

WHY IS THERE A NEED FOR PATHOLOGY-SPECIFIC MOLECULAR PET IMAGING?
Traditionally, the diagnosis of AD and non-AD dementias (eg, frontotemporal dementia [FTD], primary progressive aphasia [PPA], and parkinsonian dementias) has primarily relied on characterizing clinical phenotypes through a detailed evaluation, which includes history taking, mental status, and neurologic examination, often supplemented by neuropsychological evaluation. This process often leads a clinician to understand that a patient’s clinical phenotype matches one of several broad categories defined by the predominant cognitive domain affected, such as memory, executive function, language, visuospatial function, or socioaffective function, with each pointing to a specific associated clinical diagnosis. For example, a patient presenting with progressive episodic memory deficits is most likely to have probable AD, whereas a patient presenting with progressive language impairments is most likely to have PPA. In some patients, characteristic motor abnormalities, such as parkinsonism or motor neuron disease features, can also offer valuable diagnostic clues. A patient presenting with executive dysfunction, visuospatial impairment, visual hallucinations, and parkinsonism is likely to receive a diagnosis of dementia with Lewy bodies (DLB).
After this formulation of the most likely clinical syndrome based on predominant clinical symptoms, clinicians turn toward imaging, most often structural MR imaging. Characteristic cortical atrophy patterns seen on a T1-weighted MR image can also strengthen the original diagnostic hypothesis, such as medial temporal lobe atrophy seen in AD dementia or temporal pole atrophy seen in the semantic variant of PPA. In some settings, FDG-PET is used in a similar fashion—to support the clinical hypothesis regarding the localization of neurodegeneration. This culminates in a clinical diagnosis of AD, FTD, DLB, or another condition that is probabilistically associated with the underlying pathology or pathologies most often underlying that condition. In some cases, this clinical diagnosis with supportive MR imaging or FDG-PET findings is highly predictive of underlying pathology. For example, in a patient with clinically typical semantic variant PPA1 or progressive supranuclear palsy (PSP), the pathology is highly likely to be TDP-43 or tau-PSP, respectively. In contrast, 10% to 20% of individuals diagnosed with clinically typical AD exhibit non-AD pathologies. And a patient diagnosed with the behavioral variant of FTD is nearly equally likely to exhibit tau or TDP-43 pathology.2 The term FTD is used to indicate the clinical dementia syndrome whereas the term FTLD is used to indicate the neuropathology.
Thus, although from the 1980s through the early 2000s this approach was the traditional cutting edge of clinical practice in the diagnosis of patients with cognitive impairment, there has been a pressing need for biomarkers of underlying neuropathology.3 As there are attempts to move toward developing therapies targeting specific molecular neurodegenerative pathologies, it has become clear that molecular biomarkers for inclusion and outcome measures are needed. Attention has been increasingly called to this issue as data accumulate demonstrating that there is no simple one-to-one relationship between a clinical phenotype and the specific pathology. For example, although a majority of patients with AD with amyloid plaques and neurofibrillary tangle pathology4 present with the classic progressive episodic memory impairment, 5% of those above age 65 and up to a third of those below age 65 present with an atypical phenotype, which can include frontal variant, posterior cortical atrophy (PCA), the logopenic variant of PPA (lvPPA), and cortico-basal syndrome (CBS).5–8 Conversely, a well-defined clinical syndrome can have several possible underlying neuropathologies. For example, the diagnosis of CBS during life is notoriously inaccurate in predicting corticobasal degeneration (CBD) pathology, because patients with CBS may turn out to have tau-CBD, tau-PSP, tau-Pick disease, or AD pathology.9 As for PPA, it is still challenging to predict the pathology for the agrammatic or lvPPA subtypes.10
Amyloid PET, a specific marker of one of the core features of AD pathology—neuritic amyloid plaques—has revolutionized the field of AD since its introduction more than a decade ago.11 The recently updated diagnostic criteria for AD dementia12 explicitly incorporate molecular biomarkers of disease pathology: amyloid PET and cerebrospinal fluid (CSF) amyloid and tau. Individuals with MCI who have a positive amyloid PET scan are generally viewed as having prodromal AD and are at greater likelihood than those with a negative amyloid PET scan to progress within a few years to dementia. Clinical trials are now targeting prodromal AD patients and multiple studies have used amyloid PET imaging to demonstrate a reduction of brain amyloid levels in patients treated with anti-amyloid antibody therapy (discussed later).
In the past few years, a series of studies has also begun to demonstrate potential uses of an imaging biomarker specific to tau pathology, which holds enormous promise not only for AD but also for other forms of dementia with underlying tauopathy, such as FTD. Unfortunately, despite tremendous effort, there are still not robust leads for biomarkers of TDP-43 or α-synuclein. This article discusses the clinical utility and research applications of amyloid and tau PET imaging and how they are enhancing knowledge of the basic pathophysiologic processes underlying various dementias and accelerating efforts aiming to develop effective disease-modifying therapies.
AMYLOID PET IMAGING
Clinical Use of Amyloid PET Imaging in Patients with Cognitive Impairment
[11C]Pittsburgh compound B ([11C]PiB), the first PET radiotracer specifically binding amyloid beta (Aβ) plaques, was introduced in 2004.11 [11C]PiB retention is specific for Aβ neuritic plaque pathology; it does not bind to other protein deposits that frequently co-occur with amyloid, such as neurofibrillary tangles or α-synuclein.13 Numerous studies over the years have demonstrated its validity by showing it to be highly correlated with in vivo CSF Aβ42 as well as Aβ pathology in autopsy specimens.14 Despite the many in vivo and postmortem validation studies in its favor, one major drawback of [11C]PiB is its very short half-life of 20 minutes, limiting its use to centers with an on-site cyclotron for radiotracer production. Subsequent efforts have led to the development of a family of fluorinated compounds with longer half-lives, including [18F]florbetapir, [18F]flutametamol, and [18F]florbetaben. [18F]florbetapir was the first of these tracers to be approved by the US Food and Drug Administration in 2012, with [18F]flutametamol and [18F]florbetaben approved in 2013 and 2014, respectively. These tracers were approved after studies demonstrated that clinical readers could reliably interpret the scans in patients with a range of cognitive impairment and that scan results were sensitive and specific predictors of the presence of neuritic plaques in a subset of patients who were followed to autopsy.
As more has been learned about amyloid PET imaging in the decade since its introduction, guidelines for its clinical applicability have been extensively discussed. The use of amyloid PET imaging is considered most appropriate when patients present with cognitive impairment that could be attributed to AD pathology but where the clinician is uncertain and the confirmation of presence or absence of amyloid would change diagnostic confidence.15 In other words, in a 70-year-old patient presenting with progressive episodic memory impairment with executive dysfunction, hippocampal atrophy, and temporo-parietal hypometabolism, amyloid PET is not particularly helpful because most clinicians consider this patient likely to have typical amnesic AD dementia, and, therefore, a positive amyloid PET scan would not significantly alter the already high diagnostic confidence. On the other hand, use of amyloid PET imaging is particularly relevant for patients presenting with phenotypes such as PCA, CBS, or PPA, in which AD is one of several possible underlying pathologies,16–18 and the results of an amyloid PET scan could substantially change diagnostic confidence. In the United States, although amyloid PET imaging is FDA approved, it is not yet reimbursed by payors, limiting access. A study was recently launched that enables dementia specialists to order amyloid PET scans for Medicare subscribers over the age of 65 who meet appropriate use criteria: Imaging Dementia — Evidence for Amyloid Scanning (www.ideas-study.org). The Centers for Medicare & Medicaid Services has authorized reimbursement of the cost of scans in patients who are enrolled in this study of the clinical utility of amyloid PET imaging.
The crucial role of amyloid PET imaging can be illustrated by the case of a right-handed woman who presented at age 65 with a 6-year history of progressive language difficulties. Her speech and language profile consisted of difficulty retrieving single words and repeating sentences and phonologic errors in confrontation naming and spontaneous speech but no single word comprehension, grammar, or motor speech impairment. Memory, executive function, and visuospatial function were relatively preserved, as was comportment. Hypometabolism on FDG-PET was seen in the left temporal lobe and to a lesser degree in the left frontal lobe, with sparing of the posterior cingulate cortex. A diagnosis of lvPPA was made. Although most cases of lvPPA have underlying AD pathology, confidence was lower in this patient due to mild agrammatism, frontal involvement, and sparing of posterior cingulate cortex, raising the question of FTLD pathology. The use of amyloid PET scan, which showed increased [11C]PiB uptake most pronounced and extensive in bilateral medial frontal cortices, confirmed the presence of abundant neuritic plaques (Fig. 1). CSF analysis demonstrated reduced Aβ and elevated phospho-tau. These molecular biomarkers strongly support the likely pathologic diagnosis of AD. Although measuring CSF amyloid is a potential alternative to amyloid PET imaging as a specific molecular biomarker for AD in clinical practice,19 it is a more invasive procedure than PET imaging, which limits its use in many practice settings.
Fig. 1.

Robustly elevated amyloid PET signal as measured with florbetapir PET in a patient with lvPPA viewed from the lateral surface (left) and medial surface (right).
Amyloid PET Imaging in Preclinical Alzheimer Disease and Incidental Amyloidosis
Models of the pathophysiologic sequence of AD suggest that accumulation of Aβ occurs early in the neurodegenerative process. The plot of amyloid burden versus time is sigmoidal in shape, and the average duration from detectable levels of amyloid in vivo to levels where amyloid accumulates as plaques is estimated at approximately 15 to 20 years.20 Numerous autopsy studies have shown that cognitively normal individuals followed prospectively to autopsy may have substantial amyloid plaque pathology,21–23 especially so with increasing age.
One important implication of this observation is that it is likely that older individuals with cognitive impairment may have incidentally positive amyloid PET scans. That is, multiple pathologies in a patient with dementia become increasingly common with age. In some patients with multiple pathologies, plaques and tangles are less prominent than another neurodegenerative pathology and thus may be viewed as not likely a major contributor to symptoms.4 The direct clinical implication is that, in a patient with cognitive impairment or dementia, a positive amyloid PET scan does not immediately indicate that AD is the underlying pathophysiologic process. A positive scan does not establish a diagnosis of AD. As with any test result, the clinician has to interpret an amyloid PET result in the context of the other information available. Discussions surrounding this issue were generated in part in relation to the presence of substantial brain amyloid in individuals who are cognitively normal.
It is important to recognize that it is not yet known whether, if they are followed for a long enough period of time, most or all amyloid-positive cognitively normal individuals go on to develop AD dementia. In cognitively normal older adults, there is a spectrum ranging from individuals harboring isolated cerebral amyloidosis with no evidence of neuronal injury (stage 1), to those with amyloid and neuronal injury (stage 2), to those with these 2 biomarkers as well as subtle symptoms of cognitive decline (not yet meeting criteria for MCI; stage 3).24 This issue has recently been discussed as requiring a distinction between a cognitively normal older adult with evidence of both brain amyloid and brain tau—referred to as preclinical AD—as opposed to a cognitively normal older adult with evidence of only brain amyloid or brain tau—referred to asymptomatic at-risk for AD.25
Using Amyloid PET for Treatment Development
In addition to its clinical utility, amyloid PET imaging has revolutionized therapy development for AD. The use of amyloid PET scans as part of the inclusion criteria is now ensuring that patients enrolled in the trials have the pathophysiologic disease targeted by the therapy under study. Although these trials have so far produced disappointing results in terms of cognitive benefit, some have demonstrated a modest reduction in cerebral amyloidosis26,27 and, recently, encouraging data were reported from a phase 1b study of the monoclonal antibody aducanumab.28 After 1 year of monthly intravenous infusions of this drug, patients with prodromal or mild AD exhibited dose-dependent reductions in brain amyloid, accompanied by preliminary evidence of clinical benefit from the Clinical Dementia Rating scale and the Mini-Mental State Examination. A phase 3 trial is now in progress.
Amyloid PET Imaging in Other Dementias
Dementia with Lewy bodies and Parkinson disease with dementia
Aβ plaques, a hallmark pathology of AD, are also commonly observed in DLB and Parkinson disease with dementia (PDD).29,30 Results of PET imaging studies using amyloid radiotracers agree with neuropathologic studies. DLB patients often have high levels of amyloid deposition compared with healthy controls.31 Although fewer PDD patients have significant Aβ accumulation, some of them may still demonstrate levels of amyloid deposition in the AD range.13 Higher amyloid deposition in DLB patients has been associated with greater cognitive impairment32 and better response to cholinesterase inhibitors.33 Higher amyloid deposition in Parkinson disease (PD) patients portends faster progression to dementia.34 In addition to these associations with clinical outcomes, high cortical amyloid deposition has been shown to predict greater cortical and medial temporal lobe atrophy, anatomic changes classically associated with AD pathology.35 Taken together, these findings suggest that the amyloidosis seen in DLB and PDD is not merely incidental but signifies that α-synucleinopathy may coexist with AD pathology. Even when the presence of amyloidosis does not meet criteria for a pathologic diagnosis of AD, PD patients with amyloidosis transition faster to dementia.36 This persistent relationship between the severity of amyloid burden measured via PET imaging and cognitive impairment in PD and DLB suggests that the cognitive decline in these patients may be precipitated by a synergy between amyloid and α-synuclein.
Frontotemporal dementia
Amyloid PET imaging is usually negative in patients with the clinical syndrome of FTD13,37; therefore, it can in theory be used to exclude AD when the suspected underlying pathology is FTLD, clinically manifesting as behavioral variant FTD (bvFTD) or PPA. It is more complicated, however, in practice. When a patient clinically diagnosed with FTD has a positive amyloid scan, there are 3 possible explanations, as discussed previously: (1) the pathologic diagnosis is AD (eg, frontal variant AD38); (2) there is coexisting FTLD and AD (ie, mixed pathology) that both contribute to dementia; and (3) the amyloid in the patient is incidental with relatively low Braak-stage neurofibrillary degeneration and thus likely noncontributory to symptoms. Differentiating these possibilities may be possible with the clinical symptomatology itself and MR imaging or FDG-PET (eg, the prior probability that a patient with classical semantic variant PPA supported by MR imaging or FDG-PET has TDP-43 type C pathology is so high that a positive amyloid PET scan should be interpreted with caution). In many cases of suspected FTLD with a positive amyloid PET scan, however, an examination of CSF tau or tau PET may be helpful, which is discussed.
TAU PET IMAGING
Clinical Use of Tau PET Tracers in Alzheimer Disease
Although amyloid PET imaging has contributed tremendously to increasing diagnostic confidence in dementia patient evaluation, neuropathologic studies have long shown that the localization and magnitude of tau pathology correlate better than amyloid with neurodegeneration and clinical symptoms.39 This has been confirmed with in vivo PET studies showing that the topography of amyloid deposition does not relate to clinical symptoms, especially in atypical subtypes of AD (eg, PCA, CBS, lvPPA, and frontal variant).40–43
Motivated in part by these issues, substantial effort has been devoted to the development of radiotracers to measure tau-related neurodegeneration. Most of these efforts to date have started by identifying compounds that bind to hyperphosphorylated paired helical filament (PHF) tau in AD tissue. In the past few years, 3 main chemical classes of tau PET tracers, quinolone derivatives, benzothiazole derivatives, and benzimidazole pyrimidines, were developed and shown to selectively bind to PHF tau pathology.44 Recently, an additional 3-pyrrolo[2,3]pyridine compound has also shown promise in an in vitro study.45 Table 1 lists the individual radiotracers in each chemical class.
Table 1.
Tau radiotracers
| Chemical Class | Radiotracer |
|---|---|
| Quinolone derivatives | [18F]THK523 [18F]THK5117 [18F]THK5105 [18F]THK5351 |
| Benzothiazole derivatives | [11C]PBB3 |
| Benzimidazole pyrimidines | [18F]AV-1451 or flortaucipir (previously known as [18F]T807) [18F]T808 |
| 3-pyrrolo[2,3] pyridine | [18F]MK-6240 |
Tau PET imaging in Alzheimer disease
Of the tracers available to date, [18F]AV-1451 (previously known as [18F]T807) has generated the most published data. The first in vivo images of [18F]AV-1451 showed the expected uptake in temporal and parietal regions in patients with AD dementia and amnesic MCI likely of AD pathology.46 Subsequently, it was also shown in larger samples that the topographic distribution of [18F] AV-1451 in amnesic MCI or AD dementia, compared with normal aging, was consistent with Braak staging, with more prominent tracer binding in inferior and lateral temporoparietal cortices, parieto-occipital cortices, posterior cingulate cortices, and precuneus and less in frontal regions and primary sensorimotor cortices.47,48 Similar results have been observed in AD dementia patients using other tau PET tracers, including [18F]THK535149 and [11C]PBB3.50
As predicted by previous postmortem studies, tau pathology as indexed by [18F]AV-1451 binding also better correlated with severity and type of clinical symptoms than amyloid pathology. In typical amnesic AD dementia, [18F]AV-1451 binding in inferior temporal gyrus correlated better with clinical impairment than [11C]PiB.47 The topography of tau PET signal differentiated between atypical AD phenotypes, such as PCA, CBS, lvPPA, and behavioral/dysexecutive phenotypes, with each demonstrating a distinct topography of [18F]AV-1451 binding.51,52 The authors have made similar observations (Fig. 2).53
Fig. 2.
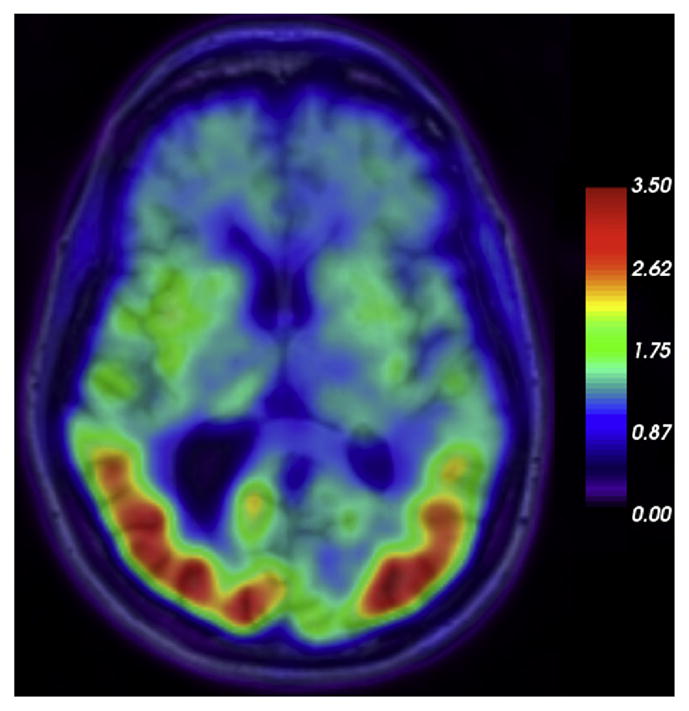
[18F]AV-1451 PET superimposed to MR imaging in a case of PCA with underlying Alzheimer dementia pathology; color bar displays legend for SUVR (standard uptake value ratios).
Ossenkoppele and colleagues51,54 also found that, in patients with atypical AD subtypes, compared with [11C]PiB, topography of [18F]AV-1451 binding colocalized better with hypometabolism measured via FDG-PET, and the degree of tracer binding correlated better with the severity of hypometabolism. The authors corroborated this finding, showing that [18F]AV-1451 topography and severity colocalized and correlated better with cortical atrophy than [11C]PiB in patients with atypical subtypes of AD.53 Based on these and other observations, not only will tau PET likely be a critical additional clinical tool in diagnosing atypical subtypes of AD with more confidence but also it will complement amyloid imaging because it colocalizes with neurodegenerative lesions predictive of clinical symptoms much better than amyloid PET imaging.
Tau PET Imaging in Frontotemporal Dementia
Tau PET also presents opportunities to improve clinical care and research for the spectrum of disorders encompassed by FTLD pathologies, which includes primary tauopathies (Pick disease, PSP, and CBD) as well as TDP-43 proteinopathies. Although some patients with FTD have autosomal-dominant genetic mutations that allow the likely pathology to be inferred, most patients with FTD have a sporadic condition, which presents important challenges in considering the possible molecular pathology. For example, bvFTD is just as likely to result from tau pathology (eg, classic Pick disease) as it is from TDP-43 pathology. Thus, an obvious and critically important goal is to determine whether tau PET imaging is useful in identifying sporadic bvFTD patients who are tau-positive versus those who are tau-negative.
For the most part, however, the radioligands developed to date have been screened against PHF-tau in AD tissue. Tau deposits in non-AD tauopathies differ in biochemical and conformational properties; radiotracers that bind to the PHF-tau found in AD may not bind as avidly to straight filaments found in PSP and CBD or to twisted filaments found in Pick disease.44 Initial postmortem studies demonstrate minimal or no binding of [18F]AV1451 to non-AD tauopathies,55,56 suggesting that current tracers will not likely generalize to FTLD. Preliminary in vivo studies, however, have also shown [18F]AV-1451 to have increased uptake in frontal and temporal cortices of patients with FTLD, a pattern expected for distribution of tauopathy in FTLD.57,58 Investigators are actively pursuing questions regarding why elevated signal has been seen in vivo but minimally or not at all in postmortem autoradiographic analyses and why elevated signal has been observed in vivo in some patients highly likely not to have a primary FTLD tauopathy (eg, semantic variant PPA). Other tracers, including [18F] THK5351, are being investigated in the FTLD spectrum as well.
Tau PET Imaging in Lewy Body Disorders
Tau PET radiotracers can also be of value in DLB and PD, in which an AD-like tauopathy frequently coexists with the primary α-syncleinopathy. One recent study examining cortical tau deposition in cognitively impaired patients with PD and DLB showed that they do have increased in vivo cortical tau accumulation compared with healthy controls as measured by [18F]AV-1451.59 Although the levels of tau radiotracer binding were lower than that seen in AD patients, the topographic distribution of tau was similar to that seen in AD. In addition, the degree of cortical tau binding in the inferior temporal gyrus and precuneus, areas known to be particularly affected in AD, correlated with severity of cognitive impairment as measured by the Mini-Mental State Examination and Clinical Dementia Rating. This is in agreement with what is predicted from prior neuropathologic studies, which have shown a relationship between cortical tau deposition with cognitive impairment in Lewy body disease.60 Even in some patients where PiB binding was low, higher than normal [18F]AV-1451 deposition was detected, suggesting that amyloid and tau pathologies may occur through independent pathways in a subset of patients with Lewy body disease.
Primary Age-Related Tauopathy
Abnormal tau also occurs in clinically normal individuals, with its prevalence increasing dramatically with age. It can be detected in the medial and inferior temporal lobe (Braak stages I/II) in 10% of 20 year olds, 80% of 60 year olds, and 95% of 80 year olds, in the absence of amyloid pathology.39,61 This pathologic entity has recently been named primary age-related tauopathy.62 It is beginning to be studied in vivo, with some evidence that elevated tau PET signal can be seen in these brain regions in people who have bio-markers consistent with low amyloid.47
FUTURE DIRECTIONS
Although the pathogenesis of tau and Aβ pathologies are thought to be partially independent processes, some investigators hypothesize that the development of Aβ pathology accelerates the low-level tauopathy that may preexist, promoting the spread of tau pathology from subcortical or archicortical regions to isocortical areas, ultimately emerging as AD dementia.63 Now that robust PET tracers are available for both pathologies, unprecedented experiments to better understand the independent and synergistic contributions of amyloid and tau in AD pathogenesis will be able to be performed. In addition, as efforts grow to develop therapeutic interventions aimed at tau pathology,64–66 tau PET imaging will be a crucial biomarker to be used in these clinical trials.
Initial studies using tau PET have shown that, compared with amyloid pathology, tau pathology better predicts the localization and severity of neurodegeneration seen on structural MR imaging and FDG-PET. Future longitudinal studies using multimodal imaging may reveal the sequence and timing of how these events (ie, appearance of amyloid and tau pathology, change in metabolism, and reduction in cortical volume) unfold over the course of the neurodegenerative process. Eventually, combining tau PET imaging with functional MR imaging and diffusion tensor imaging may allow hypotheses to be tested about how hyperphosphorylated tau pathology relates to and may spread through the complex network architecture of the brain. The use of amyloid and tau PET in conjunction with functional brain imaging in cognitively normal individuals will help illuminate the processes present in the earliest preclinical phases of AD and related disorders. Finally, tau PET imaging will also likely contribute in important ways to clinical assessment of patients with disorders of cognition. The arsenal of weapons to fight these devastating diseases has never been stronger.
KEY POINTS.
Amyloid PET imaging is a crucial biomarker for accurate diagnosis and therapy development for Alzheimer disease (AD).
Tau PET imaging will complement amyloid PET imaging for AD and serve as a valuable biomarker for non-AD tauopathies, such as frontotemporal lobar degeneration (FTLD).
Molecular PET imaging can allow examining longitudinal progression of amyloid and tau pathology in individuals who are cognitively normal or who have mild cognitive impairment (MCI) to better understand risk factors for and transition to dementia.
Multimodalstudies, including amyloidand tau PET imaging, willallow examining how molecular pathologies interact with other markers of neural integrity, such as glucose metabolism (fluorodeoxyglucose [FDG]-PET), functional connectivity or task-related activation (functional MR imaging), and gray and white matter structural integrity (structural MR imaging or diffusion tensor imaging).
Footnotes
Disclosure Statement: Dr B.C. Dickerson has received consulting fees from Merck and Piramal. Dr C. Xia has nothing to disclose.
References
- 1.Gorno-Tempini ML, Hillis A, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickerson BC, Sperling RA. Neuroimaging bio-markers for clinical trials of disease-modifying therapies in Alzheimer’s disease. NeuroRx. 2005;2(2):348–60. doi: 10.1602/neurorx.2.2.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman BT, Phelps CH, Beach TG, et al. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balasa M, Gelpi E, Antonell A, et al. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology. 2011;76(20):1720–5. doi: 10.1212/WNL.0b013e31821a44dd. [DOI] [PubMed] [Google Scholar]
- 6.Snowden JS, Stopford CL, Julien CL, et al. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex. 2007;43(7):835–45. doi: 10.1016/s0010-9452(08)70683-x. [DOI] [PubMed] [Google Scholar]
- 7.Koedam EL, Lauffer V, Van Der Vlies AE, et al. Early-versus late-onset Alzheimer’s disease: more than age alone. J Alzheimers Dis. 2010;19(4):1401–8. [Google Scholar]
- 8.Warren JD, Fletcher PD, Golden HL. The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol. 2012;8(8):451–64. doi: 10.1038/nrneurol.2012.135. [DOI] [PubMed] [Google Scholar]
- 9.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed cortico-basal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- 10.Grossman M. Primary progressive aphasia: clinico-pathological correlations. Nat Rev Neurol. 2010;6(2):88–97. doi: 10.1038/nrneurol.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 12.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villemagne VL, Ong K, Mulligan RS, et al. Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. J Nucl Med. 2011;52(8):1210–7. doi: 10.2967/jnumed.111.089730. [DOI] [PubMed] [Google Scholar]
- 14.Jack CR, Barrio JR, Kepe V. Cerebral amyloid PET imaging in Alzheimer’s disease. Acta Neuropathol. 2013;126(5):643–57. doi: 10.1007/s00401-013-1185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging task force, the society of nuclear medicine and molecular imaging, and the Alzheimer’s association. J Nucl Med. 2013;54(3):476–90. doi: 10.2967/jnumed.113.120618. [DOI] [PubMed] [Google Scholar]
- 16.Renner JA, Burns JM, Hou CE, et al. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology. 2004;63(7):1175–80. doi: 10.1212/01.wnl.0000140290.80962.bf. [DOI] [PubMed] [Google Scholar]
- 17.Ling H, O’Sullivan SS, Holton JL, et al. Does cortico-basal degeneration exist? A clinicopathological reevaluation. Brain. 2010;133(7):2045–57. doi: 10.1093/brain/awq123. [DOI] [PubMed] [Google Scholar]
- 18.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. 2008;63(6):709–19. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tapiola T, Alafuzoff I, Herukka S-K, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as bio-markers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382–9. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 20.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price JL, McKeel DW, Buckles VD, et al. Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30(7):1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price JL, Davis PB, Morris JC, et al. The distribution of tangles, plaques and related inmunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 1991;12(4):295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 23.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” alzheimer’s disease. Ann Neurol. 1999;45(3):358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 24.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323. doi: 10.1016/j.jalz.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–33. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-?? load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 28.Sevigny J, Chiao P, Bussiére T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–6. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 29.Horvath J, Herrmann FR, Burkhard PR, et al. Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat Disord. 2013;19(10):864–8. doi: 10.1016/j.parkreldis.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Colom-Cadena M, Gelpi E, Charif S, et al. Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2013;72(12):1203–12. doi: 10.1097/NEN.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 31.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71(12):903–10. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord. 2012;27(8):965–73. doi: 10.1002/mds.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graff-Radford J, Boeve BF, Pedraza O, et al. Imaging and acetylcholinesterase inhibitor response in dementia with Lewy bodies. Brain. 2012;135(8):2470–7. doi: 10.1093/brain/aws173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomperts S, Locascio J, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. 2013;80(1):85–91. doi: 10.1212/WNL.0b013e31827b1a07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimada H, Shinotoh H, Hirano S, et al. B-amyloid in lewy body disease is related to Alzheimer’s disease-like atrophy. Mov Disord. 2013;28(2):169–75. doi: 10.1002/mds.25286. [DOI] [PubMed] [Google Scholar]
- 36.Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23(3):295–7. doi: 10.1097/WAD.0b013e31819c5ef4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engler H, Santillo AF, Wang SX, et al. In vivo amyloid imaging with PET in frontotemporal dementia. Eur J Nucl Med Mol Imaging. 2008;35(1):100–6. doi: 10.1007/s00259-007-0523-1. [DOI] [PubMed] [Google Scholar]
- 38.Johnson JK, Head E, Kim R, et al. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999;56(10):1233–9. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 39.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136(3):844–58. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolk DA, Price JC, Madeira C, et al. Amyloid imaging in dementias with atypical presentation. Alzheimers Dement. 2012;8(5):389–98. doi: 10.1016/j.jalz.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabinovici GD, Jagust WJ, Furst AJ, et al. Aβ amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64(4):388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenbloom MH, Alkalay A, Agarwal N, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology. 2011;76(21):1789–96. doi: 10.1212/WNL.0b013e31821cccad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villemagne VL, Fodero-Tavoletti MT, Masters CL, et al. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14(1):114–24. doi: 10.1016/S1474-4422(14)70252-2. [DOI] [PubMed] [Google Scholar]
- 45.Hostetler ED, Walji AM, Zeng Z, et al. Preclinical characterization of 18F-MK-6240, a promising positron emission tomography (PET) tracer for in vivo quantification of human neurofibrillary tangles (NFTs) J Nucl Med. 2016;57:1599–607. doi: 10.2967/jnumed.115.171678. [DOI] [PubMed] [Google Scholar]
- 46.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-Tau Ra-dioligand [F-18]-T807. J Alzheimers Dis. 2013;34(2):457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 47.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110–9. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971–82. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harada R, Okamura N, Furumoto S, et al. 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer’s disease. J Nucl Med. 2015;57(2):1–43. doi: 10.2967/jnumed.115.164848. [DOI] [PubMed] [Google Scholar]
- 50.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79(6):1094–108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain. 2016;139(Pt 5):1551–67. doi: 10.1093/brain/aww027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ossenkoppele R, Schonhaut DR, Baker SL, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2015;77(2):338–42. doi: 10.1002/ana.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia C, Makaretz SJ, Caso C, et al. Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol. 2017 doi: 10.1001/jamaneurol.2016.5755. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138:2732–49. doi: 10.1093/brain/awv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marquié M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78(5):787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sander K, Lashley T, Gami P, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement. 2016;12(11):1116–24. doi: 10.1016/j.jalz.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 57.Ghetti B, Oblak AL, Boeve BF, et al. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dickerson B, Domoto-Reilly K, Daisy S, et al. Imaging tau pathology in vivo in Ftld: initial experience with [18F] T807 pet. Alzheimers Dement. 2014;10(Suppl 4):P115. [Google Scholar]
- 59.Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau positron emission tomographic imaging in lewy body diseases. JAMA Neurol. 2016;2129:1–8. doi: 10.1001/jamaneurol.2016.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 2008;115(4):427–36. doi: 10.1007/s00401-008-0347-5. [DOI] [PubMed] [Google Scholar]
- 61.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42(9):1681–8. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 62.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–66. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Musiek ES, Holtzman DM. Origins of Alzheimer’s disease. Curr Opin Neurol. 2012;25(6):715–20. doi: 10.1097/WCO.0b013e32835a30f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giacobini E, Gold G. Alzheimer disease therapy–moving from amyloid-β to tau. Nat Rev Neurol. 2013;9(12):677–86. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- 65.Selkoe DJ. The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann Neurol. 2013;74(3):328–36. doi: 10.1002/ana.24001. [DOI] [PubMed] [Google Scholar]
- 66.Yoshiyama Y, Lee VMY, Trojanowski JQ. Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry. 2013;84(7):784–95. doi: 10.1136/jnnp-2012-303144. [DOI] [PMC free article] [PubMed] [Google Scholar]