

Abstract
New in vitro technologies that assess neuronal excitability and the derived synaptic activity within a controlled microenvironment would be beneficial for the characterisation of compounds proposed to affect central nervous system (CNS) function. Here, a microfluidic system with computer controlled compound perfusion is presented that offers a novel methodology for the pharmacological profiling of CNS acting compounds based on calcium imaging readouts. Using this system, multiple applications of the excitatory amino acid glutamate (10 nM–1 mM) elicited reproducible and reversible transient increases in intracellular calcium, allowing the generation of a concentration response curve. In addition, the system allows pharmacological investigations to be performed as evidenced by application of glutamatergic receptor antagonists, reversibly inhibiting glutamate-induced increases in intracellular calcium. Importantly, repeated glutamate applications elicited significant increases in the synaptically driven activation of the adjacent, environmentally isolated neuronal network. Therefore, the proposed new methodology will enable neuropharmacological analysis of CNS active compounds whilst simultaneously determining their effect on synaptic connectivity.
Introduction
The burden of CNS disorders, combined with an increasingly aged population, is one of the key global health challenges of our time. Indeed, the World Health Organization has highlighted that up to 1 billion people suffer from neurological disorders and those suffering from dementia, currently exceeding 20 million, are expected to double every 20 years unless action is taken1. Despite global R&D pharmaceutical spending in brain disorders exceeding $140 billion per annum, and its growth set to continue2, early stage drug discovery has serious cost implications and high attrition rates, as investigational CNS-leads fail due to the well-known bottleneck in late stage clinical trials3. A recent meta-analysis on the clinical success rates of drugs in the development pipeline revealed that approximately, only 1 in 10 CNS active compounds eventually meets FDA approval4. The most common reasons for this level of attrition ranged from inadequate in vitro and in vivo models, to poor drug physicochemical properties and efficacy, in addition to in vivo toxicity4,5. Therefore, new technologies and methodologies for characterising CNS active compounds are a priority if healthcare and well-being are to be improved.
Early stage drug development heavily relies upon in vitro cultures, with neurons grown on coverslips prior to drug screening. However, when combined with plate-based high-throughput instrumentation, there is often failure in monitoring synaptic activity typical of the in vivo microenvironment6,7, as only single neuronal networks can be investigated with this approach. Standard laboratory in vitro techniques used to screen for CNS active compounds include electrophysiological assays, such as patch clamping and multi-electrode arrays (MEAs), as well as optical calcium (Ca2+) imaging techniques. Whilst the use of patch clamping provides exceptional temporal resolutions of neuronal activity in response to drug application8–10, it is time consuming, requires specialist training and is low throughput. Alternatively, cells can be grown across MEA substrates to obtain electrophysiological readouts across an entire neuronal network11, however the likelihood of a cell interfacing with a single electrode is low, and recordings may not detect subthreshold readings which would indicate if either excitatory or inhibitory signals were received12. The use of Ca2+ imaging instead allows repeated acute recordings of cellular activity without inducing toxicity, but the technique is typically constrained to low temporal resolutions. However, it provides single-cell spatial resolution over a wide cultured area and can be used to carry out cost-effective investigations of synaptic activity13.
The use of microfluidic procedures to model in vitro pathological conditions has gradually increased over the past decade, due to the level of control and manipulation available over the cellular microenvironment and experimental conditions14,15. These systems also show potential for use as drug discovery platforms, taking advantage of engineering techniques to facilitate high-throughput pharmacological assays, either by device design or the incorporation of perfusion systems to wash on/off multiple compounds16–19. Further advantages of microfluidic systems include low production costs, reduction in drug sample volumes, and the ability to analyse multiple, environmentally isolated but functionally connected cultures20–23, a degree of experimental capability that is lacking in conventional culturing techniques. Additionally, the transparent nature of poly-dimethylsiloxane (PDMS) based microfluidic devices make them ideal for carrying out both quantitative and qualitative pharmacological assays via Ca2+ imaging techniques20,21,24. As such, microfluidic systems are versatile tools that are well suited to perform pharmacological research15,20,21,25–27.
In this study, we present a microfluidic system that integrates computer-controlled perfusion of multiple compounds with Ca2+ imaging techniques to provide a platform for the characterisation of CNS active compounds. The novelty of this approach is its ability to simultaneously detect direct responses within a primary hippocampal culture to repeated drug applications whilst monitoring the consequent synaptic activity in an adjacent, functionally connected but environmentally isolated hippocampal culture. Hence, the proposed platform provides a novel, miniaturised solution amenable to CNS drug discovery, offering the ability to simultaneously screen both the direct effects of compounds on cells, as well as how such drugs influence communication between synaptically connected cultures.
Materials and Methods
Device fabrication and preparation
Microfluidic devices were fabricated using photo/soft-lithography techniques as described previously20, comprising of an array of microchannels between two culture chambers that are fluidically addressable via inlet/outlet wells. In brief, a two-layer microfluidic master was fabricated by spinning SU8 photoresist (3000 series, Microchem, US) onto a silicon wafer. The first layer (SU8 3010) created the microchannels (7 μm thick, 10 μm wide and 450 μm long), and the second layer (SU8 3035), the culture chambers (80 μm thick, 2 mm wide and 9.5 mm long). The silicon master was then silanized by vapour deposition of 1 H,1 H,2 H,2H-perfluorooctyl-trichlorosilane (Sigma Aldrich, UK) for 1 hour. PDMS was poured onto the master at a 10:1 ratio of base to curing agent and degassed in a vacuum desiccator prior to curing at 80 °C for 3 hours. The PDMS layer was then peeled off the master and devices were cut to the desired size, with the inlet/outlet wells (4 mm diameter) of each chamber created using a biopsy punch. Microfluidic devices were finally cleaned and irreversibly bonded to coverslips using oxygen plasma. Bonded devices were UV sterilised for 15 minutes, treated with poly-L-lysine solution (PLL, 10 μg ml−1) for 1.5 hours, and washed with sterile Neurobasal-A medium.
Microfluidic perfusion protocol
Using dual culture chamber microfluidic devices, a steady flow rate in a single culture chamber (perfused chamber; with a constant flow rate Q ch) was achieved by creating a constant hydrostatic pressure difference between its inlet and outlet wells. The adjacent culture chamber (naïve chamber) was kept in almost static fluid conditions. Polytetrafluoroethylene (PTFE) tubing (0.255 mm inner diameter, ~700 mm length, Cole-Parmer, UK) connected glass syringes to needle ports created through the PDMS wall of the inlet-outlet wells of the perfused chamber, and the fluid was actuated using syringe pumps (AL-1000, World Precision Instruments). Up to four syringe pumps were connected to one inlet well of the device for fluid injection, with only one of these syringe pumps active at any given time and set to a constant flow rate (Q I−P). Two further syringe pumps were used for fluid withdrawal at constant flow rates, one connected to the inlet (Q I−W) and one to the outlet (Q O−W) well, respectively (Fig. 1a). Syringe pumps were computer-controlled via an in-house developed MATLAB (R2015a) program and ensured reliable activation/deactivation of each flow rate at the desired times. At the beginning of the assay, the following conditions were set to achieve a steady flow rate in the perfused chamber (Q Ch):
| 1 |
| 2 |
These set of equations give rise to a self-adjusting hydrostatic pressure between the inlet and outlet wells of the perfusion chamber that reaches quickly a dynamic equilibrium. On/off alternation of the injection syringe pumps was only used to perfuse different solutions in the chamber. Due to the module of the flow rates in Eq. 1, a rapid exchange of the fluid solution in the inlet well was obtained without affecting its hydrostatic pressure and leading to a prompt injection of the new fluid solution into the chamber. The time to completely exchange a perfusate in the well was directly proportional to the inlet well’s hydraulic retention time (HRT), where the fluid volume of the well (V I−well) was replaced under a steady flow rate, resulting in HRT ≈ V I−well / Q I−P (estimated at ~25 seconds for the current setup). Therefore, for a given steady flow rate (Q Ch) in the perfusion chamber with a defined cross-section (S Ch), the time (T ex) taken for the full perfusate (i.e. 100% concentration) to travel a distance L within the chamber from the inlet well can be estimated as:
| 3 |
Under these conditions, T ex is the time required for the complete exchange of solution in the inlet well and the subsequent arrival of the new solution at an arbitrary point that is a distance L away from the inlet well within the perfused chamber. When L is half the length of the culture chamber, T ex is estimated at ~35 seconds for the current device geometry. To avoid cross contamination between the perfused and naïve chambers, the initial volume of fluid in the wells connecting the naïve and perfused chambers were adjusted to 50 μl/well and 10 μl/well, respectively. This established a hydrostatic pressure gradient across the array of microchannels that was maintained throughout the assay. Considering the ratio between the hydraulic resistance of the microchannel array and culture chambers20, the hydrostatic pressure gradient resulted in the formation of a negligible flow of vehicle from the naïve to the perfused chamber that did not influence the perfusion protocol.
Figure 1.

Development and characterisation of the microfluidic perfusion system. (a) Schematic representation of a dual chamber microfluidic device with inlet/outlet needle ports for the direct perfusion/withdrawal of fluid in/from the wells. Computer-controlled syringe pumps were used for fluid injection and withdrawal. (b) A representative image showing absence of cross contamination in the naïve chamber and microchannels during perfusion of calcein (50 µM) in the perfused chamber (dashed lines indicate the microchannel array separating the perfused chamber from the naïve chamber). (c) Fluorescent profiles in the perfused (c1) and naïve (c2) chambers, respectively upon repeated delivery of calcein solution from 3 different pumps. This demonstrated a lack of cross-contamination between the chambers and the robustness of the perfusion protocol (n = 9 devices). Data is expressed as mean ± S.E.M. (one-way ANOVA with post-hoc Tukey’s test. ***Denotes P < 0.001, relative to baseline readings).
The robustness of this perfusion model was validated experimentally using calcein as a fluorescent marker (Fig. 1b,c). Non-cultured microfluidic devices were connected to the perfusion setup, delivering deionised (DI) water through the culture chamber at a steady flow rate (4 μl min−1), with three other syringes primed with calcein (50 μM in DI water). Devices were then transferred to an inverted microscope (Axio Observer A1, Zeiss) using a 2.5× objective and the field of view was set to span an area that monitored both the microchannel array and the culture chambers. Recordings were carried out using an EMCCD camera (LucaR, Andor Technologies) at a frame rate of 2.0 Hz and an exposure time of 0.4 seconds. Time-lapsed images were obtained using Andor SOLIS.
A single region of interest (ROI) was selected for the naïve and perfused culture chamber to obtain mean fluorescent readouts on either side of the microchannel array in response to calcein perfusions. The raw data (as mean value of the ROI) obtained from individual devices was then scaled using the following equation:
| 4 |
Where x is the mean raw fluorescent data of each time trace/ROI (sampled at 2 Hz) and min and max are the absolute minimal and maximal mean fluorescent intensity values. Processed data from separate recordings across multiple devices were then averaged.
Primary hippocampal culture
Primary hippocampal cultures were prepared as described previously8,20,28. Briefly, Sprague Dawley rat pups (1–2 days old) were killed via cervical dislocation, in accordance with the UK Home Office guidelines. The brain was then removed, hippocampi dissected out and placed in dissection solution containing (in mM) NaCl 6.78, KCl 0.40, NaHCO3 2.18, NaH2PO4 0.16, MgSO4 0.15, glucose 4.5, CaCl2 0.22. The hippocampi were then incubated for 20 minutes in papain solution (1.5 mg ml−1; Sigma Alrdich, UK) diluted in dissection solution. The tissue was then dissociated via trituration in bovine serum albumin solution (BSA; 10 mg ml−1 in dissecting solution; Sigma Aldrich, UK), the cell solution was then spun down at 2000 rpm for 2 minutes and re-suspended in 1 ml of supplemented Neurobasal-A medium (L-glutamine 2 mM, B27 2% v/v; Life Technologies, UK) at a concentration of 5 × 106 cells ml−1. Cells were introduced into either one or both chambers of the device (~2.5 × 104 cells per inlet well) as required and incubated for 15 minutes before inlet/outlet wells were filled with 50 μl of supplemented media. Devices remained in a humidified incubator for up to 14 days in vitro (DIV) prior to experimentation, with media replenished every 2–3 days.
Assessment of perfusion effects on cell viability
Hippocampal cultures (12–14 DIV) in microfluidic devices were washed with a HEPES based external solution (HBS, containing, in mM: NaCl 140, KCl 5, MgCl2 2, HEPES 10, D-glucose 10, CaCl2 2; pH = 7.4; 310 mOsm). Cultured devices were connected to the perfusion setup and volumes adjusted to 50 μl and 10 μl in the wells connected to the naïve and perfused chamber, respectively. Subsequently, multiple values of steady-state flow rates (in a range of 0.4–10 μl min−1) were sustained in the perfused chambers for an hour and the cells were then stained for live/dead assessment. As control experiments, devices were also either left in stasis for an hour (Static t1h, with all wells containing 50 μl) or were stained for cell death immediately after removal from the incubator (Static t0h). Cultures were treated with propidium iodide (PI, 20 μM) & Hoechst (4.5 μM) made up in HBS for 30 minutes, then washed and allowed to recover for 15 minutes. Devices were then transferred to an inverted microscope (Axio Observer Z1, Zeiss), with bright-field and fluorescent images for each culture chamber captured using a 10× objective and a cooled CCD camera (AxioCam MRm, Zeiss), analysed with ZEN 2012 (Blue edition, Zeiss). Dead cells were identified by the co-staining of Hoechst and PI, whilst living cells were identified by the sole staining of Hoechst.
Immunocytochemistry
Microfluidic hippocampal cultures (12 DIV) were fixed and stained to identify neuronal and glial populations. Briefly, using a previously established protocol20, cultures were washed with phosphate buffered saline solution (PBS) and fixed with ice cold paraformaldehyde (4% w/v, 10 min), followed by ice cold methanol treatment (100%, 10 min). Cultures were then washed with PBS and permeabilised with Triton-X100 (0.01%, 10 min), prior to treatment with blocking solution (in % w/v; foetal bovine serum 5, BSA 1) for 1 hour to prevent non-selective binding. Cultures were incubated at 4 °C overnight with primary antibodies β-III-Tubulin (Life Technologies, UK; neuronal specific cytoskeleton marker; 1:500 dilution), glial fibrillary acidic protein (GFAP; Sigma Aldrich, UK; astrocytic marker; 1:500 dilution), synaptophysin (Millipore, UK; synaptic vesicle marker; 1:500 dilution) and MAP2 (Sigma Aldrich, UK; somatodendritic marker; 1:500 dilution). The cells were then washed with PBS, and incubated with the relevant fluorescently labelled secondary antibodies (Alexa 488 or Alexa 555; Life Technologies, UK; 1:200 dilution) for 1 hour at room temperature. Devices were then transferred to the inverted microscope (Axio Observer Z1, Zeiss), and imaged using 10×, 20× and 63× objectives with a cooled CCD camera (AxioCam MRm, Zeiss). Image analysis was performed using ZEN 2012 (Blue edition, Zeiss).
Calcium imaging
Data acquisition
Ca2+ imaging was performed on cultures using techniques described previously8,20,29. Briefly, microfluidic cultures (10–14 DIV) were loaded with the fluorescent calcium sensitive dye Fluo-4-AM (5 μM in HBS for 1 hour at room temperature) and washed with HBS (vehicle) before being transferred to an inverted microscope (Axio Observer A1, Zeiss). Devices were then connected to the perfusion setup, delivering HBS at a steady flow rate (4 μl min−1). Three other syringe pumps, primed with either glutamate (0.01–1 mM), glutamate antagonists (NBQX 20 μM, DL-AP5 100 μM, (R,S)-MCPG 500 μM) or glutamate (3 μM) in the presence of glutamate antagonists, were used to perfused drugs of interest at a steady flow rate of 4 μl min−1. Syringe pumps were primed with glutamate (3 μM), the Na+ channel blocker tetrodotoxin (TTX, 1 μM) or glutamate in the presence of TTX and the solutions were perfused at a steady flow rate of 4 μl min−1. Devices used to generate a glutamate concentration response curve were randomly assigned three separate glutamate concentrations that were perfused in order of increasing concentration. Simultaneous recordings of Ca2+ transients in the two environmentally isolated, independent cultures were obtained with a cooled EMCCD camera (LucaR, Andor Technologies) using either a 5× or 10× objective, at a frame rate of 2.0 Hz and exposure time of 0.4 s. L-glutamic acid (Tocris Bioscience, UK), the glutamate receptor antagonists (NBQX, DL-AP5 & (R,S)-MCPG; Abcam, UK) and tetrodotoxin (TTX citrate; Abcam, UK) were made up as 1000× stock solutions and diluted to final concentrations in vehicle solution on the day of experimentation. Time-lapse images were processed and analysed using Andor SOLIS, MATLAB (R2015a), and BioGraph (V2.5.6; J. Dempster, University of Strathclyde). As is standard, glutamate receptor antagonists or TTX were perfused during the resting period following the first glutamate perfusion and during the subsequent glutamate application to determine their effects on glutamate-induced Ca2+ transients. At the end of each experiment, the perfusion was stopped and KCl (25 mM final concentration) was applied to each chamber. Cells that did not elicit an immediate and sustained increase in fluorescence were excluded from analysis (see Supplementary Fig. S1).
Analysis
Regions of interest (ROI) were obtained from individual cell somas and the mean fluorescent intensity per ROI per frame was used to create a graphic readout of neuronal Ca2+ activity, with each cell’s response trace normalised to the basal value for each recording. To produce a concentration response curve, the mean peak magnitudes obtained from increasing glutamate concentrations were normalised to the mean peak magnitude obtained from the highest glutamate application (1 mM) and the concentration response curve generated using the Hill equation:
| 5 |
where RMax = maximum response observed, EC 50 = effective concentration at which 50% maximum response is obtained, and n = Hill coefficient. Otherwise, changes in neuronal activity in response to indirect stimulation from the directly perfused neural network were identified by the automated counting of Ca2+ events. The signals of each neuron obtained over the course of the assay were stitched together, filtered (moving average filter, window size 11) and normalised to the first 24 frames of the baseline recording. Events were counted on the basis of significant increases in fluorescence exceeding 7 standard deviations of the median baseline fluorescence, occurring within a moving window of 10 seconds. This provided the number of events taking place per neuron during both the basal period and glutamate perfusion of each recording, with results normalised to time and the indirect activity expressed as events/neuron/minute (ENM).
Statistics
All data is presented as mean ± S.E.M. using bar/scatter graphs and results compared using paired student’s t-tests, unpaired t-tests or one-way ANOVA with Tukey’s post-hoc comparison as appropriate, with differences considered significant when P < 0.05, or treated otherwise as non-significant (ns).
Results
A robust model for pharmacological perfusion assays
To assess the reproducibility of consecutive drug applications using the proposed system, initial experiments were performed using calcein to visualise perfusion performance and results confirmed a close match with the predicted flow behaviour (equations 1–3). The protocol developed produced a stable fluorescent plateau for the intended duration of perfusion (~30 seconds), with increasing (~35 seconds) and decreasing (~45 seconds) transients for all experiments (Fig. 1). Importantly, the absence of cross contamination between the two culture chambers was experimentally confirmed, demonstrating their fluidic isolation.
Having validated the robustness of the perfusion protocol, the effect of shear stress on the health of perfused hippocampal cultures (12–14 DIV) was examined (Fig. 2) to determine an optimal flow rate for the pharmacological study. As a benchmark, live/dead imaging was carried out on microfluidic cultures immediately after being removed from the incubator, with live cells accounting for 73.8 ± 0.01% (n = 3 devices, each device from a separate culture) of the total cell population (Fig. 2b). However, this decreased to 54.1 ± 0.9% (n = 6 devices from 3 cultures; P < 0.001, static t0h vs static t1h) of the total cell population when the devices were left at room temperature under static conditions for an hour. In contrast, constant perfusion at room temperature, with flow rates ranging from 0.4–10 μl min−1 (n = 3 devices, each device from a separate culture), revealed varying effects on cell viability after an hour, with live cells accounting for 72.6 ± 1.6% (P < 0.001 vs static t1h) of the total cell population at the optimal flow rate of 4 μl min−1 (Fig. 2b). Furthermore, the health of the directly perfused cultures was closely matched by that of the non-perfused naïve culture across all experimental conditions (non-significant, P > 0.05). Given these results, a flow rate of 4 μl min−1 was used for the remainder of this study. Finally, microfluidic cultures were stained for β-III-Tubulin, GFAP and synaptophysin revealing that only neuronal, but not astrocytic processes, were able to traverse across the microchannel array to the adjacent chamber and that synapses were formed both within and outside the microchannels (see Supplementary Fig. S2). Collectively, continuous perfusion experiments and immunocytochemistry imaging indicate that synaptically driven functional communication exists between the two hippocampal cultures, as previously reported20.
Figure 2.
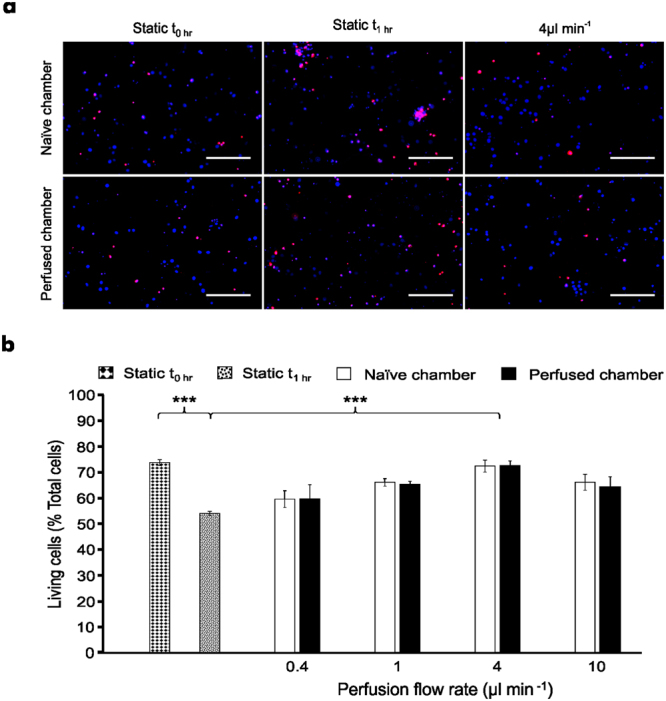
Continuous perfusion maintains cell viability between environmentally isolated neural networks. (a) Representative images obtained from perfused and naïve cultures which were stained with Hoechst (blue) and PI (red) following continuous perfusion experiments for 1 hour. Scale bar = 200 µm. (b) Microfluidic neural network viability increases in the presence of perfusion with respect to static condition after 1 hour, with an optimal flow rate of 4 μl min−1. Data are presented as mean ± S.E.M. with one-way ANOVA with post-hoc Tukey’s test performed; n = 21 devices from 4 cultures; ***denotes P < 0.001.
Perfusion permits the characterisation of CNS active compounds
As proof-of-concept work, to demonstrate the system’s ability to carry out pharmacological characterisation of neuroactive compounds, a concentration response curve was generated using the excitatory amino acid, glutamate. Perfusion of glutamate (10 nM – 1 mM) elicited reversible concentration-dependent increases in intracellular Ca2+ (Fig. 3a), resulting in a concentration response curve with an EC50 of 4.7 ± 0.6 μM (Fig. 3b).
Figure 3.

Microfluidic hippocampal cultures respond to direct glutamate (G) application in a concentration-dependent manner. (a) Ca2+ imaging trace representative of a single neuron’s response to perfusion of consecutive, increasing concentrations of glutamate (10–14 DIV). NFU = Normalised fluorescent unit. (b) Concentration response curve for glutamate (10 nm–1 mM) reveals an EC50 = 4.7 ± 0.6 μM. Data is presented as mean ± S.E.M (n ≥ 50 neurons per concentration point, curve obtained from 14 devices from 6 cultures).
Having established that our integrated perfusion system allows the generation of concentration response curves, we tested the platforms capability for the pharmacological screening of compounds. To this end, we investigated the actions of ionotropic and metabotropic glutamate receptor antagonists NBQX (20 μM), DL-AP5 (100 μM) and (R,S)-MCPG (500 μM) on glutamate (3 μM)-induced changes in intracellular Ca2+. Repeated glutamate perfusions elicited robust and reproducible increases in intracellular Ca2+ (n = 182 neurons, 4 devices, each device from a separate culture; Fig. 4a & b), which were inhibited in the presence of the glutamate antagonists to 17 ± 2.5% of the response elicited by glutamate alone (n = 219 neurons, 4 devices, each device from a separate culture; P < 0.001; Fig. 4c & d). In contrast, no significant difference in glutamate-induced increases of intracellular Ca2+ (n = 551 neurons, 3 devices, each device from a separate culture; Fig. 4e & f) were observed in the presence of TTX (1 μM).
Figure 4.

Reproducible glutamate responses are inhibited by glutamate antagonists. (a, c + e) Representative Ca2+ imaging traces of neuronal responses to repeated glutamate applications in the absence or presence of antagonists or TTX (purple trace: individual neuronal responses; red trace: average response). NFU = Normalised fluorescent unit. (b) Repeated applications of glutamate (3 μM) revealed a small but significant reduction in neuronal response during the third glutamate application. (d) The neuronal response to glutamate (3 μM) application was reversibly inhibited in the presence of glutamatergic antagonists (NBQX 20 μM, DL-AP5 100 μM & (R,S)-MCPG 500 μM). (f) The neuronal response to glutamate (3 μM) application was not significantly affected in the presence of TTX (1 μM). Data are presented as mean ± S.E.M. with one-way ANOVA with post-hoc Tukey’s test performed; n ≥ 189 neurons per application, from ≥3 devices from ≥3 separate cultures; *denotes P < 0.05 and ***denotes P < 0.001.
Modulation of neuronal activity in synaptically connected hippocampal cultures
Having previously demonstrated functional synaptic connectivity across two environmentally isolated hippocampal cultures following a single drug application20, here changes in postsynaptic responses in the naïve chamber were examined as a consequence of repeated glutamate (3 μM) applications in the directly perfused chamber (Fig. 5, see Supplementary Video S1).
Figure 5.

Repeated glutamate application induces increased neuronal activity in synaptically connected hippocampal cultures. (a) Ca2+ imaging traces representative of activity obtained from individual neurons in the directly perfused (trace D1) and naïve (traces I1–I3) culture chambers, demonstrating functional synaptic communication between independent hippocampal cultures. (b) Significant increases in the number of neuronal calcium events were observed in the naïve chamber, with respect to the basal (B) activity, in response to the three glutamate (G) applications in the perfused chamber. (c) Significant increases in the number of neuronal calcium events were observed in the naïve chamber, with respect to the basal (B) activity, in response to glutamate applications (G) in the absence and presence of glutamate antagonists in the perfused chamber. (d) Basal activity in the naïve chamber was significantly increased in the presence of TTX in the perfused chamber. Further changes in the number of neuronal calcium events were not observed following glutamate (G) application in the presence of TTX. Data are presented as mean ± S.E.M. with paired student’s t-test performed; n = ≥ 222 neurons per application, ≥3 devices from ≥3 separate cultures; ns denotes P > 0.05, *denotes P < 0.05, **denotes P < 0.01, ***denotes P < 0.001 and #denotes P < 0.05.
During the first glutamate application, neuronal activity within the naïve chamber significantly increased from a baseline of 0.16 ± 0.04 ENM to 0.38 ± 0.04 ENM (P < 0.001; n = 222 neurons; Fig. 5b). Consistent with this, the second glutamate application also induced a significant increase in neuronal activity from 0.17 ± 0.04 ENM to 0.44 ± 0.04 ENM during glutamate application (P < 0.001). Similarly, the final glutamate application elicited a significant increase in activity, from 0.27 ± 0.05 ENM to 0.4 ± 0.05 ENM (P > 0.05). In the presence of glutamate antagonists in the perfused chamber, neuronal activity was unaltered in the naïve chamber (Fig. 5c). A significant elevation in neuronal activity in the naïve chamber was observed following glutamate application in the presence of glutamate antagonists, from a baseline of 0.19 ± 0.03 ENM to 0.42 ± 0.04 ENM (P < 0.001; n = 350 neurons; Fig. 5c. In contrast, baseline activity in the naïve chamber was significantly increased from 0.10 ± 0.02 ENM to 0.22 ± 0.03 ENM (P < 0.01; B1 vs B2; n = 381 neurons; Fig. 5d) with the presence of TTX in the directly perfused chamber, whereas glutamate application in the presence of TTX did not induce any further increases in neuronal activity in the naïve chamber (Fig. 5d). Finally, in order to confirm that changes in neuronal activity were unidirectional, hippocampal cultures were grown in one chamber only and glutamate perfused in the non-cultured chamber. Repeated application of glutamate (3 μM) in the non-cultured chamber resulted in no changes in neuronal activity in the cultured chamber (see Supplementary Fig. S3).
Discussion
This study presents the first successful integration of microfluidic perfusion with Ca2+ imaging techniques into a platform that allows the pharmacological characterisation of CNS active compounds using primary hippocampal cultures. The novelty of this approach is in examining simultaneously both the direct response to drug application (i.e. neuronal excitability) and the consequent alterations in neuronal activity in synaptically connected but environmentally isolated primary hippocampal cultures (i.e. induced synaptic activity), over customisable and repeatable conditions.
In all the experiments, the diffusive transport of compounds could be neglected with respect to their convective transport in the perfused chamber and microchannel array, as the Péclet number30 was always >100 and the Reynolds number was always <1. Absence of cross-contamination between chambers was experimentally confirmed using fluorescent imaging and the results were in line with previously established models20,31–33. Importantly, interchangeable perfusion of different solutions through a single culture chamber was achieved without disturbance in the flow. This was obtained by rapidly replacing the liquid volume inside the inlet open well of the perfusion chamber, selecting flow rate values that were much greater than that created in the perfused chamber. The open well system, in combination with carefully selected values of the flow rates (equations 1–2), generated a self-adjusting hydrostatic pressure difference between inlet and outlet wells of the perfused chamber that maintained a constant flow rate in the perfused chambers for the duration of the experiment. If the flow rate assigned to the inlet well had been the same as that of the perfused chamber, then the time taken to completely change perfusates would be significantly longer (~5 minutes), which highlights the need for the faster flow rate at the inlet well. This approach enabled a quick fluid transition without creating a pulsatile flow when exchanging buffer and drug solutions. Further improvements to the platform could be made by replacing the syringe pump setup with a pressure-driven, valve-controlled perfusion system, thus extending the number of drugs that can be tested in the system, as well as reducing the size and number of external instrumentation needed for active flow control.
Microfluidic perfusion has been extensively applied to a variety of cell types and culture conditions, including both brain slices and tissue preparations18,34–38. A previous approach has been reported where a single perfusion pump was used to exert a negative pressure across an isolated axonal compartment, consequently withdrawing drug solution (applied in situ) from a reservoir to modulate synaptic activity and protein expression37. The main limitations of that setup were the requirement for regular, manual drug application throughout the experimental procedure and that the flow of perfusate was not selectively applied to an environmentally isolated culture. Alternatively, a model for perfusion has also been described in which neural networks were continuously perfused with automated pumps for up to 70 days38, with the cells grown on a microelectrode array within a microscope onstage incubator. Whilst constant simultaneous morphological imaging and electrophysiological readout of neuronal activity was monitored over weeks, providing exceptional spatiotemporal resolutions, the protocol is time and resource heavy. Interestingly, these reports have shown that continuous perfusion alters synaptic activity and may be beneficial to the health of primary neuronal cultures34–36. Previous reports have shown how 2D vs 3D cultures, co-culture conditions and shear stress may influence gene expression, production/transport/removal of nutrients and molecular secretions39–42. Our findings suggest that an optimal steady flow rate confers neuroprotective properties not only to a single perfused primary culture, but also to an adjacent, synaptically connected culture. Recently, we have revealed evidence of neuroprotection against spreading toxicity between neighbouring synaptically connected but environmentally isolated hippocampal cultures21, and previous studies have proposed suppression of excitotoxicity via interneurons in response to mechanically-induced astrocytic release of ATP43–45. However, the exact mechanisms underlying this observation in the present study remain unclear.
Ca2+ imaging is a powerful and cost-effective technique that can be used to provide optical readouts of both spontaneous and induced cellular activity8,29,46. When combined with the precise spatial and temporal fluid flow control achieved using microfluidic systems, high-throughput and miniaturised assays can be performed. Whilst neuropharmacological studies have used Ca2+ imaging to perform high-throughput pharmacological analysis47–50, they are either limited in their fine control of stimulus delivery or often require user intervention to perform in situ drug delivery. Furthermore, such conventional systems, which integrate Ca2+ imaging techniques to observe neuronal excitability following compound delivery, lack neighbouring environmentally isolated cultures, thus limiting the understanding of a drug’s effect on the synaptic connectivity between cultures.
In the present study, we demonstrate proof-of-concept analysis of both neuronal excitability and consequent synaptic connectivity. First, a concentration response curve was generated by obtaining reproducible responses to a range of glutamate concentrations, revealing an EC50 of 4.7 μM, which compares well to that reported in the literature51–53. Additionally, the neuronal response to glutamate perfusion in the absence or presence of glutamatergic receptor antagonists was examined, revealing reversible inhibition of glutamate-induced transient increases in intracellular Ca2+. These assays highlight that the microfluidic perfusion platform described here can be utilised for the pharmacological profiling of CNS active compounds. Furthermore, our miniaturised system shows for the first time, repeatable assessment of induced network synaptic activity in addition to pharmacological profiling of directly applied compounds in a microfluidic format. Indeed, neuronal activity in the naïve hippocampal culture was increased in response to multiple glutamate applications but, strikingly, this led to an increased level of basal neuronal activity prior to the final glutamate application. Whilst the final glutamate application still induced an increase in neuronal activity with respect to baseline, repeated synaptically driven signalling appears to induce an excited state of activity in the naïve culture, which gradually becomes resistant to further stimulation. This observation mimics the increased responsiveness of neurons that undergo synaptic plasticity54 and in this case, increased responsiveness similar to that observed in long term potentiation (LTP). Whilst we have not probed the underlying mechanisms in the present study, these results are in line with previous studies in which LTP has been investigated in cultured hippocampal neurons using both electrophysiological and Ca2+ imaging techniques55–57. Basal neuronal activity within the naïve chamber was unaltered in the presence of glutamate antagonists whereas increases were observed following glutamate application in their presence. Given that glutamate-induced Ca2+ responses in the perfused chamber were significantly reduced, but not abolished, in the presence of the antagonists, the increases in the naïve chamber are likely to be induced via the residual responses observed in the perfused chamber. In contrast, basal activity in the naïve chamber was significantly enhanced in the presence of TTX in the perfused chamber. Given that our experiments, in which cultures were grown in one chamber only, revealed that the glutamate-induced increase in neuronal activity is unidirectional, this result indicates that TTX application in the perfused chamber may be impairing an inhibitory tone that controls neuronal activity within the naïve chamber. As primary hippocampal cultures grown under our experimental conditions contain approximately 6% GABAergic neurons58, these will exert a strong inhibitory control over excitatory neurotransmission within the cultures. Indeed, application of GABAergic antagonists has been used to mimic epileptiform-like activity in hippocampal cultures59,60, hence the inhibition of GABAergic neuronal firing by TTX in the perfused chamber would result in the increased activity in the naïve chamber due to decreased GABAergic tone extending from the perfused to the naïve chamber. Furthermore, glutamate induced increases in neuronal activity were abolished during the co-application of glutamate and TTX, despite increases in intracellular calcium in neurons within the perfused chamber. Collectively, these results demonstrate that changes in neuronal activity within the naïve culture chamber are driven by functional, synaptic communication from neurons in the perfused chamber.
Conclusions
We have developed a novel platform that integrates microfluidic perfusion and Ca2+ imaging techniques for studying neuromodulation and synaptic connectivity between primary hippocampal cultures. We show that both direct neuronal excitability and the consequent synaptic communication between synaptically connected but environmentally isolated cultures can be simultaneously monitored, providing hundreds of readouts from each device. In addition, with the recent surge in interest in the use of human stem cell derived neurons to investigate CNS disorders61,62, the platform described in the present study may offer new avenues to examine the underlying causes of CNS disease and allow potential novel therapeutics to be tested in a cost-effective, miniaturised manner.
Data availability
Access to all data underpinning this publication is restricted to a request only basis. More information and contact details are available from the University of Strathclyde at 10.15129/5a018c9b-9458-4e3f-8a92-23657abcd44e.
Electronic supplementary material
Acknowledgements
The authors would like to thank the National Centre for The Replacement, Refinement and Reduction of Animals in Research (NC3Rs PhD studentship, NC/L00139X/1) and the University of Strathclyde for financial support.
Author Contributions
C.M., G.R. and M.Z. designed the microfluidic system and developed the microfluidic perfusion assay; C.M. and T.B. planned the pharmacological assays; C.M. and G.R. performed the experiments and analysed the data; All authors wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-15950-0.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World Health Organization. Neurological Disorders: Public Health Challenges. 10.1001/archneurol.2007.19 (World Health Oragnization, 2008).
- 2.EvaluatePharma. World Preview 2018: Embracing the Patent Cliff. (2012).
- 3.Skripka-Serry, J. The great neuro-pipeline ‘brain drain’ (and why Big Pharma hasn’t given up on CNS disorders). Drug Discovery World (2013).
- 4.Hay, M. Clinical development success rates for investigational drugs. Nat Biotechnol32, (2014). [DOI] [PubMed]
- 5.Waring MJ, et al. An analysis of the attrition of pharmaceutical companies. Nat. Publ. Gr. 2015;14:475–486. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- 6.Shuler ML, Hickman JJ. Toward in vitro models of brain structure and function. Proc. Natl. Acad. Sci. USA. 2014;111:13682–13683. doi: 10.1073/pnas.1414484111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsaioun K, Bottlaender M, Mabondzo A. ADDME – Avoiding Drug Development Mistakes Early: central nervous system drug discovery perspective. BMC Neurol. 2009;9(Suppl 1):S1. doi: 10.1186/1471-2377-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gan J, Greenwood SM, Cobb SR, Bushell TJ. Indirect modulation of neuronal excitability and synaptic transmission in the hippocampus by activation of proteinase-activated receptor-2. Br. J. Pharmacol. 2011;163:984–994. doi: 10.1111/j.1476-5381.2011.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martina M, et al. Recordings of cultured neurons and synaptic activity using patch-clamp chips. J. Neural Eng. 2011;8:34002. doi: 10.1088/1741-2560/8/3/034002. [DOI] [PubMed] [Google Scholar]
- 10.Dunlop J, Bowlby M, Peri R, Vasilyev D, Arias R. High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008;7:358–368. doi: 10.1038/nrd2552. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Sulser A, et al. Hippocampal neuron firing and local field potentials in the in vitro 4-aminopyridine epilepsy model. J. Neurophysiol. 2012;108:2568–2580. doi: 10.1152/jn.00363.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spira ME, Hai A. Multi-electrode array technologies for neuroscience and cardiology. Nat. Nanotechnol. 2013;8:83–94. doi: 10.1038/nnano.2012.265. [DOI] [PubMed] [Google Scholar]
- 13.Grienberger C, Konnerth A. Imaging Calcium in Neurons. Neuron. 2012;73:862–885. doi: 10.1016/j.neuron.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–73. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 15.Park JW, Kim HJ, Kang MW, Jeon NL. Advances in microfluidics-based experimental methods for neuroscience research. Lab Chip. 2013;13:509–21. doi: 10.1039/c2lc41081h. [DOI] [PubMed] [Google Scholar]
- 16.Hung PJ, Lee PJ, Sabounchi P, Lin R, Lee LP. Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays. Biotechnol. Bioeng. 2005;89:1–8. doi: 10.1002/bit.20289. [DOI] [PubMed] [Google Scholar]
- 17.Mcmillan KS, Boyd M, Zagnoni M. Transitioning from multi-phase to single-phase microfluidics for long-term culture and treatment of multicellular spheroids. Lab Chip. 2016;16:3548–3555. doi: 10.1039/C6LC00884D. [DOI] [PubMed] [Google Scholar]
- 18.Kim L, Toh Y-C, Voldman J, Yu H. A practical guide to microfluidic perfusion culture of adherent mammalian cells. Lab Chip. 2007;7:681–694. doi: 10.1039/b704602b. [DOI] [PubMed] [Google Scholar]
- 19.Young EWK, Beebe DJ. Fundamentals of microfluidic cell culture in controlled microenvironments. Chem Soc Rev. 2010;39:1036–1048. doi: 10.1039/b909900j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robertson G, Bushell TJ, Zagnoni M. Chemically induced synaptic activity between mixed primary hippocampal co-cultures in a microfluidic system. Integr Biol. 2014;6:636–644. doi: 10.1039/C3IB40221E. [DOI] [PubMed] [Google Scholar]
- 21.Samson AJ, Robertson G, Zagnoni M, Connolly CN. Neuronal networks provide rapid neuroprotection against spreading toxicity. Sci. Rep. 2016;6:33746. doi: 10.1038/srep33746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanagasabapathi TT, Ciliberti D, Martinoia S, Wadman WJ, Decré MMJ. Dual-compartment neurofluidic system for electrophysiological measurements in physically segregated and functionally connected neuronal cell culture. Front. Neuroeng. 2011;4:13. doi: 10.3389/fneng.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor AM, Rhee SW, Tu CH, Cribbs DH, Cotman CW. Microfluidic Multicompartment Device for. Neuroscience Research. 2010;19:1551–1556. doi: 10.1021/la026417v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wevers NR, et al. High-throughput compound evaluation on 3D networks of neurons and glia in a microfluidic platform. Sci. Rep. 2016;6:38856. doi: 10.1038/srep38856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park J, et al. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab Chip. 2014;15:141–50. doi: 10.1039/C4LC00962B. [DOI] [PubMed] [Google Scholar]
- 26.Honegger T, et al. Microfluidic neurite guidance to study structure-function relationships in topologically-complex population-based neural networks. Sci. Rep. 2016;6:28384. doi: 10.1038/srep28384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda S, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015;6:8490. doi: 10.1038/ncomms9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ledgerwood CJ, Greenwood SM, Brett RR, Pratt JA, Bushell TJ. Cannabidiol inhibits synaptic transmission in rat hippocampal cultures and slices via multiple receptor pathways. Br. J. Pharmacol. 2011;162:286–294. doi: 10.1111/j.1476-5381.2010.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50:714–725. doi: 10.1016/j.neuropharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 30.Patankar, S. V. Numerical Heat Transfer and Fluid Flow. (CRC Press, 1980).
- 31.Kunze A, et al. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS. Integr. Biol. (Camb). 2013;5:964–75. doi: 10.1039/c3ib40022k. [DOI] [PubMed] [Google Scholar]
- 32.Taylor aM, Menon S, Gupton SL. Passive microfluidic chamber for long-term imaging of axon guidance in response to soluble gradients. Lab Chip. 2015;15:2781–2789. doi: 10.1039/C5LC00503E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor, A. M. et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. 2, 0–4 (2007). [DOI] [PMC free article] [PubMed]
- 34.Rambani K, Vukasinovic J, Glezer A, Potter SM. Culturing thick brain slices - An interstitial 3D microperfusion system for enhanced viability. J. Neurosci. Methods. 2009;180:243–254. doi: 10.1016/j.jneumeth.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullen DK, Vukasinovic J, Glezer A, Laplaca MC. Microfluidic engineered high cell density three-dimensional neural cultures. J. Neural Eng. 2007;4:159–72. doi: 10.1088/1741-2560/4/2/015. [DOI] [PubMed] [Google Scholar]
- 36.Killian NJ, Vernekar VN, Potter SM, Vukasinovic J. A device for long-term perfusion, imaging, and electrical interfacing of brain tissue in vitro. Front. Neurosci. 2016;10:135. doi: 10.3389/fnins.2016.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor AM, Dieterich DC, Ito HT, Kim Sa, Erin M. Manipulation of Synapses. 2010;66:57–68. doi: 10.1016/j.neuron.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saalfrank D, et al. Incubator-independent cell-culture perfusion platform for continuous long-term microelectrode array electrophysiology and time-lapse imaging. R. Soc. Open Sci. 2015;2:150031. doi: 10.1098/rsos.150031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dirks B, Hanke J, Krieglstein J, Stock R, Wickop G. Studies on the Linkage of Energy Metabolism and Neuronal Activity in the Isolated Perfused Rat Brain. J. Neurochem. 1980;35:311–317. doi: 10.1111/j.1471-4159.1980.tb06266.x. [DOI] [PubMed] [Google Scholar]
- 40.Ivanov A, Zilberter Y. Critical state of energy metabolism in brain slices: The principal role of oxygen delivery and energy substrates in shaping neuronal activity. Front. Neuroenergetics. 2011;3:9. doi: 10.3389/fnene.2011.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller LJ, Gillette MU. New perspectives on neuronal development via microfluidic environments. Trends Neurosci. 2012;35:752–761. doi: 10.1016/j.tins.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Helm MW, van der Meer AD, Eijkel JCT, van den Berg A, Segerink LI. Micro fluidic organ-on-chip technology for blood- brain barrier research. Tissue Barriers. 2016;4:e1142493. doi: 10.1080/21688370.2016.1142493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guthrie PB, et al. ATP Released from Astrocytes Mediates Glial Calcium Waves. J. Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang JM, et al. ATP Released by Astrocytes Mediates Glutamatergic Activity-Dependent Heterosynaptic Suppression. Neuron. 2003;40:971–982. doi: 10.1016/S0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- 45.Bowser DN. ATP Excites Interneurons and Astrocytes to Increase Synaptic Inhibition in Neuronal Networks. J. Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward MW, Kushnareva Y, Greenwood S, Connolly CN. Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J. Neurochem. 2005;92:1081–1090. doi: 10.1111/j.1471-4159.2004.02928.x. [DOI] [PubMed] [Google Scholar]
- 47.Doolen S, Blake CB, Smith BN, Taylor BK. Peripheral nerve injury increases glutamate-evoked calcium mobilization in adult spinal cord neurons. Mol. Pain. 2012;8:56. doi: 10.1186/1744-8069-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teichert RW, et al. Functional profiling of neurons through cellular neuropharmacology. Proc. Natl. Acad. Sci. USA. 2012;109:1388–95. doi: 10.1073/pnas.1118833109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsch J, Ventimigli D, Bargmann CI, Albrecht DR. High-throughput imaging of neuronal activity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2013;110:E4266–E4273. doi: 10.1073/pnas.1318325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi N, Takahara Y, Ishikawa D, Matsuki N, Ikegaya Y. Functional multineuron calcium imaging for systems pharmacology. Anal. Bioanal. Chem. 2010;398:211–218. doi: 10.1007/s00216-010-3740-6. [DOI] [PubMed] [Google Scholar]
- 51.Haak LL. Metabotropic glutamate receptor modulation of glutamate responses in the suprachiasmatic nucleus. J Neurophysiol. 1999;81:1308–17. doi: 10.1152/jn.1999.81.3.1308. [DOI] [PubMed] [Google Scholar]
- 52.Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009;29:12045–58. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anson LC, Chen PE, Wyllie DJ, Colquhoun D, Schoepfer R. Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J. Neurosci. 1998;18:581–589. doi: 10.1523/JNEUROSCI.18-02-00581.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 55.Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 2004;7:126–35. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- 56.Ivenshitz M, Segal M. Simultaneous NMDA-Dependent Long-Term Potentiation of EPSCs and Long-Term Depression of IPSCs in Cultured Rat Hippocampal Neurons. J. Neurosci. 2006;26:1199–1210. doi: 10.1523/JNEUROSCI.2964-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molnár E. Long-term potentiation in cultured hippocampal neurons. Semin. Cell Dev. Biol. 2011;22:506–513. doi: 10.1016/j.semcdb.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Benson DL, Watkins FH, Steward O, Banker G. Characterization of GABAergic neurons in hippocampal cell cultures. J. Neurocytol. 1994;23:279–95. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- 59.Colombi I, Mahajani S, Frega M, Gasparini L, Chiappalone M. Effects of antiepileptic drugs on hippocampal neurons coupled to micro-electrode arrays. Front. Neuroeng. 2013;6:10. doi: 10.3389/fneng.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Popko J, Krogh KA, Thayer SA. Epileptiform stimulus increases Homer 1a expression to modulate synapse number and activity in hippocampal cultures. J. Neurophysiol. 2013;109:1494–504. doi: 10.1152/jn.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srikanth P, Young-Pearse TL. Stem cells on the brain: modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J. Neurogenet. 2015;28:5–29. doi: 10.3109/01677063.2014.881358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu DX, Marchetto MC, Gage FH. Therapeutic translation of iPSCs for treating neurological disease. Cell Stem Cell. 2013;12:678–688. doi: 10.1016/j.stem.2013.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Access to all data underpinning this publication is restricted to a request only basis. More information and contact details are available from the University of Strathclyde at 10.15129/5a018c9b-9458-4e3f-8a92-23657abcd44e.