-3190-7 [DOI] [PubMed] [Google Scholar]
- 66.Ito T, Jensen RT. Imaging in multiple endocrine neoplasia type 1: recent studies show enhanced sensitivities but increased controversies. Int J Endocr Oncol 2016;3:53-66. 10.2217/ije.15.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morgat C, Velayoudom-Cephise FL, Schwartz P, et al. Evaluation of (68)Ga-DOTA-TOC PET/CT for the detection of duodenopancreatic neuroendocrine tumors in patients with MEN1. Eur J Nucl Med Mol Imaging 2016;43:1258-66. 10.1007/s00259-016-3319-3 [DOI] [PubMed] [Google Scholar]
- 68.Curtis C. Genomic profiling of breast cancers. Curr Opin Obstet Gynecol 2015;27:34-9. 10.1097/GCO.0000000000000145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579-86. 10.1200/JCO.2012.45.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res 2014;20:1698-705. 10.1158/1078-0432.CCR-13-2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reinert T, Scholer LV, Thomsen R, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016;65:625-34. [DOI] [PubMed] [Google Scholar]
- 72.Oberg K, Modlin I, DeHerder W, et al. Biomarkers for Neuroendocrine Tumor Disease: A Delphic Consensus assessment of Multianalytes, Genomics, Circulating Cells and Monoanalytes. Lancet Oncol 2015;16:e435046. [Google Scholar]
- 73.Khan MS, Tsigani T, Rashid M, et al. Circulating tumor cells and EpCAM expression in neuroendocrine tumors. Clin Cancer Res 2011;17:337-45. 10.1158/1078-0432.CCR-10-1776 [DOI] [PubMed] [Google Scholar]
- 74.Miller HC, Frampton AE, Malczewska A, et al. MicroRNAs associated with small bowel neuroendocrine tumours and their metastases. Endocr Relat Cancer 2016;23:711-26. 10.1530/ERC-16-0044 [DOI] [PubMed] [Google Scholar]
- 75.Li SC, Essaghir A, Martijn C, et al. Global microRNA profiling of well-differentiated small intestinal neuroendocrine tumors. Mod Pathol 2013;26:685-96. 10.1038/modpathol.2012.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li SC, Khan M, Caplin M, et al. Somatostatin Analogs Treated Small Intestinal Neuroendocrine Tumor Patients Circulating MicroRNAs. PLoS One 2015;10:e0125553. 10.1371/journal.pone.0125553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gilad S, Lithwick-Yanai G, Barshack I, et al. Classification of the four main types of lung cancer using a microRNA-based diagnostic assay. J Mol Diagn 2012;14:510-7. 10.1016/j.jmoldx.2012.03.004 [DOI] [PubMed] [Google Scholar]
- 78.Rapa I, Votta A, Felice B, et al. Identification of MicroRNAs Differentially Expressed in Lung Carcinoid Subtypes and Progression. Neuroendocrinology 2015;101:246-55. 10.1159/000381454 [DOI] [PubMed] [Google Scholar]
- 79.Singh R, Ramasubramanian B, Kanji S, et al. Circulating microRNAs in cancer: Hope or hype? Cancer Lett 2016;381:113-21. 10.1016/j.canlet.2016.07.002 [DOI] [PubMed] [Google Scholar]
- 80.Muscarella LA, D'Alessandro V, la Torre A, et al. Gene expression of somatostatin receptor subtypes SSTR2a, SSTR3 and SSTR5 in peripheral blood of neuroendocrine lung cancer affected patients. Cell Oncol (Dordr) 2011;34:435-41. [DOI] [PubMed] [Google Scholar]
- 81.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 82.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 83.Modlin I, Drozdov I, Kidd M. The Identification of gut neuroendocrine tumor disease by multiple synchronous transcript analysis in blood. PLoS One 2013;8:e63364. 10.1371/journal.pone.0063364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kidd M, Drozdov I, Modlin I. Blood and tissue neuroendocrine tumor gene cluster analysis correlate, define hallmarks and predict disease status. Endocr Relat Cancer 2015;22:561-75. 10.1530/ERC-15-0092 [DOI] [PubMed] [Google Scholar]
- 85.Walenkamp A, Crespo G, Fierro Maya F, et al. Hallmarks of gastrointestinal neuroendocrine tumours: implications for treatment. Endocr Relat Cancer 2014;21:R445-60. 10.1530/ERC-14-0106 [DOI] [PubMed] [Google Scholar]
- 86.Modlin I, Drozdov I, Alaimo D, et al. A multianalyte PCR blood test outperforms single analyte ELISAs for neuroendocrine tumor detection Endocr Relat Cancer 2014;21:615-28. 10.1530/ERC-14-0190 [DOI] [PubMed] [Google Scholar]
- 87.Modlin IM, Frilling A, Salem RR, et al. Blood measurement of neuroendocrine gene transcripts defines the effectiveness of operative resection and ablation strategies. Surgery 2016;159:336-47. 10.1016/j.surg.2015.06.056 [DOI] [PubMed] [Google Scholar]
- 88.Cwikla JB, Bodei L, Kolasinska-Cwikla A, et al. Circulating transcript analysis (NETest) in GEP-NETs treated with Somatostatin Analogs defines Therapy. J Clin Endocrinol Metab 2015;8. [DOI] [PubMed] [Google Scholar]
- 89.Pavel M, Jann H, Prasad V, et al. NET Blood Transcript Analysis defines the Crossing of the Clinical Rubicon: When Stable Disease becomes Progressive. Neuroendocrinology 2017;104:170-82. [DOI] [PubMed] [Google Scholar]
- 90.Lewczuk A, Min Chung K, Kolasinska-Cwikla A, et al. editors. Blood Gene Transcript Analysis Diagnoses Bronchopulmonary NETs and Identifies Progressive Disease. ENETs; 2016; Barcelona: Neuroendocrinology. [Google Scholar]
- 91.Dracopoli NC, Boguski MS. The Evolution of Oncology Companion Diagnostics from Signal Transduction to Immuno-Oncology. Trends Pharmacol Sci 2017;38:41-54. [DOI] [PubMed] [Google Scholar]
- 92.Filosso P, Roffinella M, Lewczuk A, et al. Utility of a circulating NET mRNA gene signature for defining bronchopulmonary neuroendocrine neoplasia and evaluating surgical efficacy. Interactive Journal for Cardiovascular and Thoracic Surgery 2017;Suppl:A27. [Google Scholar]