

Abstract
Increased permeability of the intestinal barrier is proposed as an underlying factor for obesity-associated pathologies. Consumption of high fat diets (HFD) is associated with increased intestinal permeabilization and increased paracellular transport of endotoxins which can promote steatosis and insulin resistance. This study investigated whether dietary (-)-epicatechin (EC) supplementation can protect the intestinal barrier against HFD-induced permeabilization and endotoxemia, and mitigate liver damage and insulin resistance. Mechanisms leading to loss of integrity and function of the tight junction (TJ) were characterized. Consumption of a HFD for 15 weeks caused obesity, steatosis, and insulin resistance in male C57BL/6J mice. This was associated with increased intestinal permeability, decreased expression of ileal TJ proteins, and endotoxemia. Supplementation with EC (2–20 mg/kg body weight) mitigated all these adverse effects. EC acted modulating cell signals and the gut hormone GLP-2, which are central to the regulation of intestinal permeability. Thus, EC prevented HFD-induced ileum NOX1/NOX4 upregulation, protein oxidation, and the activation of the redox-sensitive NF-κB and ERK1/2 pathways. Supporting NADPH oxidase as a target of EC actions, in Caco-2 cells EC and apocynin inhibited tumor necrosis alpha (TNFα)-induced NOX1/NOX4 overexpression, protein oxidation and monolayer permeabilization. Together, our findings demonstrate protective effects of EC against HFD-induced increased intestinal permeability and endotoxemia. This can in part underlie EC capacity to prevent steatosis and insulin resistance occurring as a consequence of HFD consumption.
Abbreviations: ALT, alanine aminotransferase; AMPK, AMP activated protein kinase; EC, (-)-epicatechin; ERK1/2, extracellular signal-regulated kinase; GTT, glucose tolerance test; GLP-2, glucagon-like peptide-2; HFD, high fat diet; HNE, 4-hydroxynonenal; ITT, insulin tolerance test; LPS, lipopolysaccharides; MCP-1, monocyte chemoattractant protein-1; NAFLD, nonalcoholic fatty liver disease; NOS2, nitric oxide synthase 2; T2D, type 2 diabetes; TEER, transepithelial electrical resistance; TJ, tight junction; TNFα, tumor necrosis factor alpha; MLCK, myosin light chain kinase
Keywords: Intestinal permeability, (-)-Epicatechin, Steatosis, Insulin resistance, Endotoxemia, NADPH oxidase
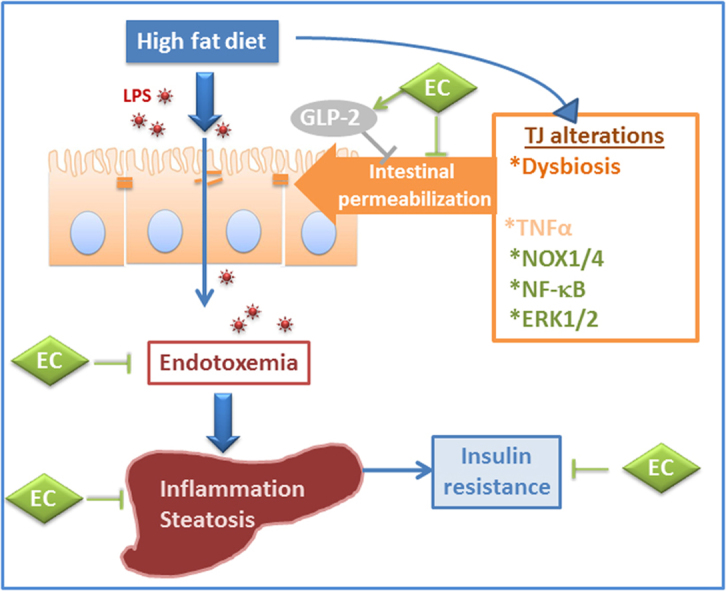
Graphical abstract
Highlights
-
•
(-)-Epicatechin (EC) inhibits high fat diet (HFD)-induced steatosis and insulin resistance in mice.
-
•
EC inhibits HFD-induced intestinal permeabilization and endotoxemia.
-
•
EC preserves ileum tight junction composition and function.
-
•
EC mitigates the activation of redox sensitive signals that promote permeabilization.
-
•
EC effects at the gastrointestinal tract could underlie its beneficial systemic actions.
1. Introduction
Steatosis and insulin resistance are among the major and more severe consequences of obesity. Nonalcoholic fatty liver disease (NAFLD) is more frequent in individuals with diabetes, higher body mass index, and those frequently consuming fast foods [1]. One of the hypotheses on the mechanisms linking obesity and Western-style diets with NAFLD and insulin resistance is the associated increase in intestinal permeability [2], [3]. In this regard, it is proposed that an increased transfer of bacterial lipopolysaccharides (LPS) from the gut lumen into the circulation can cause tissue inflammation and damage, which in the liver would lead to steatosis and insulin resistance [3], [4], [5].
The intestinal barrier is composed of a single layer of intestinal epithelial cells sealed by the tight junctions (TJs). TJs modulate intestinal permeability by regulating the paracellular transport of water and ions. They also constitute the first line of defense against the entry of noxious bacteria/bacterial toxins (e.g. lipopolysaccharide (LPS)) and toxins/antigens present in food. Once transported via the paracellular route, they can initiate local inflammation, and once in the circulation they can also promote systemic tissue inflammation and damage [6], [7], [8]. Increased intestinal permeability is observed in different pathologies including celiac disease and inflammatory bowel diseases, and is present in obesity and type 2 diabetes (T2D) [3], [5], [9], [10]. Consumption of high fat diets (HFD) also causes intestinal permeabilization, impairs mucosal defenses [11], and alters the composition of the intestinal microbiota [11]. In murine models, these intestinal alterations have been attributed to the dietary fat per se [12], being present before the development of obesity and insulin resistance [11].
(-)-Epicatechin (EC) is a flavan-3-ol abundant in human diet [13]. Cumulative evidence has shown an improvement of insulin sensitivity in both humans and rodents upon consumption of EC or EC-containing foods [14], [15], [16], [17], [18], [19]. We previously observed that EC protects rodents from dietary high fructose- and high fat-induced insulin resistance [18], [19]. EC prevented the increased liver triglyceride deposition associated with high fructose consumption in rats [18]. Although EC mitigates events involved in hepatic and adipose tissue insulin resistance (i.e. NADPH oxidase activation, oxidative stress, altered redox signaling, inflammation and endoplasmic reticulum stress) [18], [19], [20], [21], the primary mechanisms involved in EC protective effects remain unknown.
In vitro, EC mitigated the permeabilization of Caco-2 intestinal cell monolayers induced by tumor necrosis factor alpha (TNFα) [22]. The underlying protective mechanisms of EC involved the prevention of TNFα-mediated NADPH oxidase activation, increased superoxide anion production, and altered TJ protein expression and organization [22]. Given the above, we hypothesize that EC can mitigate HFD-induced steatosis and insulin resistance by protecting the intestine from permeabilization. Thus, this study investigated the capacity of EC to prevent liver fat deposition and inflammation in HFD-fed mice, and its relationship to EC capacity to modulate intestinal permeability and the associated endotoxemia. We characterized the effects of EC on major events regulating intestinal permeability, including TJ protein expression, NADPH oxidases and modulatory redox sensitive signaling cascades (NF-κB and extracellular signal-regulated kinase (ERK1/2)), glucagon-like peptide-2 (GLP-2), and dysbiosis. The actions of EC on NADPH oxidases NOX1 and NOX4, protein oxidation, and intestinal permeability were further characterized in Caco-2 cell monolayers. EC improved insulin sensitivity and mitigated the development of steatosis in HFD-fed mice. This was associated with the prevention of HFD-induced intestinal TJ disruption, permeabilization and endotoxemia. EC protected TJs in part by preventing intestinal NOX1/NOX4 upregulation, protein oxidation, and the activation of the redox-sensitive NF-κB and ERK1/2 signaling cascades, and increasing plasma GLP-2. The beneficial effects of EC at the gastrointestinal tract could in part underlie its capacity to improve insulin sensitivity, steatosis, and other obesity-associated pathologies.
2. Materials and methods
2.1. Materials
Cholesterol and triglycerides concentrations were determined using kits purchased from Wiener Lab Group (Rosario, Argentina). Glucose levels were measured using a kit purchased from Sigma-Aldrich Co (St. Louis, MO). Concentrations of insulin, alanine transaminase (ALT), GLP-2, and bile acids were determined using kits purchased from Crystal Chem Inc (Downers Grove, IL). Endotoxin levels were determined using a kit from Lonza (Basel, Switzerland). Antibodies for β-actin (#12620), monocyte chemoattractant protein-1 (MCP-1) (#2029), TNFα (#119487), phospho (Ser536) p65 (#3033), p65 (#3987), phospho (Thr172) AMPKα (#2535), AMPKα (#5832), phospho (Thr202/Tyr204) ERK (#4370), and ERK (#9102) were obtained from Cell Signaling Technology (Danvers, MA,). Antibodies for F4/80 (sc-25830), heterogeneous nuclear ribonucleoprotein (hnRNP) (sc-32301), HSC-70 (SC-1059), nitric oxide synthase 2 (NOS2) (sc-649), and NOX4 (sc-21860) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for ZO-1 (33-9100), occludin (71-1500), and claudin-1 (71-7800) were from Invitrogen (Carlsbad, CA). Antibodies for 4-hydroxinonenal (HNE) (ab46545) and NOX1 (ab55831) were from Abcam Inc. (Cambridge, MA). PVDF membranes were obtained from BIO-RAD (Hercules, CA, USA). The Enhanced chemiluminescence (ECL) Western blotting system was from Thermo Fisher Scientific Inc. (Piscataway, NJ). Apocynin, VAS-2780, EC, fluorescein isothiocyanate (FITC)-dextran (4 kDa) and all other chemicals were purchased from Sigma-Aldrich Co (St. Louis, MO).
2.2. Animals and animal care
All procedures were in agreement with standards for the care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures were administered under the auspices of the Animal Resource Services of the University of California, Davis. Experimental protocols were approved before implementation by the University of California, Davis Animal Use and Care Administrative Advisory Committee.
Healthy male C57BL/6J mice (20–25 g) (9–10 mice/group) were fed for 15 weeks either: A- a diet containing approximately 10% total calories from fat (Control) (TD.06416, Harlan Laboratories, Madison, WI), B- a diet containing approximately 60% total calories from fat (lard) (HF) (TD.06414, Harlan Laboratories, Madison, WI), C- the control diet supplemented with 20 mg EC/kg body weight (CE), and D- the HFD supplemented with 2 (HFE2), 10 (HFE10) or 20 (HFE20) mg EC/kg body weight. EC-containing diets were prepared every two weeks to account for changes in body weight and food intake, and to prevent potential EC degradation. All diets were stored at −20 °C until use. The highest amount of EC supplemented has been found to improve insulin resistance in rats fed high fructose levels [18] and in mice fed a HFD [19]. In comparison to EC intake in human populations [23], the highest EC amounts supplemented are relatively high. However, they can be reached by supplementation or consumption of select EC-rich fruits/vegetables and derivatives [13].
Body weight and food intake were measured weekly throughout the study as previously described [19]. After 15 weeks on the dietary treatments, mice were euthanized by cervical dislocation. Blood was collected from the abdominal aorta into heparinized tubes, and plasma collected after centrifugation at 3000g for 10 min at room temperature. Tissues were dissected and flash frozen in liquid nitrogen and then stored at −80 °C for further analysis.
2.3. Metabolic measurements
For insulin tolerance tests (ITT), mice were fasted for 4 h and injected intraperitoneally with 1 U/kg body weight human insulin (Novolin R U-100, Novo Nordisk Inc, Princeton NJ). Blood glucose values were measured before and at 15, 30, 60 and 120 min post-injection. For glucose tolerance tests (GTT), overnight fasted mice were injected with d-glucose (2 g/kg body weight), and blood glucose was measured before and at 15, 30, 60, and 120 min post-injection. For both tests glucose levels were measured using a glucometer (Easy Plus II, Home Aid Diagnostics Inc, Deerfield Beach, FL). Total cholesterol, triglycerides, glucose, insulin, GLP-2 and endotoxin concentrations, and alanine transaminase activity were determined following manufacturer's guidelines.
2.4. Intestinal permeability
Intestinal permeability was measured after 13 weeks on the diets as described previously [24] with minor modifications. Mice were fasted for 4 h then gavaged with fluorescein isothiocyanate FITC-dextran 4 kDa (200 mg/kg body weight). After 90 min, 100 μl of blood were collected from the tip of the tail vein. The blood was kept in the dark and centrifuged at 3000g for 10 min at room temperature, and the serum collected. Serum aliquots (20 μl) and a standard curve of FITC-dextran were plated in 96-well plates and diluted to 200 μl with 0.9% (w/v) NaCl. Fluorescence was measured using a microplate spectrofluorometer (Wallac 1420 VICTOR2™, PerkinElmer Life Science, Waltman, USA) at λexc: 485 nm and λem: 520 nm.
2.5. Caco-2 cell culture and assessment of monolayer permeability
Caco-2 cells (at passages 3 through 15) were cultured as previously described [25]. Briefly, cells were used 21 d after reaching confluence to allow for differentiation into intestinal epithelial cells. All the experiments were performed in serum- and phenol red-free MEM.
Monolayer permeability was assessed measuring the transepithelial electrical resistance (TEER) and the paracellular transport of FITC-dextran (4 kDa) as described [25]. Briefly, cells were grown on transwell inserts (12 mm, 0.4 µm pore polyester membranes) in 12-well plates (0.3 × 106 cells/transwell), and monolayers were used when TEER values were between 350–450 Ω cm2. TEER was measured using a Millicell-ERS Resistance System (Millipore, Bedford, MA) and calculated as: TEER = (Rm – Ri) × A (Rm, transmembrane resistance; Ri, intrinsic resistance of a cell-free media; A, membrane surface area in cm2). For the experiments, Caco-2 cell monolayers were preincubated for 24 h with interpheron-γ (10 ng/ml) to upregulate the TNFα receptor. Monolayers were then incubated in the presence of 1 µM EC or apocynin added to the upper compartment and incubated for 30 min. Subsequently, cells were incubated in the absence or the presence of TNFα (5 ng/ml) added to the lower compartment, and cells were further incubated for 6 h. For TEER assessment, incubation media were removed from the upper and lower compartments, cells rinsed with HBSS 1X, and the same solution was added to both compartments.
The paracellular transport of FITC-dextran was measured after the 6 h incubation by adding 100 μM FITC-dextran (final concentration) to the upper compartment. After 3.5 h incubation, 100 μl of the medium in the lower compartment were collected, diluted with 100 μl HBSS 1X, and fluorescence was measured at λexc: 485 nm and λem: 520 nm.
2.6. Western blot analysis
Tissue and cell total homogenates and nuclear fractions were prepared as previously described [18], [21], [26]. Aliquots of total homogenates or nuclear fractions containing 25–40 μg protein were denatured with Laemmli buffer, separated by reducing 7.5–12.5% polyacrylamide gel electrophoresis, and electroblotted onto PVDF membranes. Membranes were blocked for 1 h in 5% (w/v) bovine serum albumin and subsequently incubated in the presence of the corresponding primary antibodies (1:500–1:1000 dilution) overnight at 4 °C. After incubation for 90 min at room temperature in the presence of the corresponding secondary antibodies (HRP conjugated) (1:10,000 dilution) the conjugates were visualized by ECL system using a Phosphoimager 840 (Amersham Pharmacia Biotech. Inc., Piscataway, NJ).
2.7. Histological analyses
The liver was removed and samples fixed overnight in 4% (w/v) neutralized paraformaldehyde buffer solution. Samples were subsequently washed twice in phosphate buffer saline solution, dehydrated, and then embedded in paraffin for histological analysis. Sections with a thickness of 5 µm were obtained from paraffin blocks and placed on glass slides. Hematoxylin and eosin staining was performed following standard procedures. Sections were examined using an Olympus BX51 microscope (Olympus America Inc., Center Valley, PA, USA). Hepatic histological examination was performed using the histological scoring system for NAFLD as described by Kleiner et al. [27]. Three randomly selected fields per animal were assessed blindly by an independent pathologist who did not know the identities of the study groups. All liver specimens were analyzed using Pro Plus 5.1 software (Media Cybernetics, Rockville, MD).
2.8. Immunohistochemistry
Ileum samples were dissected out, fixed in 4% (w/v) solution of paraformaldehyde in PBS overnight, rinsed with PBS and stored in 70% (v/v) ethanol. Samples were embedded in paraffin and 5 µm sections were obtained. Once deparaffinized, sections were processed for antigen retrieval by incubation in 10 mM sodium citrate buffer (pH 6.0) containing 0.05% (v/v) Tween 20 at 95 °C for 10 min, washed twice with 0.1% (v/v) Triton X-100 in 0.1 M PBS, blocked for 50 min in 10% (v/v) donkey serum in 0.1% (v/v) Triton X-100 in 0.1 M PBS, and incubated overnight at 4 °C with primary antibodies for occludin (1:200), ZO-1 (1:100) or claudin-1 (1:200). Sections were washed in PBS and incubated for 2 h at room temperature with Cy2- or Cy3-conjugated donkey anti-mouse or anti-rabbit IgG (1:500) (Jackson ImmunoResearch Co. Laboratories West Grove, PA). After immunostaining, cell nuclei were stained with Hoechst 33342 and sections were imaged using an Olympus FV 1000 laser scanning confocal microscope (Olympus, Japan). Olympus Fluoview version 4.0 software was used to merge images. Four slices per animal and four animals from each group were analyzed. The integrated optical density was measured using Image Pro Plus 5.1 software (Media Cybernetics, Rockville, MD) and expressed per area. Three randomly selected fields were measured per animal for each antibody and experimental condition.
2.9. Determination of NADPH oxidase activity
NADPH oxidase activity was measured using a lucigenin-enhanced chemiluminescence assay in membrane fractions from differentiated Caco-2 cells incubated in the absence or the presence of TNFα (5 ng/ml). For the isolation of membrane fractions, cells were homogenized in Krebs Buffer (20 mM HEPES, 119 mM NaCl, 4.7 mM KCl, 1 mM MgSO4, 0.4 mM NaH2PO4, 0.15 mM Na2HPO4 and 1.25 mM CaCl2) containing 1 mM PMSF, and Roche proteases inhibitor cocktail (Roche, Switzerland) and centrifuged at 800g for 10 min at 4 °C. The supernatant was subsequently centrifuged at 100,000g for 60 min at 4 °C, the pellet was collected (membrane fraction) and resuspended in Krebs buffer. Aliquots of membrane fractions (30 μg of protein) were added with or without 1 μM epicatechin or apocynin, and subsequently with 5 μM lucigenin and 50 μM NADPH. The reaction was followed under temperature-controlled conditions (37 °C). Light emission was measured every 30 s for 20 min using a Biotek Synergy H1 plate reader (BioTek Instruments, Inc.,Winooski, VT, USA) in the chemiluminescence mode. Results were expressed as the difference between the areas under the curve in the absence and in the presence of the NADPH oxidase inhibitor VAS-2780 (1 μM).
2.10. Microbiota analysis
Cecum content samples were collected in sterile tubes for subsequent investigation of the microbiome through high throughput sequencing. Samples were stored at −80 °C until processing for DNA extraction. Genomic DNA was extracted from cecal samples using the Zymo Research Fecal DNA Miniprep Kit per the manufacturer's instructions (Zymo Research, Irvine, CA, USA). The V4 region of the 16S rRNA gene was amplified by targeted barcoded primers F515 (5′-NNNNNNNNGTGTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACHVGGGTWTCTAAT-3′) as previously described [28]. Amplicons were then pooled and purified with the QIAquick PCR Purification Kit (Qiagen, Germantown, MD, USA) and taken to the UC Davis Genome Center DNA Technologies Sequencing Core for library preparation and paired-end sequenced on an Illumina Miseq. PEAR [29] was used to merge the paired end reads and they were subsequently demultiplexed using the FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Cutadapt was then used to trim off barcodes and primers from reads [30]. Read quality filtering, OTU picking using the implemented swarm method, filtering the OTU table, rarefaction, and beta diversity data analysis was carried out within the QIIME software package (University of Colorado, Boulder, CO, USA. version 1.9.1) [31]. Swarm was used within the QIIME package as the operational taxonomic unit clustering method [32]. Beta diversity metrics were calculated based on Unifrac distances.
2.11. Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) using Statview 5.0 (SAS Institute Inc., Cary, NC). Fisher least significance difference test was used to examine differences between group means. A P value < 0.05 was considered statistically significant. Data are shown as mean ± SE.
3. Results
3.1. EC supplementation improves insulin sensitivity and dyslipidemia in HFD-fed mice
As previously observed [19], consumption of a HFD for 15 weeks caused obesity, dyslipidemia and insulin resistance in C57BL/6J mice (Table 1). Body weight of HFD-fed mice was 40% higher than controls (C and CE), and comparable to the three HFD-fed and EC-supplemented groups. Plasma cholesterol and triglyceride levels were 43% and 63% higher, respectively, in the HF group compared to controls, and both were decreased only in the HFE10 and HFE20 groups.
Table 1.
Metabolic parameters.
| Parameter | C | CE | HF | HFE2 | HFE10 | HFE20 |
|---|---|---|---|---|---|---|
| Body weight (g) | 31.6 ± 0.9a | 33.0 ± 0.7a | 44.0 ± 1.6b | 40.9 ± 2.2b | 42.1 ± 2.6b | 39.8 ± 1.9b |
| Food intake (g/d) | 3.5 ± 0.19a | 4.1 ± 0.05b | 2.8 ± 0.17c | 3.0 ± 0.07c | 2.8 ± 0.09c | 2.8 ± 0.12c |
| Fasted glucose (mg/dl) | 96 ± 4a | 85 ± 12a | 161 ± 8b | 134 ± 9c | 137 ± 15b,c | 130 ± 7c |
| Fasted insulin (ng/ml) | 0.15 ± 0.04a | 0.22 ± 0.10a | 0.87 ± 0.23b | 0.38 ± 0.11a | 0.47 ± 0.09a | 0.37 ± 0.07a |
| Total cholesterol (mg/dl) | 119 ± 6a | 128 ± 5a,d | 170 ± 6b | 156 ± 10b,c | 146 ± 7c,d | 136 ± 10a,c |
| TG (mg/dl) | 45.4 ± 2.1a | 55.4 ± 3.8c,d | 73.8 ± 3.6b | 62.5 ± 3.2c | 49.9 ± 3.2a,d | 62.7 ± 1.3c |
Results are shown as means ± SE and are the average of 5-8 animals/group. Values having different superscripts are significantly different; (p < 0.05, one way ANOVA).
Dietary EC supplementation (2–20 mg/kg body weight) attenuated the hyperglycemia and hyperinsulinemia caused by HFD consumption (Table 1). Chronic consumption of HFD was associated with decreased insulin sensitivity and decreased glucose tolerance as evidenced by insulin (ITT) and glucose (GTT) tolerance tests, respectively (Fig. 1A and B). The area under the curve for the ITT in HF mice was 31% and 71% higher, and for the GTT, 54% and 73% higher, respectively, than in C and CE groups. The ITT area under the curve was similar in the HFE2, and significantly lower (26% and 44%, respectively) in the HFE10 and HFE20 compared to the C group. At all concentrations tested, EC supplementation mitigated HFD-mediated increase in the GTT area under the curve. The above results confirm the capacity of dietary EC to improve insulin sensitivity in HFD-fed C57BL/6J mice even at the lowest concentration tested (2 mg/kg body weight). However, although most of the protective effects were observed in all the range of concentrations assessed (2–20 mg/kg body weight), the highest EC concentration was that consistently effective in all the different parameters tested. Thus, in most of the following experiments we have evaluated select parameters only in the groups supplemented with 20 mg/kg body weight (CE, HFE20).
Fig. 1.

Effects of EC supplementation on metabolic parameters in HFD-fed mice. A- ITT and B- GTT (the corresponding area under the curve (A.U.C) is shown in the right panels), were performed on weeks 9 and 11 on the diets, respectively. Mice were fed a control diet (empty circles and empty bars), the control diet supplemented with 20 mg EC/kg body weight (blue circles and dashed bars), a HFD (HF) (black triangles and bars), or the HFD supplemented with 2 (HFE2), 10 (HFE10), or 20 (HFE20) mg EC/kg body weight (grey triangles and bars). Results are shown as means ± SE and are the average of 5–8 animals/group. Values having different symbols are significantly different (p < 0.05, one way ANOVA).
3.2. EC supplementation attenuates HFD-induced steatosis and inflammation
The effects of EC on liver steatosis and inflammation triggered by the consumption of a HFD were assessed by measuring liver triglyceride content using a biochemical assay, inflammation markers by Western blot, and histology. Liver triglyceride content was 38% higher in HF mice. Values for EC-supplemented mice were similar to controls (Fig. 2A). Accordingly, hematoxylin-eosin stained liver sections showed higher NAFLD activity score in liver from HF mice compared to C, CE and HFE20 mice (Fig. 2C). Although not statistically different, the CE group showed a trend (p < 0.07) for higher NAFLD activity score than the C group. Steatosis-associated cell damage was also evaluated by measuring plasma alanine aminotransferase (ALT) activity in plasma. In HF mice we observed 48% and 68% higher activity of plasma ALT compared to the C and CE groups (Fig. 2B). Supplementation with 20 mg EC/kg body weight prevented this increase. Values for HFE2 and HFE20 mice were not significantly different to those of controls and HF.
Fig. 2.

Effects of EC supplementation on steatosis and hepatic inflammation. Mice were fed a control diet (empty bars), the control diet supplemented with 20 mg EC/kg body weight (dashed bars), a HFD (HF) (black bars), or the HFD supplemented with 2 (HFE2), 10 (HFE10), or 20 (HFE20) mg EC/kg body weight (grey bars). On week 15 on the corresponding diets the following parameters were measured: A-liver triglyceride content, B- plasma alanine amino transferase (ALT) activity, C- fat liver deposition and NAFLD activity score (NAS) evaluated by hematoxylin/eosin tissue staining, D-proteins involved in inflammation: MCP-1, TNFα, NOS2 and F4/80 protein levels were measured by Western blot. Bands were quantified and values referred to β-actin levels (loading control). Results for HF, HFE20 and CE were referred to control group values (C). Results are shown as mean ± SE of 5–8 animals/group. A,B- Values having different symbols are significantly different; C,D- #Significantly different from all other groups; (p < 0.05, one way ANOVA test).
We next evaluated the capacity of EC to mitigate HFD-induced liver inflammation by measuring hepatic protein levels of the chemokine MCP-1, the cytokine TNFα, NOS2, and the macrophage marker F4/80 by Western blot. In HF mice we observed 81%, 210%, 40% and 58% higher levels of MCP-1, TNFα, NOS2 and F4/80, respectively, compared to the C group. Supplementation with EC (HFE20) mitigated the effects of the high fat feeding on the expression of all the tested proteins (Fig. 2D).
3.3. EC supplementation prevents intestinal permeabilization and endotoxemia in HFD-fed mice
Increased intestinal permeability and the associated increase in bacterial endotoxins in the bloodstream are significant contributors to HFD- and obesity-associated steatosis. Thus, we investigated the effects of EC supplementation on HF-induced permeabilization and increased plasma endotoxin levels. We measured intestinal permeability using FITC-dextran after 13 weeks on the corresponding diets. The intestinal paracellular permeability to FITC-dextran was 100% higher in the HF group compared to controls and all the EC-supplemented groups (Fig. 3A). Plasma endotoxin levels were 63% higher in HF mice than in controls. EC supplementation either partially (HFE10) or significantly (HFE20) mitigated endotoxemia (Fig. 3B). Plasma endotoxin levels positively correlated with plasma ALT activity (r: 0.84, p = 0.04), and with the area under the curve corresponding to the GTT (r: 0.89, p = 0.02).
Fig. 3.

Effects of EC on epithelial barrier permeabilization induced by HFD consumption in mice and by TNFα in Caco-2 cells. A,C- Mice were fed a control diet (empty bars), the control diet supplemented with 20 mg EC/kg body weight (dashed bars), a HFD (HF) (black bars), or the HFD supplemented with 2 (HFE2), 10 (HFE10), or 20 (HFE20) mg EC/kg body weight (grey bars). Intestinal permeability was evaluated by measuring at week 13 FITC-dextran permeability (A), and at week 15 plasma endotoxin (B) and TNFα (C) concentrations. Results are shown as mean ± SE of 5–8 animals/group. D- FITC-dextran transport and D- TEER in Caco-2 cells. Polarized cells were incubated for 6 h at 37 °C in the absence of additions (control, C); or after addition of 5 ng/ml TNFα to the lower chamber in the absence (TNF) or the presence of 1 μM EC or 1 μM apocynin (Apo) added to the upper chamber. Results are shown as mean ± SE of 3 independent experiments. A,C-E- #Significantly different from all other groups; B-Values having different symbols are significantly different; (p<0.05,one way ANOVA test).
3.4. EC prevents TNFα-induced permeabilization of Caco-2 cell monolayers
TNFα is a major inducer of intestinal barrier permeabilization. TNFα plasma concentration was 2.3-fold higher in HF mice compared to controls, and EC supplementation caused only a partial prevention (47%) of this increase (Fig. 3C). Thus, using Caco-2 monolayers as an in vitro model of intestinal barrier, we further assessed the capacity of EC to protect the monolayer from TNFα-induced permeabilization. For this purpose, we measured the FITC-dextran paracellular transport and the monolayer transepithelial electrical resistance (TEER). TNFα (5 ng/ml), added to the lower chamber, caused a 1.1-fold increase in FITC-dextran transport (Fig. 3D) and a 31% decrease in TEER (Fig. 3E) in Caco-2 cell monolayers. These changes were prevented by simultaneous incubation of cells with EC (1 μM) or with apocynin (1 μM) added to the upper chamber.
3.5. EC supplementation attenuates HFD-induced alterations in TJ protein expression
An increased intestinal permeability can be due to impaired TJ structure and/or function. To investigate the potential protective effects of EC on HFD-mediated alterations of TJs, we examined the expression of the TJ proteins ZO-1, occludin, and claudin-1 in mouse ileum by Western blot and by immunofluorescence. ZO-1, occludin and claudin-1 protein levels assessed by Western blot were 64%, 55% and 33% lower, respectively, in the ileum from HF mice compared to controls (Fig. 4A). Supplementation of the HF mice with 20 mg EC/kg body weight prevented the decrease in ZO-1, occludin and claudin-1. Similar trends were observed when TJ proteins were evaluated by immunofluorescence confocal microscopy (Fig. 4B and C).
Fig. 4.

Effects of EC supplementation on HFD-induced alterations in ileum tight junction protein expression. Mice were fed a control diet (empty bars), the control diet supplemented with 20 mg EC/kg body weight (dashed bars), a HFD (HF) (black bars), or the HFD supplemented with 20 mg (HFE20) EC/kg body weight (grey bars). A- TJ proteins occludin, claudin-1 and ZO-1, expression was measured by Western blot. Bands were quantified and values referred to HSC70 levels (loading control). Results for HF, HFE20 and CE were referred to control group values (C). Results are shown as mean ± SE of 8 animals/treatment. B- Representative images for immunohistochemistry and confocal microscopy for occludin and ZO-1 (red fluorescence), and claudin-1 (green fluorescence). Hoechst was used for nuclear counterstaining (blue). Bar: 50 µm C- Fluorescence intensity was measured as described in methods and results are shown as mean ± SE of 4 animals/treatment. A,C- #Significantly different from all other groups; (p < 0.05, one way ANOVA test).
3.6. Mechanisms underlying the protective effects of EC on HFD-induced increased intestinal permeability
Intestinal permeability can be regulated by different cell signaling pathways (ERK1/2, AMPK and NF-κB), some of which are susceptible to redox regulation. GLP-2 is a major regulator of epithelial monolayer permeability. To assess signaling activation we measured the phosphorylation of ERK1/2, NF-κB (p65) and AMPK, and the nuclear presence of p65 by Western blot (Fig. 5A and B). Higher levels of ERK1/2, p65, and AMPK phosphorylation were observed in HF, but not in HFE20 mice when compared to controls (Fig. 5A). In agreement with the pattern of p65 phosphorylation in total homogenates, nuclear p65 levels were significantly higher (98%) in the HF group compared to controls and HFE20 mice (Fig. 5B). Plasma GLP-2 levels were 1- and 1.5-fold higher in CE and HFE20 mice, respectively, compared to the C group (Fig. 5C). In HF mice, plasma GLP-2 concentration was not statistically different compared to the control groups. Significantly, plasma GLP-2 levels in both EC-supplemented groups (CE, HFE20) were significantly higher than in control mice.
Fig. 5.

Effects of EC supplementation on HFD-induced alterations on signaling events that regulate intestinal permeability. Mice were fed a control diet (empty bars), the control diet supplemented with 20 mg EC/kg body weight (dashed bars), a HFD (HF) (black bars), or the HFD supplemented with 20 mg EC/kg body weight (HFE20) (grey bars). A- Phosphorylation levels of ERK1/2 (Thr202/Tyr204), p65 (Ser536) and AMPK (Thr172) in total ileum homogenates, and B- p65 and hnRNP levels in ileum nuclear fractions were measured by Western blot. Bands were quantified and values referred to either (A) the non-phosphorylated protein form or (B) hnRNP levels. Results for HF, HFE20 and CE were referred to control group values (C). C- Plasma GLP-2 concentration was measured using a commercial kit. A-C- Results are shown as mean ± SE of 6–8 animals/treatment. A,B- #Significantly different from all other groups; C- Values having different symbols are significantly different; (p<0.05, one way ANOVA test).
Oxidative stress could contribute to intestinal permeabilization upon HFD consumption. Thus, we measured protein levels of ileum NADPH oxidases NOX1 and NOX4, and 4-hydroxynonenal (HNE)-protein adducts as parameter of oxidative stress (Fig. 6A). NOX4 and NOX1 levels were 50% higher in the ileum from HF mice than in controls, and this was prevented by EC supplementation (Fig. 6B). 4-HNE-protein adducts in the molecular weight corresponding to actin were 24% higher in HF mice than in all other groups. To assess the potential involvement of NADPH oxidases on intestinal permeabilization, we measured the effects of TNFα, EC, and apocynin on the expression of NOX1 and NOX4, and on NADPH oxidase activity in Caco-2 cells. TNFα caused a 79% and 52% increase in NOX1 and NOX4 expression, respectively (Fig. 6B). Both EC and apocynin (1 μM) prevented both increases. Supporting a condition of increased oxidant production and oxidative stress, TNFα caused higher (38%) levels of 4-HNE-protein adducts in Caco-2 cells, which were prevented by EC and apocynin. At 1 μM concentration, both EC and apocynin added to Caco-2 membrane fractions inhibited NADPH oxidase activity (Fig. 6C). The above experiments support the capacity of EC to inhibit NADPH oxidase both at the level of expression and activity.
Fig. 6.

Effects of EC on the upregulation of intestinal NADPH oxidases and protein oxidation induced by HFD consumption in mice and by TNFα in Caco-2 cells. Total protein levels of NOX4, NOX1, and HNE-protein adducts (MW: 40 kDa) were measured by Western blot in: A- the ileum of mice fed a control diet (empty bars), the control diet supplemented with 20 mg EC/kg body weight (dashed bars), a HFD (HF) (black bars), or the HFD supplemented with 20 mg EC/kg body weight (HFE20) (grey bars); B- Caco-2 cells incubated for 6 h at 37 °C in the absence of additions (control, C) (empty bars); or after addition of 5 ng/ml TNFα in the absence (TNF) (dark blue bars) or the presence of 1 μM EC (TNF + EC) (light blue bars) or 1 μM apocynin (Apo) (grey bars). Bands were quantified and values referred to β-actin or HSC70 levels (loading controls). Results were referred to control group values (C). C- In vitro effects of 1 μM EC and apocynin (Apo) on NADPH oxidase activity were measured in membrane fractions isolated from Caco-2 cells incubated without or with 5 ng/ml TNFα for 6 h. Results are shown as mean ± SE of A- 6–8 animals/treatment, and B,C- 4 independent experiments. #Significantly different from all other groups (p < 0.05, one way ANOVA test).
3.7. EC supplementation does not affect HFD-induced dysbiosis
As expected, the chronic consumption of a HFD caused dysbiosis in mice. Principal component analysis of cecum microbiota showed clearly separated clusters for the control and HF groups. EC supplementation did not normalize the observed microbiota cluster segregation (Fig. 7).
Fig. 7.

Effects of EC on HFD-induced changes in cecal microbiota. Changes in cecal microbiota assessed by clustering of samples based on diet. Principal coordinate analysis (PCA) was performed based on the weighted UniFrac distance matrix generated from sequencing fecal 16S rRNA gene in cecum samples from mice fed the corresponding diets for 15 weeks. The X-axis represents the tertiary coordinate, the Y-axis represents the secondary coordinate. Axis numbering represents the relative distance between samples based on the weighted UniFrac distance matrix.
4. Discussion
This study presents evidence that EC supplementation mitigates intestinal permeabilization and endotoxemia induced by HFD consumption in mice. EC protected the integrity of the TJ and modulated signaling pathways that contribute to TJ normal function, including NOX1/NOX4 upregulation, oxidative stress, and NF-κB and ERK1/2 activation. The involvement of NADPH oxidase in EC-mediated protection of barrier integrity in HFD-fed mice was supported by evidence in Caco-2 cells. The beneficial effects of EC supplementation on intestinal permeability could protect the liver from endotoxin-induced damage. This may explain EC capacity to improve the metabolic profile in HFD-induced obesity and T2D [19], [20].
The link between steatosis and a “leaky gut” is supported by a large body of evidence associating NAFLD to different conditions with increased intestinal permeability, including cardiometabolic disorders, obesity [2], [3], and inflammatory bowel diseases [33]. We currently observed that the chronic consumption of a HFD led to steatosis, liver inflammation (high tissue chemokines and cytokines, NOS2, and macrophage F4/80 protein levels) and damage (high ALT plasma activity) in mice. This can in part explain previous findings showing impaired hepatic response to insulin in HFD mice [19]. EC improved systemic glucose homeostasis, and mitigated HFD-induced liver triglyceride accumulation, inflammation and ALT release. Accordingly, in a rat model of high fructose-induced T2D, EC supplementation also prevented hepatic triglyceride accumulation and insulin resistance [18]. Overfeeding is recognized as a contributor of NAFLD [34]. Although EC can prevent steatosis in two models of overfeeding (high fat and high fructose diets) in mice, the underlying mechanisms are unknown. Evidence that EC can preserve Caco-2 cell monolayer permeability in an in vitro model of inflammation [22], points to the intestinal barrier as a potential target of EC beneficial actions on steatosis, and hepatic/systemic insulin resistance.
A permeable intestinal epithelium allows the paracellular transport of bacterial and dietary antigens leading to local and systemic inflammation [5], [7]. For example, endotoxemia occurs upon overfeeding in men [35], as a consequence of consuming a HFD in T2D patients [36], and correlates with energy intake in healthy individuals [37]. One consequence of increased endotoxemia is liver inflammation, steatosis and insulin resistance. In support of this mechanism, all these symptoms manifest in mice i.p. injected with LPS for four weeks [5]. We observed that EC prevented HFD-induced barrier permeabilization and increased circulating endotoxin. Accordingly, an EC-containing cocoa extract decreased endotoxemia in high fat fed mice [38]. Suggesting a potential link between a leaky gut and liver dysfunction upon HFD consumption, we observed that plasma endotoxin concentrations positively correlated with plasma ALT activity (parameter of liver damage), and GTT area under the curve (parameter of glucose tolerance).
The protective capacity of EC on intestinal permeability is in part due to the preservation of TJ structure. HFD consumption caused a decreased expression of the TJ proteins occludin, claudin-1 and ZO-1 in the ileum, which was prevented by EC supplementation. Among several factors, TNFα is a central trigger of TJ structural/functional disruption. EC partially mitigated HFD-induced increase in circulating TNFα which can partially explain the protective action of EC on barrier function. This is in agreement with previous observations showing that EC protects Caco-2 monolayers from TNFα-induced decrease in ZO-1 expression, alteres ZO-1 subcellular distribution, and increases paracellular transport [22]. NADPH oxidase is activated when TNFα binds to its receptor, and increased O2.- production contributes to TNFα-induced TJ disruption [22]. EC mitigated HFD-induced increased expression of NOX1 and NOX4 and protein oxidation in the ileum. Significantly, we also observed that the permeabilization of Caco-2 monolayers by TNFα was associated with increased NOX1 and NOX4 expression and protein oxidation. As previously observed in other tissues [18], [20], [39], [40], EC mitigated NADPH oxidase overexpression (NOX1 and NOX4) in HFD-fed mice and Caco-2 cells, and inhibited the enzyme activity in cells. In vitro experiments showing the parallel action of a NADPH oxidase inhibitor (apocynin) and EC preventing TNFα-induced permeabilization and NADPH oxidase overexpression and activity, strongly support ileum epithelial NADPH oxidase as a significant target for the protective actions of EC on HFD-induced intestinal barrier permeabilization in mice. Both NOX1 and NOX4 have κB sites in their promoters, and NF-κB is a central regulator of both NOX1 and NOX4 expression [41]. Thus, a decrease in cellular O2.- /H2O2 production as a consequence of NADPH oxidase inhibition, and subsequent inhibition of NF-κB activation, can explain the observed prevention by EC and apocynin of HFD- and TNFα-induced overexpression of both NOX1 and NOX4. On the other hand, it has been argued that apocynin is not a NADPH oxidase inhibitor unless the dimer is formed. However, apocynin inhibits NADPH oxidase in vitro both as monomer and as dimer, with an IC50 4-fold lower for the dimer than for the monomer [40]. The low (1 μM) concentration of apocynin that inhibited TNFα-induced Caco-2 monolayer permeabilization suggests that the involved mechanism is primarily the inhibition of NADPH oxidase rather than a direct antioxidant action. Overall, although the role of NADPH oxidase on TJ structure/function regulation is not completely understood, an increase in O2.-/H2O2 production could increase intestinal permeability through the oxidation of TJ structural and modulatory proteins and, as described below, the activation of select redox-sensitive signals.
Different signaling cascades are involved in the regulation of TJ assembly. AMPK plays a key role in promoting the assembly of TJs [42], [43]. On the other hand, AMPK is also important for nutrient absorption [44]. In our model, AMPK activation by HFD consumption seems to be related to the local management of nutrient absorption and metabolism rather than to the regulation of intestinal permeabilization. Very importantly, TNFα activates pathways that increase TJ permeability, including NF-κB and ERK1/2. NF-κB regulates the transcription of myosin light chain kinase (MLCK) [45], [46], which increases TJ permeability by phosphorylating the myosin light chain protein. ERK1/2 activation increases TJ permeability by regulating the expression of TJ proteins [47] and promoting MLCK transcription via the ETS domain-containing protein ELK-1 [48]. Thus, the above results are consistent with an upstream regulatory effect of EC (i.e. TNFα-triggered NADPH oxidase activation) in the inhibition of NF-κB and ERK1/2 activation and ultimately TJ disruption.
Different factors/events are proposed to contribute to the intestinal permeabilization triggered by HFD consumption including fat per se, an increase in luminal bile acids, and alterations in the microbiota [49]. In fact, consumption of HFDs with different fat sources leads to increased intestinal permeability in mice [50]. Dysbiosis, including increased Bacteroides and decreased Firmicutes, decreased bacterial diversity and altered representation of bacterial genes, is observed in obese individuals [51], [52]. Consumption of high energy foods, obesity and dysbiosis are linked to steatosis and NAFLD [24], [53], [54], [55]. Although the microbiota can be central to energy metabolism, intestinal permeability, endotoxemia and steatosis; mitigation of dysbiosis by EC through changes in microbiota composition does not seem to be the mechanism involved in the observed EC beneficial effects. This is based on the lack of effects of EC on the overall microbiota changes observed as a consequence of HFD consumption.
GLP-2 is a 33 aminoacid peptide that is generated from the cleavage of proglucagon. GLP-2 is involved in different aspects of intestinal physiology, including trophic actions and decreased barrier paracellular transport [56]. GLP-2 also protects the liver from steatosis. In this regard, mice fed a HFD and injected with an antagonist to the GLP-2 receptor showed worsening of triglyceride hepatic deposition and steatosis degeneration [57]. EC supplementation increased plasma GLP-2 levels both in control and HFD-fed mice. Thus, EC-mediated plasma GLP-2 increase could contribute to the mitigation of steatosis by both preserving the structure/function of TJs in HF mice, and also decreasing hepatic triglyceride deposition.
In summary, consumption of a HFD leads to intestinal permeabilization, steatosis, hepatic inflammation and insulin resistance in mice. EC supplementation mitigates all these adverse effects. HFD-induced endotoxemia, secondary to increased intestinal permeabilization can be in part involved in the development of steatosis. Indeed, HFD consumption caused alterations in TJ composition, and affected events involved in intestinal permeabilization, including dysbiosis and increased plasma TNFα. In the ileum, HFD consumption triggered a cascade of redox-related events: upregulation of NOX1/NOX4, protein oxidation, activation of NF-κB and ERK1/2, and TJ disruption. EC protection of barrier function is in part due to its capacity to preserve normal TJ composition in part by preventing NOX1/NOX4 upregulation, inhibiting NF-κB and ERK1/2, and increasing GLP-2 expression. Current results support the concept that dietary EC might be important in mitigating the adverse effects of Western style diets.
Acknowledgements
This work was supported by the NIFA-USDA (CA-D*-xxx-7244-H) (P.I.O.). A.B is funded by NIH/NIDDK [R00DK100736]. Research in the F.G.H laboratory is funded by NIH grants R01DK090492 and R01DK095359. P.I.O is a honorary researcher from CONICET, Argentina.
References
- 1.Williams C.D., Stengel J., Asike M.I., Torres D.M., Shaw J., Contreras M. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Damms-Machado A., Louis S., Schnitzer A., Volynets V., Rings A., Basrai M. Gut permeability is related to body weight, fatty liver disease, and insulin resistance in obese individuals undergoing weight reduction. Am. J. Clin. Nutr. 2017;105:127–135. doi: 10.3945/ajcn.116.131110. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff S.C., Barbara G., Buurman W., Ockhuizen T., Schulzke J.D., Serino M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189. doi: 10.1186/s12876-014-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geurts L., Neyrinck A.M., Delzenne N.M., Knauf C., Cani P.D. Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benef. Microbes. 2014;5:3–17. doi: 10.3920/BM2012.0065. [DOI] [PubMed] [Google Scholar]
- 5.Cani P.D., Amar J., Iglesias M.A., Poggi M., Knauf C., Bastelica D. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 6.Horton F., Wright J., Smith L., Hinton P.J., Robertson M.D. Increased intestinal permeability to oral chromium ( Cr) -EDTA in human Type 2 diabetes. Diabet. Med.: a J. Br. Diabet. Assoc. 2013 doi: 10.1111/dme.12360. [DOI] [PubMed] [Google Scholar]
- 7.Piya M.K., Harte A.L., McTernan P.G. Metabolic endotoxaemia: is it more than just a gut feeling? Curr. Opin. Lipidol. 2013;24:78–85. doi: 10.1097/MOL.0b013e32835b4431. [DOI] [PubMed] [Google Scholar]
- 8.Spruss A., Kanuri G., Stahl C., Bischoff S.C., Bergheim I. Metformin protects against the development of fructose-induced steatosis in mice: role of the intestinal barrier function. Lab. Investig. 2012;92:1020–1032. doi: 10.1038/labinvest.2012.75. [DOI] [PubMed] [Google Scholar]
- 9.Keita A.V., Soderholm J.D. The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterol. Motil.: Off. J. Eur. Gastrointest. Motil. Soc. 2010;22:718–733. doi: 10.1111/j.1365-2982.2010.01498.x. [DOI] [PubMed] [Google Scholar]
- 10.Biasi F., Leonarduzzi G., Oteiza P.I., Poli G. Inflammatory bowel disease: mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013;19:1711–1747. doi: 10.1089/ars.2012.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomas J., Mulet C., Saffarian A., Cavin J.B., Ducroc R., Regnault B. High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA. 2016 doi: 10.1073/pnas.1612559113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T., Hara H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr. Metab. 2010;7:19. doi: 10.1186/1743-7075-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harnly J.M., Doherty R.F., Beecher G.R., Holden J.M., Haytowitz D.B., Bhagwat S. Flavonoid content of U.S. fruits, vegetables, and nuts. J. Agric. Food Chem. 2006;54:9966–9977. doi: 10.1021/jf061478a. [DOI] [PubMed] [Google Scholar]
- 14.National Cholesterol Education Program National Heart, Lung, and Blood Institute, Third Report of the National Cholesterol Education Program (NECP) Expert Panel on Detection, Evaluation and Treatments of High Blood Cholesterol in Adults (Adult Treatments Panel III) Executive Summary, National Institutes of Health NIH Publication No. 01-3670, May 2001.
- 15.Shrime M.G., Bauer S.R., McDonald A.C., Chowdhury N.H., Coltart C.E., Ding E.L. Flavonoid-rich cocoa consumption affects multiple cardiovascular risk factors in a meta-analysis of short-term studies. J. Nutr. 2011;141:1982–1988. doi: 10.3945/jn.111.145482. [DOI] [PubMed] [Google Scholar]
- 16.Grassi D., Desideri G., Necozione S., Lippi C., Casale R., Properzi G. Blood pressure is reduced and insulin sensitivity increased in glucose-intolerant, hypertensive subjects after 15 days of consuming high-polyphenol dark chocolate. J. Nutr. 2008;138:1671–1676. doi: 10.1093/jn/138.9.1671. [DOI] [PubMed] [Google Scholar]
- 17.Davison K., Coates A.M., Buckley J.D., Howe P.R. Effect of cocoa flavanols and exercise on cardiometabolic risk factors in overweight and obese subjects. Int. J. Obes. 2008;32:1289–1296. doi: 10.1038/ijo.2008.66. [DOI] [PubMed] [Google Scholar]
- 18.Bettaieb A., Vazquez-Prieto M.A., Lanzi C.R., Miatello R.M., Haj F.G., Fraga C.G. (-)-Epicatechin mitigates high fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free Radic. Biol. Med. 2014;72:247–256. doi: 10.1016/j.freeradbiomed.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cremonini E., Bettaieb A., Haj F.G., Fraga C.G., Oteiza P.I. (-)-Epicatechin improves insulin sensitivity in high fat diet-fed mice. Arch. Biochem. Biophys. 2016;599:13–21. doi: 10.1016/j.abb.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettaieb A., Cremonini E., Kang H., Kang J., Haj F.G., Oteiza P.I. Anti-inflammatory actions of (-)-epicatechin in the adipose tissue of obese mice. Int. J. Biochem. Cell Biol. 2016;81:383–392. doi: 10.1016/j.biocel.2016.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vazquez-Prieto M.A., Bettaieb A., Haj F.G., Fraga C.G., Oteiza P.I. (-)-Epicatechin prevents TNFalpha-induced activation of signaling cascades involved in inflammation and insulin sensitivity in 3T3-L1 adipocytes. Arch. Biochem. Biophys. 2012;527(2):113–118. doi: 10.1016/j.abb.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Contreras T.C., Ricciardi E., Cremonini E., Oteiza P.I. (-)-Epicatechin in the prevention of tumor necrosis alpha-induced loss of Caco-2 cell barrier integrity. Arch. Biochem. Biophys. 2015;573:84–91. doi: 10.1016/j.abb.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Vogiatzoglou A., Mulligan A.A., Lentjes M.A., Luben R.N., Spencer J.P., Schroeter H. Flavonoid intake in European adults (18 to 64 years) PLoS One. 2015;10:e0128132. doi: 10.1371/journal.pone.0128132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cani P.D., Bibiloni R., Knauf C., Waget A., Neyrinck A.M., Delzenne N.M. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 25.Cremonini E., Mastaloudis A., Hester S.N., Verstraeten S.V., Anderson M., Wood S.M. Anthocyanins inhibit tumor necrosis alpha-induced loss of Caco-2 cell barrier integrity. Food Funct. 2017;8(8):2915–2923. doi: 10.1039/c7fo00625j. [DOI] [PubMed] [Google Scholar]
- 26.Aimo L., Mackenzie G.G., Keenan A.H., Oteiza P.I. Gestational zinc deficiency affects the regulation of transcription factors AP-1, NF-kappaB and NFAT in fetal brain. J. Nutr. Biochem. 2010;21:1069–1075. doi: 10.1016/j.jnutbio.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 28.Frese S.A., Parker K., Calvert C.C., Mills D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome. 2015;3:28. doi: 10.1186/s40168-015-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J., Kobert K., Flouri T., Stamatakis A. PEAR: a fast and accurate Illumina paired-end read merger. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. [Google Scholar]
- 31.Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Lozupone C.A., Turnbaugh P.J. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahe F., Rognes T., Quince C., de Vargas C., Dunthorn M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ. 2014;2:e593. doi: 10.7717/peerj.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chao C.Y., Battat R., Al Khoury A., Restellini S., Sebastiani G., Bessissow T. Co-existence of non-alcoholic fatty liver disease and inflammatory bowel disease: a review article. World J. Gastroenterol. 2016;22:7727–7734. doi: 10.3748/wjg.v22.i34.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yki-Jarvinen H. Nutritional modulation of non-alcoholic fatty liver disease and insulin resistance. Nutrients. 2015;7:9127–9138. doi: 10.3390/nu7115454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laugerette F., Alligier M., Bastard J.P., Drai J., Chanseaume E., Lambert-Porcheron S. Overfeeding increases postprandial endotoxemia in men: inflammatory outcome may depend on LPS transporters LBP and sCD14. Mol. Nutr. Food Res. 2014;58:1513–1518. doi: 10.1002/mnfr.201400044. [DOI] [PubMed] [Google Scholar]
- 36.Harte A.L., Varma M.C., Tripathi G., McGee K.C., Al-Daghri N.M., Al-Attas O.S. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care. 2012;35:375–382. doi: 10.2337/dc11-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amar J., Burcelin R., Ruidavets J.B., Cani P.D., Fauvel J., Alessi M.C. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- 38.Gu Y., Yu S., Park J.Y., Harvatine K., Lambert J.D. Dietary cocoa reduces metabolic endotoxemia and adipose tissue inflammation in high-fat fed mice. J. Nutr. Biochem. 2014;25:439–445. doi: 10.1016/j.jnutbio.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calabro V., Piotrkowski B., Fischerman L., Vazquez Prieto M.A., Galleano M., Fraga C.G. Modifications in nitric oxide and superoxide anion metabolism induced by fructose overload in rat heart are prevented by (-)-epicatechin. Food Funct. 2016;7:1876–1883. doi: 10.1039/c6fo00048g. [DOI] [PubMed] [Google Scholar]
- 40.Steffen Y., Gruber C., Schewe T., Sies H. Mono-O-methylated flavanols and other flavonoids as inhibitors of endothelial NADPH oxidase. Arch. Biochem. Biophys. 2008;469:209–219. doi: 10.1016/j.abb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Manea A., Tanase L.I., Raicu M., Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 2010;396:901–907. doi: 10.1016/j.bbrc.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L., Li J., Young L.H., Caplan M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA. 2006;103:17272–17277. doi: 10.1073/pnas.0608531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng B., Cantley L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA. 2007;104:819–822. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bland M.L., Lee R.J., Magallanes J.M., Foskett J.K., Birnbaum M.J. AMPK supports growth in Drosophila by regulating muscle activity and nutrient uptake in the gut. Dev. Biol. 2010;344:293–303. doi: 10.1016/j.ydbio.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma T.Y., Iwamoto G.K., Hoa N.T., Akotia V., Pedram A., Boivin M.A. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 46.Ye D., Ma T.Y. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J. Cell Mol. Med. 2008;12:1331–1346. doi: 10.1111/j.1582-4934.2008.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T., Yoshinaga N., Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 2011;286:31263–31271. doi: 10.1074/jbc.M111.238147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Sadi R., Guo S., Ye D., Ma T.Y. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am. J. Pathol. 2013;183:1871–1884. doi: 10.1016/j.ajpath.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreira A.P., Texeira T.F., Ferreira A.B., Peluzio Mdo C., Alfenas Rde C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012;108:801–809. doi: 10.1017/S0007114512001213. [DOI] [PubMed] [Google Scholar]
- 50.Murakami Y., Tanabe S., Suzuki T. High-fat diet-induced intestinal hyperpermeability is associated with increased bile acids in the large intestine of mice. J. Food Sci. 2016;81:H216–H222. doi: 10.1111/1750-3841.13166. [DOI] [PubMed] [Google Scholar]
- 51.Turnbaugh P.J., Hamady M., Yatsunenko T., Cantarel B.L., Duncan A., Ley R.E. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishra A.K., Dubey V., Ghosh A.R. Obesity: an overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metab.: Clin. Exp. 2016;65:48–65. doi: 10.1016/j.metabol.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 53.Alisi A., Ceccarelli S., Panera N., Nobili V. Causative role of gut microbiota in non-alcoholic fatty liver disease pathogenesis. Front. Cell. Infect. Microbiol. 2012;2:132. doi: 10.3389/fcimb.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spencer M.D., Hamp T.J., Reid R.W., Fischer L.M., Zeisel S.H., Fodor A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140:976–986. doi: 10.1053/j.gastro.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bashiardes S., Shapiro H., Rozin S., Shibolet O., Elinav E. Non-alcoholic fatty liver and the gut microbiota. Mol. Metab. 2016;5:782–794. doi: 10.1016/j.molmet.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dube P.E., Brubaker P.L. Frontiers in glucagon-like peptide-2: multiple actions, multiple mediators. Am. J. Physiol. Endocrinol. Metab. 2007;293:E460–E465. doi: 10.1152/ajpendo.00149.2007. [DOI] [PubMed] [Google Scholar]
- 57.Baldassano S., Amato A., Caldara G.F., Mule F. Glucagon-like peptide-2 treatment improves glucose dysmetabolism in mice fed a high-fat diet. Endocrine. 2016;54:648–656. doi: 10.1007/s12020-016-0871-3. [DOI] [PubMed] [Google Scholar]