

Abstract
Flavorings-related lung disease is a potentially disabling disease of food industry workers associated with exposure to the α-diketone butter flavoring, diacetyl (2,3-butanedione). To investigate the hypothesis that another α-diketone flavoring, 2,3-pentanedione, would cause airway damage, rats that inhaled air, 2,3-pentanedione (112, 241, 318, or 354 ppm), or diacetyl (240 ppm) for 6 hours were sacrificed the following day. Rats inhaling 2,3-pentanedione developed necrotizing rhinitis, tracheitis, and bronchitis comparable to diacetyl-induced injury. To investigate delayed toxicity, additional rats inhaled 318 (range, 317.9—318.9) ppm 2,3-pentanedione for 6 hours and were sacrificed 0 to 2, 12 to 14, or 18 to 20 hours after exposure. Respiratory epithelial injury in the upper nose involved both apoptosis and necrosis, which progressed through 12 to 14 hours after exposure. Olfactory neuroepithelial injury included loss of olfactory neurons that showed reduced expression of the 2,3-pentanedione–metabolizing enzyme, dicarbonyl/L-xylulose reductase, relative to sustentacular cells. Caspase 3 activation occasionally involved olfactory nerve bundles that synapse in the olfactory bulb (OB). An additional group of rats inhaling 270 ppm 2,3-pentanedione for 6 hours 41 minutes showed increased expression of IL-6 and nitric oxide synthase-2 and decreased expression of vascular endothelial growth factor A in the OB, striatum, hippocampus, and cerebellum using real-time PCR. Claudin-1 expression increased in the OB and striatum. We conclude that 2,3-pentanedione is a respiratory hazard that can also alter gene expression in the brain.
In May 2000, an occupational medicine physician reported the clinical diagnosis of severe bronchiolitis obliterans in eight workers at a Missouri microwave popcorn plant.1, 2, 3 An investigation at the plant revealed that employees had an increased rate of airway obstruction and that the prevalence of obstruction increased with increased exposure to diacetyl (2,3-butanedione), a dicarbonyl compound.2 Although several other hazardous chemicals were known causes of bronchiolitis obliterans,4, 5, 6, 7, 8, 9 they were notably absent in the air of that workplace.2 However, a potential etiological role for diacetyl was suggested by its chemical structure. In diacetyl, the carbonyl groups are adjacent to each other, placing diacetyl into the class of compounds known as α-dicarbonyl compounds, compounds that are often chemically reactive.10, 11, 12, 13, 14, 15 Diacetyl was used in microwave popcorn production because it imparts the flavor and aroma of butter to foods. It can be a natural component of foods, including butter, but may also be present as a component of natural and artificial flavorings. The new workplace disease became known as popcorn workers' lung, popcorn lung, or flavorings-related lung disease.16, 17, 18
Toxicologic pathology studies provided insight into the etiological characteristics of flavorings-related lung disease. Inhalation of butter flavoring vapors or the vapors of diacetyl alone caused necrosis of airway epithelial cells in exposed rats and mice.19, 20, 21 Damage to airway epithelium was a critical finding because damage to airway epithelium is believed to be the cause of bronchiolitis obliterans, the disease seen in the first sentinel cases of flavorings-related lung disease.19, 22, 23, 24 Investigation of the inhalation dosimetry of diacetyl led to the development of a hybrid computational fluid dynamic–physiologically based pharmacokinetic model that described diacetyl uptake during short-term exposures in the rat and human respiratory tract.25, 26 The dosimetry study provided an explanation for the observation that diacetyl caused predominantly upper airway damage in rodents, whereas flavorings-related lung disease in workers predominantly affected the deep lung: during short-term exposures, diacetyl damaged airway epithelium when critical concentrations were achieved in the target cells.26 In a mouth-breathing, lightly exercising worker, the diacetyl tissue concentration in the bronchioles is estimated to be >40-fold greater than the concentration in the bronchioles of a nose-breathing rat in an inhalation chamber.25
In the past 5 years, additional human studies have provided further converging evidence implicating diacetyl in causing flavorings-related lung disease. Additional cases of flavorings-related lung disease have been found in microwave popcorn workers and in workers manufacturing diacetyl itself.17, 25, 27
For many occupational exposures, a useful control strategy is the substitution of a safer agent to replace one that is hazardous (National Institute for Occupational Safety and Health, http://www.cdc.gov/niosh/topics/flavorings, last accessed April 14, 2012). However, the number of potential flavorings is limited by taste receptors and by the need for flavorings to either be approved food additives or generally recognized as safe when present as intended in food.28, 29 An additional concern is that biological receptors that control aroma and taste are incompletely described and may tightly constrain the chemical structures that are perceived as producing a specific flavor and smell, such as butter.30, 31, 32, 33 The Flavor and Extract Manufacturers Association of the United States conducts surveys of users and manufacturers of flavorings that record the category of use (eg, as a flavoring or an antimicrobial) and the weight used for specific compounds.34 Such surveys occur at intervals of several years and will not necessarily reflect a recent shift in use caused by substitution for another flavoring agent. In addition, the formulations used to impart flavor are generally proprietary. Thus, it is not known how often specific substitutes for diacetyl are used in flavoring formulations.
Recently, dicarbonyl flavoring compounds of unknown respiratory toxicity have been identified in workplace air.35 These include three compounds that impart the flavor of butter to foods: 2,3-pentanedione (PD), 2,3-hexanedione, and 2,3-heptanedione. Each of these flavorings is an α-dicarbonyl compound, raising the concern that these close structural relatives of diacetyl may taste like diacetyl and may have comparable toxicity. PD is particularly structurally similar to diacetyl in that it is simply one methylene group longer. As with diacetyl, PD is a volatile α-dicarbonyl compound that can occur naturally in foods.36, 37, 38, 39, 40 It can also be added to food as a synthetic flavoring.28, 29, 41 Historically, PD has received less use as a flavoring than diacetyl, and it is estimated that 4590 pounds of PD was used for flavoring in 2005.34 In addition to the structural similarity to diacetyl, PD is structurally similar to the β-diketone, 2,4-pentanedione, in which the carbonyl groups are two carbons away from each other. 2,4-Pentanedione is a compound that is neurotoxic.42, 43
During final preparation of this article, 2-week PD exposures were reported to cause bronchiolitis obliterans–like lesions in rats, which were characterized by bronchial and bronchiolar fibrosis.44 Therefore, in this study, we investigated the hypothesis that inhaled PD is a respiratory hazard with comparable short-term toxicity to diacetyl. We further investigated the hypothesis that 2,3-pentanedione, as with 2,4-pentanedione, is neurotoxic. In this study, we found that short-term PD inhalation has respiratory toxicity that is comparable to diacetyl. However, it also causes necrosis and apoptosis in the olfactory neuroepithelium, activation of caspase 3 in axons in olfactory nerve bundles, and, consistent with neurotoxicity, up-regulation of brain IL-6, inducible nitric oxide synthase (Nos)-2, and claudin-1 (Cldn1) transcripts. Finally, by using immunofluorescence, we found that the greatest toxicity in the olfactory neuroepithelium involved olfactory neurons that had reduced expression of the major PD-metabolizing system, dicarbonyl/L-xylulose reductase (DCXR),45 whereas the sustentacular cells that had elevated DCXR expression were relatively spared. However, the PD-exposed sustentacular cells were disorganized and immunoreactive DCXR was sometimes aggregated within cytoplasmic vacuoles, suggesting loss of the protective apical DCXR seen in control rats.
Materials and Methods
Animals
Research was conducted with approval and guidance from the Institutional Animal Care and Use Committee in a barrier animal facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Male Hla(SD)CVF rats (Hilltop Lab Animals, Scottdale, PA), weighing 325 to 390 g (at exposure), were housed two per cage in individually ventilated microisolator units supplied with high-efficiency particulate air–filtered laminar flow air (Thoren Caging Systems, Hazleton, PA). Rats were provided with irradiated Harlan Teklad Global 18% protein rodent diet (Harlan Teklad, Madison, WI) and tap water ad libitum in shoe box cages with autoclaved α-Dri virgin cellulose chips (Shepherd Specialty Papers, Watertown, TN) and hardwood β-chips (NEPCO, Warrensburg, NY) for bedding. Rats were acclimatized for at least 7 days before exposure.
Experimental Design
The dose-response experiment was conducted as per the experimental design, as outlined in Table 1. The design included four groups of rats exposed to PD, one group of rats exposed to 240-ppm diacetyl, and two air control groups. All exposures were for 6 hours. Rats were individually housed in wire cages during the exposures, and six rats were simultaneously exposed at each exposure concentration. Half of the rats from one of the air-exposed groups were randomly assigned to the neurotoxicology study (described later), and half were assigned to the respiratory tract dose-response study. This resulted in an n of 6 for the diacetyl group and each of the PD exposed groups. The air control group had an n of 9. The 240-ppm concentration of diacetyl was chosen to minimize potential stress to the rats because nasal, but not intrapulmonary, changes occur at that dose.19 Clinical observations of the rats were made as the rats were removed from the exposure chamber, at intervals during the next 18 to 20 hours, and were last made immediately before sacrifice.
Table 1.
2,3-Pentanedione 6-Hour Dose-Response Experimental Design
| Compound | Target concentration (ppm) | Actual concentration (ppm)⁎ |
|---|---|---|
| Air | NA | NA |
| Diacetyl | 240 | 240.3 ± 0.26 |
| 2,3-Pentanedione | 120 | 111.7 ± 0.12 |
| 2,3-Pentanedione | 240 | 241.2 ± 0.15 |
| 2,3-Pentanedione | 320 | 318.4 ± 0.17 |
| 2,3-Pentanedione | 360 | 354.2 ± 0.20 |
NA, not applicable.
Expressed as mean ± SE.
For the neurotoxicity experiment, rats inhaled control air for 6 hours or PD (mean concentration, 270 ppm) for 6 hours 41 minutes.
For the time course experiment, the target concentration of 320 ppm was used for 6-hour PD inhalation exposures, the lowest concentration of PD that produced clinical signs by 18 hours after exposure in the dose-response experiment. PD-exposed rats were sacrificed immediately (0 to 2), 12 to 14, and 18 to 20 hours after exposure (Table 2).
Table 2.
Target and Actual Concentrations of 2,3-Pentanedione as a Time-Weighted Average in the Time Course Experiment
| Postexposure time point (hours) | Compound | Target concentration (ppm) | Actual concentration (ppm)⁎ |
|---|---|---|---|
| 0 | 2,3-Pentanedione | 320 | 318.9 ± 0.21 |
| 12 | 2,3-Pentanedione | 320 | 318.7 ± 0.21 |
| 18 | 2,3-Pentanedione | 320 | 317.9 ± 0.19 |
| 18 | Air | NA | NA |
NA, not applicable.
Expressed as mean ± SE.
Exposures
To avoid rodent stress associated with nose-only exposures,46 all exposures were whole-body inhalation exposures. The rats were exposed to PD or diacetyl vapors in a glass and stainless steel whole-body inhalation chamber that was modified and developed for butter-flavoring exposures.20 During exposures, the rats were housed in 7 × 5 × 3-in3 individual cages within the chamber. The PD (product 241962-100G; Sigma-Aldrich, St. Louis, MO) and diacetyl (product B85307-100ML; Sigma-Aldrich) were administered by injecting the liquid from a computer-controlled syringe pump (210 kDa; Scientific Inc., Holliston, MA) through a septum and into a heated cross section of stainless steel tubing with a diluent airstream (20 L/minute). The external surface of the tubing was heated to 150°C, which resulted in an air temperature of 70°C at the point in the cross section where the liquid PD or diacetyl was injected and vaporized. The air-vapor mix then passed into the exposure chamber, and the flavoring concentration was measured with a volatile organic compound (VOC) meter (PGM-7600; RAE Systems, San Jose, CA). As organic vapors passed by an UV high-energy photon source lamp within the VOC meter, they were photoionized, and the ejected electrons were measured as a current. VOC readings were used in a feedback loop that adjusted the flow of the syringe pump to maintain the concentration levels at a constant, user-defined value. The temperature within the inhalation chamber was regulated between 27.0°C and 27.8°C to replicate the generation of butter-flavoring vapors in the workplace.17 Relative humidity was controlled between 30% and 40% for animal comfort. Data acquisition and control software was developed to display exposure information, save pertinent data, and control system parameters.
To confirm a uniform concentration distribution throughout the exposure chamber, test runs were conducted with three VOC meters. One VOC meter was connected to the sampling port where concentration measurements were usually conducted during exposures. The other two VOC meters were placed in the animal cages within the animal's breathing zones. These VOC meters were rotated to different cages after each test run to check for biases. Results indicated negligible concentration differences at various locations in the chamber.
The VOC meters were calibrated weekly, with either diacetyl or PD, using a custom system that delivered user-defined air temperatures, relative humidity, and flavoring concentrations. The absolute humidity during calibration was matched to that during exposures to account for humidity effects on VOC measurements. Gravimetric filter measurements, a scanning mobility particle sizer (SMPS; TSI, St Paul, MN), and an aerodynamic particle sizer (TSI) were used to determine whether any of the vapor was in an aerosol form. All measurements indicated a negligible aerosol concentration.
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Immediately after euthanasia, the brain was removed and bisected into right and left hemispheres. Brain regions [olfactory bulb (OB), striatum (STR), hippocampus (HIP), and cerebellum (CER)] from the right hemisphere of each animal were dissected (within 3 to 5 minutes of euthanasia), stabilized in RNALater (Applied Biosystems, Foster City, CA), and frozen at −75°C until RNA isolation. The left hemisphere was preserved in 10% buffered formalin for neuropathological analysis to be performed in future follow-up studies. The brain tissues (OB, STR, HIP, or CER) were homogenized in Tri Reagent (Molecular Research Center, Inc., Cincinnati, OH), and the aqueous phase was separated with MaXtract High Density gel (Qiagen, Valencia, CA). Total RNA from the aqueous phase was then isolated using RNeasy minispin columns (Qiagen), and concentrations were determined with a NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE). First-strand cDNA synthesis was performed using total RNA (1 μg), random hexamers, and MultiScribe Reverse Transcriptase (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems) in a 20-μL reaction. Real-time PCR amplification was performed using the 7500 Real-Time PCR System (Applied Biosystems) in combination with TaqMan chemistry. Specific primers and FAM dye-labeled TaqMan MGB probe sets [TaqMan Gene Expression Assays for rat IL-6, Nos2, Cldn1, occludin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1)] were procured from Applied Biosystems and used according to the manufacturer's recommendation. All PCR amplifications (40 cycles) were performed in a total volume of 25 μL, containing 1-μL cDNA, 1.25 μL of the specific TaqMan Gene Expression Assay, and 12.5 μL of TaqMan Universal master mix (Applied Biosystems, Foster City, CA). Sequence detection software (version 1.7; Applied Biosystems) results were exported as tab-delimited text files and imported into Microsoft Excel (Microsoft Corporation, Redmond, WA) for further analysis. After normalization to β-actin, relative quantification of gene expression was performed using the CT method, as described by the manufacturer (Applied Biosystems; User Bulletin 2). The values are expressed as fold change over air-exposed controls.
Histopathological Data
Rats were euthanized by an overdose of pentobarbital (Sleepaway; Fort Dodge Animal Health, Fort Dodge, IA). In the dose-response experiment, lungs were fixed by airway perfusion with 10% neutral-buffered formalin. Tissues in the time course experiment were fixed by intravascular perfusion with 4% paraformaldehyde–0.1% cacodylate buffer to provide adequate fixation of brains for neuropathological findings while also fixing the lungs. After fixation, tissues were collected and stored in 10% neutral-buffered formalin. Lung and trachea were processed the day of necropsy, whereas brain and nasal tissues remained in 10% neutral-buffered formalin for at least a week. Nasal tissues were decalcified in 13% formic acid. Lung, trachea, and nasal sections were embedded in paraffin and stained with H&E.
TUNEL Assays
The TUNEL assay on nasal section T1 was conducted as previously described.47
Fluorescence Assays
Tissue sections were placed on ProbeOn Plus slides (Fisher Scientific, Pittsburgh, PA) and deparaffinized in xylene and alcohols, and antigenicity was retrieved by microwave heating in 1 mmol/L EDTA at a pH of 8 for lung and brain and a pH of 7 for nose (nose sections fall off the slides at a pH of 8). After rinsing in distilled water, non-specific staining for all slides was blocked by incubation for 2 hours in 10% donkey serum (Jackson ImmunoResearch Corp, West Grove, PA). For DCXR immunofluorescent stains, the slides were pretreated with 5% IgG-free bovine serum albumin before the serum blocking step.
For all immunofluorescent stains, at least one negative control slide was run with each immunofluorescent staining run with the primary antibodies replaced by non-immune IgG from the same species. Normal mouse IgG (SC-2025; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was the negative control for mouse monoclonal antibodies. Normal/pre-immune rabbit IgG was the negative control for rabbit polyclonal antibodies (Alpha Diagnostic, San Antonio, TX). Normal/pre-immune goat IgG was the negative control for goat polyclonal antibodies (Alpha Diagnostic).
E-cadherin and activated caspase 3 double-label immunofluorescence was performed by incubating at 4°C overnight with a 1:100 dilution of mouse anti–E-cadherin (BD Biosciences, San Jose, CA) and a 1:200 dilution of rabbit anti-activated caspase 3 (antibody AF835; R&D Systems, Minneapolis, MN). Secondary antibodies were donkey anti-mouse Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 594 (Molecular Probes/Invitrogen, Eugene, OR).
Glucose transporter 1 and activated caspase 3 double-label immunofluorescence was performed by incubating overnight at 4°C with a 1:200 dilution of mouse anti-glucose transport 1 (Abcam, Cambridge, MA) and a 1:200 dilution of rabbit activated caspase 3 antibody (AF835; R&D Systems). Secondary antibodies were donkey anti-mouse Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 594 (Molecular Probes/Invitrogen).
Olfactory marker protein (OMP) and activated caspase 3 double-label immunofluorescence was performed as per a previously published enzymatic immunohistochemical (IHC) assay for activated caspase 3 and OMP,48 which was adapted for immunofluorescence. Slides were incubated overnight at 4°C with a 1:100 dilution of goat anti-olfactory marker protein (Wako, Richmond, VA) and a 1:150 dilution of rabbit activated caspase 3 antibody (AF835; R&D Systems). Secondary antibodies were donkey anti-rabbit Alexa Fluor 488 and donkey anti-goat Alexa Fluor 594. This assay was run a second time for slides in the time course experiment to ensure reproducibility, and a negative control slide was run for each slide in the repeat assay.
DCXR/E-cadherin and DCXR/OMP double-label immunofluorescence was performed using a polyclonal rabbit anti-DCXR antibody that was well characterized by the manufacturer for IHC detection of DCXR in formalin-fixed human tissue (Prestige Antibodies Powered by Atlas Antibodies, affinity-isolated antibody, HPA023371; Sigma-Aldrich, St. Louis, MO); this antibody was selected based on its ability to differentiate DCXR expression in wild-type and DCXR knockout mice in preliminary work (data not shown). Slides from rats inhaling PD at concentrations overtly cytotoxic to olfactory neuroepithelium (318 and 354 ppm) or air in the time course experiment were evaluated for DCXR expression in olfactory neuroepithelium.
Rabbit anti-DCXR antibody was diluted 1:6.25 with filter-sterilized 1.5 times calcium and magnesium-free PBS containing 1% IgG-free bovine serum albumin and then mixed with an equal volume of either goat anti-OMP or mouse anti-E-cadherin and incubated overnight at 4°C. The secondary antibodies for demonstrating DCXR, E-cadherin, and OMP were donkey anti-rabbit Alexa Fluor 594, donkey anti-mouse Alexa Fluor 488, and donkey anti-goat Alexa Fluor 488, respectively.
The DCXR/OMP double label allowed red DCXR expression to be distinguished between cells that did not stain with OMP and green cells (neurons) that did stain with OMP. The DCXR/E-cadherin double label demonstrated all epithelial intercellular junctions and the vasculature in green because of the presence of E-cadherin recognized by the primary antibody or endogenous rat immunoglobulins recognized by the secondary antibody. The DCXR–E-cadherin double label, therefore, provided general context for DCXR and specifically allowed demonstration of the excretory ducts of the Bowman's glands.
FluoroJade B staining was used to identify degenerating neurons. Deparaffinized sections of brain and nose were treated with potassium permanganate for 20 minutes and then stained with a 1:25 dilution of FluoroJade B (Millipore, Billerica, MA) in 0.1% acetic acid for an additional 20 minutes, following the manufacturer's protocol.
Statistics
The statistical analyses for this study were conducted using SAS/STAT software, version 9.1 of the SAS System for Windows (SAS Institute, Inc., Cary, NC). Histopathological data were analyzed using Kruskal-Wallis nonparametric tests, followed by pairwise comparisons using the Wilcoxon rank-sum test. In the dose-response experiment, semiquantitative histopathological scores for each region were analyzed for the effect of exposure dose on the pathological score. In the time course experiment, semiquantitative histopathological scores for each region were analyzed for the effect of time after exposure on the pathological score. Real-time PCR data from each brain region were converted to fold-change values and analyzed using analysis of variance. Data were log-transformed to meet the assumptions of the statistical test. Data are presented as fold-change values. All differences were considered significant at P ≤ 0.05.
Results
Dose-Response Experiment
Clinical Evaluations
To monitor potential distress, rats were observed visually for any adverse respiratory or other clinical signs. Rats in obvious distress were euthanized as soon as practical. In rats inhaling 318-ppm PD, clinical signs were not present immediately after exposure. Clinical signs were present in four of six rats at 17 to 20 hours after exposure, but were generally subtle. However, at 18 to 20 hours after exposure, one rat had episodic mouth breathing; and on auscultation, inspiratory sounds were harsh, consistent with upper airway changes. Auscultation detected faint inspiratory harshness in two additional rats. One rat had normal lung sounds but appeared to have increased respiratory frequency at rest in his cage.
In rats inhaling 354-ppm PD, clinical signs were not present immediately or 4 hours after exposure. Consistent with delayed toxicity, clinical signs were present in four of six rats at 15 hours 50 minutes after exposure and in five of six rats by 18 hours after exposure. Clinical signs were variable, but included periods of mouth breathing (three rats), slightly harsh inspiratory sounds on auscultation (three rats), increased swallowing (one rat), and decreased lung sounds (one rat). In one rat, subtle mouth breathing was noted at 15 hours 50 minutes and with time became more apparent. To minimize potential distress, this rat was euthanized. Although euthanized as soon as clinically indicated, the time point for this rat was 15 minutes before the scheduled sacrifice time.
No clinical signs were observed in rats exposed to PD at <318 ppm, in rats exposed to 240-ppm diacetyl, or in the air-exposed rats.
Necrotizing Rhinitis, Tracheitis, and Bronchitis in Rats Inhaling PD
In the nose, histopathological findings in control rats were within normal limits, with changes limited to a few foci of neutrophilic to neutrophilic and eosinophilic rhinitis in two rats (Figure 1A). Rats inhaling PD developed histopathological lesions in the nose that were characterized by apoptosis and necrosis of respiratory, transitional, and olfactory epithelium (Table 3). At the lowest exposure concentration, significant changes were restricted to the first nasal section (T1), where the morphological changes were principally characterized by cell detachment, pyknosis, karyorrhexis, and karyolysis. At higher-exposure concentrations, nasal epithelial necrosis was the predominant and characteristic response to PD exposure in the respiratory and transitional epithelium (Figure 1B). A predominantly neutrophilic inflammatory response consistently accompanied the nasal epithelial necrosis at higher-exposure concentrations, with many neutrophils within exudates, but variable infiltration of the necrotic epithelium. The olfactory neuroepithelium of air-exposed rats consistently displayed normal morphological characteristics (Figure 1C). In the olfactory neuroepithelium of the PD-exposed rats, the histopathological changes were principally characterized by apoptosis and multifocal invagination (Figure 1D). In PD-exposed rats, changes in the olfactory neuroepithelium were most frequently seen in nasal section T2, but were seen in level T3 in four of six rats exposed to the highest PD concentration (Table 4). Morphologically, similar changes were seen in the olfactory neuroepithelium of diacetyl-exposed rats. However, with diacetyl exposure, the number of affected rats and the severity of histopathological changes were comparable in levels T2 and T3 (Table 4). Histopathological changes in the respiratory epithelium of rats inhaling 240-ppm diacetyl were comparable to those seen in rats inhaling 241.2-ppm PD (Table 3).
Figure 1.

Histopathological changes in nasal epithelium after PD exposure. A: Normal transition epithelium of the maxilloturbinate in an air control rat. B: Necrotizing rhinitis of the maxilloturbinate in a PD-exposed rat is characterized by necrosis of the epithelium, with few infiltrating neutrophils. C: Normal neuroepithelium of an air-exposed control rat. D: The neuroepithelium of a rat after inhaling 318-ppm PD shows loss of cells, cells with pyknotic nuclei, and foci of invagination. Scale bar = 20 μm.
Table 3.
Necrotizing Rhinitis, Tracheitis, and Bronchitis in Rats Inhaling 2,3-Pentanedione and Diacetyl
| Exposure | Concentration | Nose score⁎ |
Trachea score⁎ | Bronchus score⁎ | |||
|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | ||||
| Air | NA | 0 ± 0 (0/9)† | 0 ± 0 (0/9)† | 0 ± 0 (0/9) | 0 ± 0 (0/9) | 0 ± 0 (0/9)† | 0.22 ± 0.22 (1/9) |
| Diacetyl | 240.3 ± 0.26 | 7.33 ± 0.21 (6/6)‡ | 4.00 ± 0.86 (5/6)‡ | 0.33 ± 0.33 (1/6) | 1.33 ± 0.84 (0/6) | 3.00 ± 0.73 (5/6)‡ | 0.67 ± 0.67 (1/6) |
| 2,3-Pentandione | 111.7 ± 0.12 | 4.17 ± 0.17 (6/6)†‡ | 1.33 ± 0.84 (2/6)† | 0 ± 0 (0/6) | 0 ± 0 (0/6) | 0.80 ± 0.80 (1/5) | 0.33 ± 0.33 (1/6) |
| 2,3-Pentandione | 241.2 ± 0.15 | 7.83 ± 0.17 (6/6)‡ | 5.33 ± 0.21 (6/6)‡ | 1.33 ± 0.84 (2/6) | 0.5 ± 0.5 (1/6) | 2.00 ± 1.15 (2/4)‡ | 0.80 ± 0.80 (1/5) |
| 2,3-Pentandione | 318.4 ± 0.17 | 8.33 ± 0.21 (6/6)†‡ | 7.33 ± 0.42 (6/6)†‡ | 5.5 ± 0.22 (6/6)†‡ | 4.33 ± 0.67 (6/6)†‡ | 7.50 ± 0.43 (6/6)†‡ | 2.67 ± 0.84 (4/6)‡ |
| 2,3-Pentandione | 354.2 ± 0.20 | 8.33 ± 0.21 (6/6)†‡ | 7.17 ± 0.31 (6/6)†‡ | 6.0 ± 0.36 (6/6)†‡ | 6.5 ± 0.5 (6/6)†‡ | 8.67 ± 0.21 (6/6)†‡ | 4.0 ± 0 (6/6)†‡ |
Histopathologic changes were diagnosed as necrotizing if cell death was a major component of the histopathologic change. Tissues evaluated were lined by respiratory or transitional epithelium. The olfactory epithelium was scored separately (see Table 4).
NA, not applicable.
Data are expressed as mean ± SE of the pathology scores, followed parenthetically by the number of affected rats/number of rats exposed. The specific nasal sections are designated sequentially (T1, T2, T3, and T4), starting with the most anterior section.
Significantly different from the group inhaling 240-ppm diacetyl (2,3-butanedione; P ≤ 0.05).
Significantly different from the air control group (P ≤ 0.05).
Table 4.
Necrotizing Rhinitis in the Olfactory Epithelium of Rats Inhaling 2,3-Pentanedione and 2,3-Butanedione
| Exposure | Concentration | Nose score⁎ |
||
|---|---|---|---|---|
| T2 | T3 | T4 | ||
| Air | NA | 0 ± 0 (0/9)† | 0 ± 0 (0/9)† | 0 ± 0 (0/9) |
| Diacetyl | 240.3 ± 0.26 | 2.33 ± 1.05 (3/6)‡ | 2.67 ± 1.20 (3/6)‡ | 0 ± 0 (0/6) |
| 2,3-Pentanedione | 111.7 ± 0.12 | 0 ± 0 (0/6) | 0 ± 0 (0/6) | 0 ± 0 (0/6) |
| 2,3-Pentanedione | 241.2 ± 0.15 | 4.00 ± 0.86 (5/6)‡ | 0 ± 0 (0/5)§ | 0 ± 0 (0/6) |
| 2,3-Pentanedione | 318.4 ± 0.17 | 6.17 ± 0.48 (6/6)†‡ | 1.60 ± 1.60 (1/5)§ | 0.83 ± 0.83 (1/6) |
| 2,3-Pentanedione | 354.2 ± 0.20 | 5.50 ± 0.43 (6/6)†‡ | 4.17 ± 1.40 (4/6)‡ | 0 ± 0 (0/6) |
Histopathologic changes were diagnosed as necrotizing if cell death was a major component of the histopathologic change.
NA, not applicable.
Data are expressed as mean ± SE of the pathology scores, followed parenthetically by the number of affected rats/number of rats exposed. The specific nasal sections are designated sequentially (T1, T2, T3, and T4), starting with the most anterior section. Olfactory epithelium is not present in section T1, and it is, therefore, not included in this table.
Significantly different from the group inhaling 240-ppm diacetyl (P ≤ 0.05).
Significantly different from the air control group (P ≤ 0.05).
Inadequate fixation of the dorsal portions of the nose by immersion in 10% neutral-buffered formalin prevented assessment of olfactory epithelial damage in one animal.
In the trachea and lung of control rats, tissues were within normal histological limits. Exposure-related changes in the lung of PD-exposed rats were restricted to the mainstem bronchus, were subtle, and were characterized by minimal individual cell apoptosis, which was statistically significant in the two highest-dose groups (Table 3 and Figure 2).
Figure 2.

Histopathological changes in the mainstem bronchus in a rat after inhaling 354 ppm. Changes are subtle and limited to a few cells with pyknosis or karyorrhexis (arrow) within the respiratory epithelium. Scale bar = 20 μm.
Time Course of PD-Induced Epithelial and Neuronal Injury
Because clinical evaluation in the dose-response experiment suggested delayed toxicity, rats exposed to 319-ppm PD were sacrificed at three different times after exposure: 0 to 2, 12 to 14, and 18 to 20 hours. As with the dose-response study, histopathological changes in PD-exposed rats were characterized by necrotizing rhinitis, tracheitis, and bronchitis (Table 5). Respiratory epithelial necrosis was most severe in the first two sections of the nose. Consistent with the delayed toxicity noted clinically, the pathological score in these first two nasal sections significantly increased during the first 12 to 14 hours after exposure (Table 5).
Table 5.
Time Dependency of Necrotizing Rhinitis, Tracheitis, and Bronchitis in Rats Inhaling 318-ppm 2,3-Pentanedione
| Time after exposure (hours) | Nose score⁎ |
Trachea score⁎ | Mainstem bronchus score⁎ | |||
|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | |||
| Air (18–20) | 0 ± 0 (0/6)†‡ | 0 ± 0 (0/6)†‡ | 0 ± 0 (0/6)†‡ | 0 ± 0 (0/6)†‡ | 0 ± 0 (0/6)†‡ | 0 ± 0 (0/5)†‡ |
| 2,3-Pentanedione (0–2) | 4.33 ± 0.21 (0/6)‡§ | 3.33 ± 0.80 (5/6)‡§ | 4.17 ± 0.16 (6/6)§ | 2.67 ± 0.84 (4/6)§ | 5.0 ± 0 (6/6)‡§ | 4.5 ± 0.29 (4/4)§ |
| 2,3-Pentanedione (12–14) | 8.00 ± 0.00 (6/6)†§ | 5.83 ± 0.17 (6/6)†§ | 4.17 ± 0.16 (6/6)§ | 2.50 ± 0.88 (4/6)§ | 4.0 ± 0 (6/6)†§ | 4.0 ± 0 (5/5)§ |
| 2,3-Pentanedione (18–20) | 7.67 ± 0.21 (6/6)†§ | 5.00 ± 0.26 (6/6)‡§ | 1.03 ± 0.42 (4/6)†‡§ | 1.33 ± 0.84 (2/6) | 3.5 ± 0.72 (5/6)†§ | 2.0 ± 1.16 (2/4) |
The exposure concentration was 318 ppm at each time point (range, 317.9—318.9 ppm). Morphologic changes were diagnosed as necrotizing if cell death was a major component of the histopathologic change. Tissues evaluated were lined by respiratory or transitional epithelium. The olfactory epithelium was scored separately (see Table 6).
Data are expressed as mean ± SE of the pathology scores, followed parenthetically by the number of affected rats/number of exposed rats examined. In all exposures, six rats were exposed. However, the mainstem bronchus was missing from a few sections. The specific nasal sections are designated sequentially (T1, T2, T3, and T4), starting with the most anterior section.
Significantly different from 0 to 2 hours after exposure (P ≤ 0.05).
Significantly different from 12 to 14 hours after exposure (P ≤ 0.05).
Significantly different from the air control group (P ≤ 0.05).
No changes were seen in H&E-stained sections or Luxol fast blue/PAS-stained sections of olfactory neuroepithelium from air control rats or in the olfactory neuroepithelium of rats sacrificed immediately after 6 hours' inhalation of 319-ppm PD (Table 6 and Figure 3A). However, apoptosis and foci of invagination were seen in the olfactory neuroepithelium at 12 hours (four of six rats) and 18 hours (two of six rats) after PD inhalation. Luxol fast blue/PAS staining demonstrated a consistent reduction in the amount of PAS-positive material in Bowman's glands in section T2 at 12 and 18 hours after exposure, and the sites of invagination within the olfactory neuroepithelium could often be localized near the excretory ducts of the Bowman's glands (Figure 3B).
Table 6.
Time Dependency of Necrotizing Rhinitis in the Olfactory Epithelium of Rats Inhaling 318-ppm 2,3-Pentanedione
| Time after exposure (hours) | Nose score⁎ |
||
|---|---|---|---|
| T2 | T3 | T4 | |
| Air (18–20) | 0 ± 0 (0/6)† | 0 ± 0 (0/6) | 0 ± 0 (0/6) |
| 2,3-Pentanedione (0–2) | 0 ± 0 (0/6)† | 0 ± 0 (0/6) | 0 ± 0 (0/6) |
| 2,3-Pentanedione (12–14) | 3.33 ± 1.12 (4/6)‡§ | 1.83 ± 1.17 (2/6) | 0 ± 0 (0/6) |
| 2,3-Pentanedione (18–20) | 2.00 ± 1.26 (2/6) | 0 ± 0 (0/6) | 0 ± 0 (0/6) |
The exposure concentration was 318 ppm at each time point (range, 317.9—318.9 ppm). Histopathologic changes were diagnosed as necrotizing if cell death was a major component of the histopathologic change.
Data are expressed as mean ± SE of the pathology scores, followed parenthetically by the number of affected rats/number of rats exposed. The specific nasal sections are designated sequentially (T1, T2, T3, and T4), starting with the most anterior section. Olfactory epithelium is not present in section T1, and it is, therefore, not included in this table.
Significantly different from 12 to 14 hours after exposure (P ≤ 0.05).
Significantly different from the air control group (P ≤ 0.05).
Significantly different from 0 to 2 hours after exposure (P ≤ 0.05).
Figure 3.

Luxol fast blue/PAS staining of the olfactory neuroepithelium. A: In an air-exposed control rat, the Bowman's glands contain PAS-positive secretory granules (dashed arrows), and their excretory ducts (solid arrows) traverse the basement membrane. In the lumen, small amounts of PAS-positive material are seen. B: In a PD-exposed rat from the 12-hour postexposure group, the lamina propria in this focus is devoid of PAS-positive Bowman's glands. A focus of invagination (asterisk) is associated with the greatest loss of cellularity in the olfactory neuroepithelium and lies above the excretory duct of the Bowman's gland, which traverses the basement membrane (solid arrow). In the nasal airway lumen, the amount of PAS-positive material is increased and is aggregated. Scale bar = 20 μm.
No neuropathologic alterations were seen in the brains of PD-exposed rats in H&E-stained sections or in sections stained by dual immunofluorescence for glucose transporter-1 and activated caspase-3.
Time Course of Apoptosis in PD-Induced Death of Respiratory Epithelium
PD-induced apoptotic cell death of respiratory epithelial cells was confirmed by both TUNEL assay and immunofluorescent staining for activated caspase 3 and E-cadherin in the maxilloturbinate from section T1. PD-induced apoptosis in respiratory epithelium was quantified via cell counts in the maxilloturbinate in section T1 in a TUNEL assay and was confirmed by morphometry for caspase 3 activation in the maxilloturbinate. Apoptosis was significantly increased at all time points, as assessed by caspase 3 activation or TUNEL. With the activated caspase 3 assay, but not with the TUNEL assay, apoptosis was significantly greater at 12 and 18 hours, as opposed to immediately after exposure (Table 7).
Table 7.
Apoptosis in the Lining Epithelium of the Maxilloturbinate in Section T1, Quantified by Two Different Methods at Three Different Time Periods after Inhaling 318-ppm PD (n = 6 in Each Group)
| Variable | Air | Time after 2,3-pentanedione (hours) |
||
|---|---|---|---|---|
| 0 | 12 | 18 | ||
| TUNEL apoptotic cell count | 0.00 ± 0.00 | 15.33 ± 6.873⁎ | 34.50 ± 13.62⁎ | 26.17 ± 7.78⁎ |
| Activated caspase-3 morphometry | 0.10 ± 0.10 | 0.33 ± 0.21⁎ | 2.87 ± 0.98⁎† | 2.47 ± 1.19⁎† |
Data are given as affected cell number (mean ± SE cell count/mm basement membrane) and as the area of caspase-3 expression (μm2) in the airway epithelium above each μm of basement membrane. The exposure concentration was 318 ppm at each time point (range, 317.9—318.9 ppm).
Significantly different from the air control group (P ≤ 0.05).
Significantly different from 0 to 2 hours after exposure (P < 0.05).
Dose-Response Study
Olfactory Neurotoxicity
To identify whether PD caused neurotoxicity in olfactory neurons, nasal section T2 was stained using dual-label immunofluorescence for OMP, a specific marker for olfactory neurons; and activated caspase 3, a marker for apoptosis. Activated caspase 3 could be demonstrated within the olfactory neuroepithelium. Caspase 3 activation was rare in the neuroepithelium of air-exposed rats (Figure 4A). Caspase 3 activation varied from rare to occasional in rats inhaling 320- to 360-ppm PD.
Figure 4.
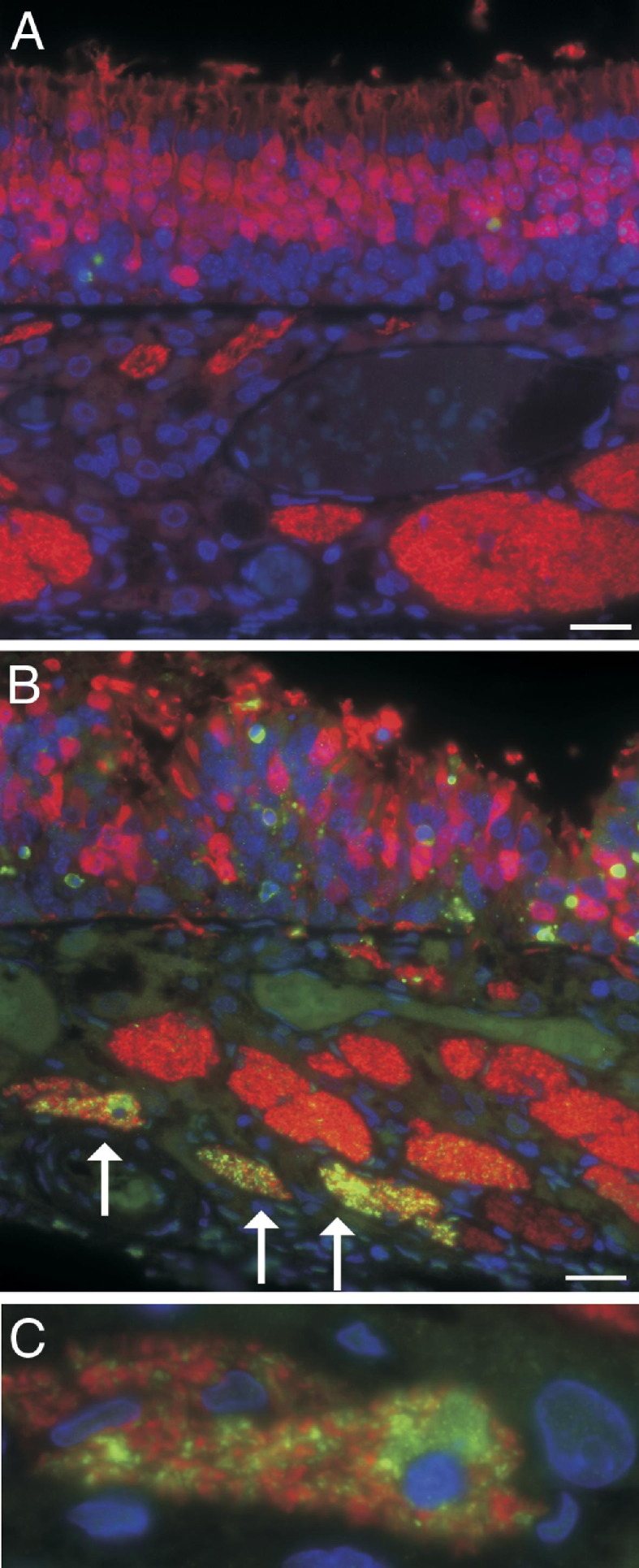
Immunofluorescence for activated caspase 3 (green) demonstrates cells in the execution stage of apoptosis. Immunofluorescence for OMP (red) demonstrates neurons. A: Olfactory neuroepithelium of an air-exposed control rat demonstrates numerous well-organized neuroepithelial cells (red) within the epithelium, with rare apoptotic cells (green). Beneath the neuroepithelium, olfactory nerve bundles also express OMP and stain red but show no evidence of apoptosis. B: Olfactory neuroepithelium from a rat after 6-hour inhalation exposure to 354-ppm PD. Within the neuroepithelium, the number of olfactory neurons (red) is reduced; the olfactory neurons are disorganized; and many olfactory neurons have variations in size and swelling of dendrites, consistent with degenerative changes. In the lamina propria, some olfactory nerve bundles do not contain axons with activated caspase 3, whereas other olfactory nerve bundles contain many apoptotic axons (arrows). C: A higher magnification of an olfactory nerve bundle from B showing activated caspase 3 in axons and a cell morphologically consistent with a macrophage containing apparently phagocytized cellular debris of axons with caspase 3 activation. Scale bar = 20 μm.
The OMP staining in these sections demonstrated the olfactory neurons and allowed clear visualization of PD-associated morphological changes within the neuroepithelium and the olfactory nerve bundles. In rats inhaling 240-ppm diacetyl or ≥240-ppm PD, OMP immunofluorescence demonstrated irregular spacing of olfactory neurons, small olfactory neurons, loss of sustentacular cells, and swelling and shortening of dendrites of olfactory neurons. Foci of disorganized neurons were most prominent near foci of invagination of the olfactory neuroepithelium. In the most severely affected rat, activation of caspase 3 in multiple axons was demonstrated within specific olfactory nerve bundles, with the most severely affected nerve bundle associated with a macrophage expressing activated caspase 3 (Figure 4, B and C).
Relationship between Olfactory Neurotoxicity and DCXR Expression
The olfactory neuroepithelium contained immunoreactive DCXR, which was distinguishable from negative controls and from expression in the respiratory epithelium, which has lower levels of DCXR expression.26 Within the olfactory neuroepithelium, DCXR was expressed in cells in an exposure-dependent manner.
In the olfactory neuroepithelium of air-exposed rats, DCXR was in the cytoplasm of sustentacular cells and, to a lesser extent, basal cells. By using OMP as the second label for dual-label immunofluorescence, DCXR expression in olfactory neurons and in the lamina propria was indistinguishable from that in negative controls (Figure 5, A–F). After inhalation of 320- or 360-ppm PD, the olfactory neurons and sustentacular cells were markedly disorganized relative to controls, leading to a loss of the apical barrier of DCXR, which characterized the control neuroepithelium (Figure 6A). In the most extensively damaged neuroepithelium, neurons and sustentacular cells detached (Figure 6B). In some less affected foci, DCXR immunoreactivity changed and was sequestered as an intensely immunofluorescent lining of apical cytoplasmic vacuoles in sustentacular cells (Figure 6C). By using E-cadherin as a second label for dual-label immunofluorescence, the ducts of the Bowman's glands did not contain DCXR detectable by immunofluorescence in either air-exposed (Figure 7A) or PD-exposed (Figure 7B) rats.
Figure 5.

Dual-label confocal immunofluorescence for DCXR and OMP in the olfactory neuroepithelium of nasal section T2 in an air-exposed control rat. A: DCXR expression (red) is restricted to the olfactory neuroepithelium. B: Adding a second label for OMP (green) demonstrates that the OMP-positive neurons are in cells without demonstrable DCXR. C: Negative control showing low levels of autofluorescence similar to what is seen in the lamina propria and olfactory neurons in A and B. D: Higher magnification of DCXR expression in the apical cytoplasm of sustentacular cells showing a line of the diketone metabolizing enzyme at the air interface. E: OMP (green) demonstrates that the dendrites and cell bodies of neurons have little or no DCXR. F: High-magnification negative control showing low levels of autofluorescence. Scale bar = 10 μm.
Figure 6.

PD-induced changes in dual-label immunofluorescence for DCXR (red) and OMP (green) in nasal section T2. A: In the air-exposed olfactory neuroepithelium, DCXR is abundant in the apical cytoplasm of sustentacular cells at the air interface. B: PD-induced cytotoxicity in the olfactory neuroepithelium disrupts the continuity of the layer of DCXR (red) at the air interface and increases exposure of neurons to the air interface. Sustentacular cells and neurons are detaching (arrows). Inset: UV fluorescence is added to demonstrate DAPI-stained nuclei in detaching cells. C: Representative focus demonstrating aggregation of DCXR (red) into vacuoles in the apical cytoplasm of sustentacular cells after inhaling 360-ppm PD. Scale bar = 20 μm.
Figure 7.

Dual-label immunofluorescence for DCXR (red) and E-cadherin (green). A: In the air-exposed olfactory neuroepithelium, DCXR is abundant in the apical cytoplasm of sustentacular cells, but no detectable DCXR is in the excretory duct (arrow) of the Bowman's glands. B: In the PD-exposed neuroepithelium, a focus of severe cytotoxicity is localized to the excretory duct (arrow) of the Bowman's glands, which does not contain DCXR. Scale bar = 20 μm.
Time Course Experiment with Confirmation of Olfactory Neuronal Injury
In the time course experiment, as in the dose-response study, dual-label immunofluorescence for activated caspase 3 and OMP in nasal section T2 demonstrated rare apoptosis in air-exposed rats, with increased amounts of apoptosis in the neuroepithelium of PD-exposed rats (Figure 8A). Caspase 3 activation varied among the PD-exposed animals but clearly played a role in cell death of the olfactory neuroepithelium in some rats (Figure 8A). The OMP staining demonstrated morphological changes within the cell bodies and dendrites of olfactory neurons (Figure 8B). The morphological changes in olfactory nerve cell bodies and dendrites were equivocal to subtle immediately after exposure, but variations in the size and shape of olfactory nerves, clustering of olfactory nerves, and shortening and swelling of dendrites were clearly visualized by OMP staining at the 12- and 18-hour postexposure time points. Within the olfactory neuroepithelium, caspase 3 activation was most frequently seen in cells without OMP, suggesting that these were the sustentacular cells. In the olfactory nerve bundles that contained the axons of the olfactory nerves, caspase 3 activation was absent in air-exposed rats, was equivocal in a few axons in one of six rats immediately after exposure, was clearly demonstrated in all six rats at 12 hours after exposure, and was clearly demonstrated in three of six rats at 18 hours after exposure. Caspase 3 activation was expected in axons because of apoptosis in the olfactory neuroepithelium, which contained the cell bodies of neurons, but frequently involved many axons localized to a few olfactory bundles located near completely unaffected olfactory nerve bundles (Figure 8B), which served as internal negative controls. Rare caspase 3 activation was observed in cells morphologically consistent with macrophages that were within olfactory nerve bundles. Caspase 3 activation was never observed in the olfactory nerve bundles of air-exposed controls.
Figure 8.

A: Dual-label immunofluorescence for activated caspase 3 (green) and OMP (red). Apoptosis is demonstrated by caspase 3 activation, and olfactory neurons are demonstrated by expression of OMP. In this rat from the 12-hour PD postexposure group, apoptosis and invagination of the olfactory neuroepithelium are clearly demonstrated. Olfactory nerve bundles (dashed arrows) that contain the axons of the olfactory nerves are beneath the olfactory neuroepithelium. The respiratory epithelium (solid arrows) does not contain olfactory neurons. Scale bar = 50 μm. B: Confocal microscopic image of dual-label immunofluorescence for activated caspase 3 (green) and OMP (red) in the olfactory neuroepithelium in a rat from the 12-hour PD postexposure group. Olfactory neuron dendrites are often swollen or shortened (solid arrows), and apoptosis of axons in an olfactory nerve bundle is demonstrated by expression of activated caspase 3 (dashed arrow). Scale bar = 10 µm.
FluoroJade B staining confirmed olfactory neurotoxicity in section T2 at all time points. FluoroJade B staining was seen in the olfactory neuroepithelium in section T2 in all but one PD-exposed rat but was absent from the neuroepithelium of air-exposed control rats (Figure 9A). FluoroJade B staining in the olfactory nerves of PD-exposed rats was localized to the junction of the dendrite and olfactory neuron cell body (Figure 9B).
Figure 9.

FluoroJade B staining for degenerating neurons stains olfactory neurons of PD-exposed, but not control, rats. A: Olfactory neuroepithelium of nasal section T2 in an air-exposed control rat. B: Olfactory neuroepithelium of nasal section T2 in a PD-exposed rat. C: Olfactory neuroepithelium of nasal section T4 in a PD-exposed rat. Scale bar = 20 μm.
FluoroJade B staining was demonstrated in the olfactory neurons of the neuroepithelium in nasal section T4 in four of six rats at 0 hours after exposure, five of six rats at 12 hours after exposure, and three of six rats at 18 hours after exposure (Figure 9C). Olfactory neurons in nasal section T4 of air-exposed rats were consistently FluoroJade B negative. Unlike the neurons in the olfactory neuroepithelium, neurons in the OB of nasal section T4 and neurons in brain sections did not stain with FluoroJade B in either air- or PD-exposed rats. In H&E-stained sections of brain, no morphological alterations were detected in air- or PD-exposed rats. Dual-label immunofluorescent staining of brain sections for activated caspase 3 and the brain vascular marker, glucose transporter-1, did not demonstrate any PD-associated changes in apoptosis and/or the vasculature of the brain (data not shown).
Central Neurotoxicity in PD-Exposed Rats
Rats inhaling PD (270 ppm, 6 hours 41 minutes) exhibited brain region–specific changes in expression of inflammatory mediators and blood-brain barrier (BBB)–related markers by 24 hours after exposure. Specifically, PD induced the expression of IL-6, inducible Nos2, and Cldn1 transcripts and reduced the expression of vascular endothelial growth factor A (Vegf-A) in discrete brain areas (Figure 10, A–D). Small, but significant, increases in the mRNA expression of IL-6 were seen in the OB (twofold, P = 0.0024), STR (2.7-fold, P = 0.0017), HIP (2.3-fold, P = 0.0003), and CER (1.7-fold, P = 0.0013) (Figure 10A). PD also significantly enhanced the expression of inducible Nos2 in the OB (5.9-fold, P = 0.0001), STR (17-fold, P = 0.0001), HIP (4.9-fold, P = 0.032), and CER (5.0-fold, P = 0.0067) (Figure 10B). PD-mediated alterations in the expression of the BBB marker, Cldn1, were observed only in the OB (3.7-fold, P = 0.0001) and STR (4.6-fold, P = 0.0001), but not in the HIP or CER (Figure 10C). The expression of other BBB markers, including occludin, ICAM-1, and VACM1, were not altered in any of the brain areas examined (data not shown). The expression of the angiogenesis, vascular permeability, and neuroprotective factor Vegf-A was significantly decreased in the OB (to 68% of control, P = 0.0003), STR (to 61% of control, P = 0.0041), HIP (to 53% of control, P = 0.0005), and CER (to 57% of control, P = 0.0007) (Figure 10D).
Figure 10.
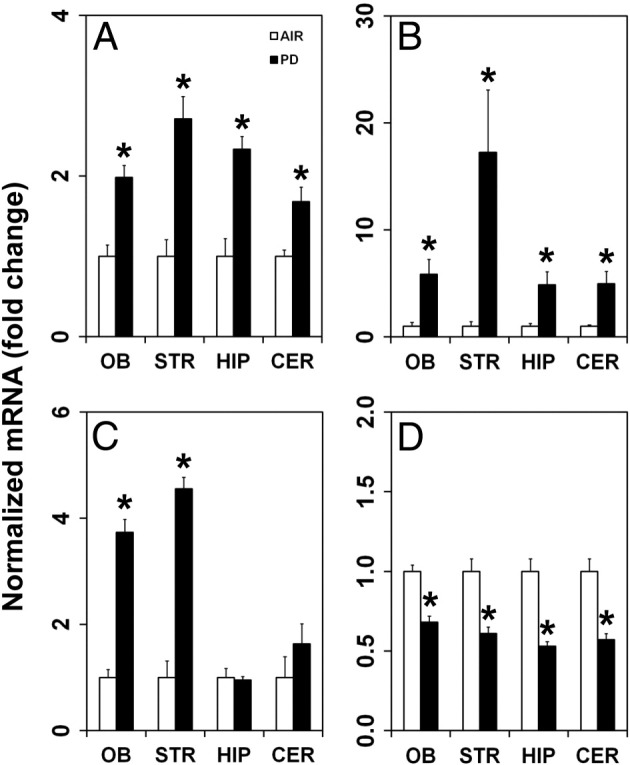
Inhalation of PD (270 ppm, 6 hours 41 minutes) alters mRNA expression in discrete brain areas. A: The inflammatory mediator IL-6 is induced in the OB, STR, HIP, and CER. B: The inflammatory mediator Nos2 is up-regulated in the OB, STR, HIP, and CER. C: Cldn1 is increased in the OB and STR, but not in the HIP or CER. D: Vegf-A expression decreases in the OB, STR, HIP, and CER. AIR denotes control group exposed to air. Asterisk denotes significant change from air-exposed controls.
Discussion
Our study demonstrates that PD, as with diacetyl, damages airway epithelium and suggests that additional α-dicarbonyl flavorings are potentially toxic to airway epithelium. This finding is important because damage to the airway epithelium of the bronchiole is believed to be the underlying cause for bronchiolitis obliterans.22, 24 Our study also provides evidence that is consistent with the established recommendation that substitution of flavorings in foods should use agents only where there is evidence that the substitute is less toxic than the agent it replaces (National Institute for Occupational Safety and Health, http://www.cdc.gov/niosh/topics/flavorings, last accessed April 14, 2012).
The PD exposures selected for this study were similar to the concentrations used in previous short-term inhalation toxicity studies of diacetyl in the rat.19 The lowest concentration was similar to the highest concentration measured as a time-weighted average in a workplace.2 The highest concentration used was designed to be threefold greater than the lowest exposure and less than a third of the peak potential exposure concentration for diacetyl, which was considered to be the concentration in the headspace of mixing vats in a workplace.2
Because rats are obligate nasal breathers, inhaled PD enters the respiratory tract through the nose. Within the nose, airflow may travel through a ventral or a dorsal pathway. Most airflow (approximately 88%) is in the ventral pathway.26, 49 Respiratory epithelium lines most of the ventral airflow pathway in the nose and also lines the bronchi and bronchioles of the intrapulmonary airways, although the respiratory epithelium of the intrapulmonary airways is shorter and includes many Clara cells. At these exposure concentrations, PD caused epithelial damage predominantly in the upper airways of rats, rather than in the bronchioles that are the major target site in flavorings-related lung disease in humans. This is similar to the pattern of acute airway injury seen in rodents inhaling vapors of diacetyl or a diacetyl-containing butter flavoring mixture.19, 20, 21 The anatomical location of respiratory epithelial injury appears to be explained by dose to the respiratory epithelium, and the anatomical location of cytotoxic doses for the respiratory epithelium can differ between species. For diacetyl, computational fluid dynamic–physiologically based pharmacokinetic models have been developed that indicate that lightly exercising workers have much higher concentrations of diacetyl in bronchioles than do rats.26, 50 Computational fluid dynamic–physiologically based pharmacokinetic models have not been developed for PD, but a similar pattern of interspecies differences is likely, given its structural similarity to diacetyl. In our study, histopathological evidence of airway epithelial injury in H&E sections was most apparent in the airway respiratory and transitional epithelium. Thus, as with diacetyl, PD has remarkable respiratory epithelial cytotoxicity. Comparative studies of airway reactivity changes in rats after diacetyl and pentanedione exposure are ongoing, and preliminary findings have been published in abstract form.51
By using TUNEL assays and activated caspase 3 immunofluorescence to augment the evaluation of H&E sections, we confirmed the histopathological impression that apoptotic cell death was a component of PD-induced airway epithelial injury. Although physiological apoptosis can be a controlled process that avoids inflammation associated with necrotic cell death,52, 53 apoptotic cells that are not rapidly phagocytized contribute to inflammation and fibrosis.54, 55 While recognizing that modifiers may be needed, the Society of Toxicologic Pathologists recommends that toxicologic pathologists use the term necrosis for the diagnosis of all forms of cell death seen in tissue sections.56 As was recently reviewed, secondary necrosis is a typical outcome for apoptotic cells that are not phagocytosed.57
In addition to cytotoxicity in the respiratory and transitional epithelium, PD caused cytotoxicity in the olfactory neuroepithelium. In the nose, the dorsal airflow pathway receives a small, but significant, percentage of the airflow (approximately 12%).26, 49 Much of the dorsal pathway is lined by olfactory neuroepithelium, which is histologically and functionally distinct from respiratory epithelium.58 The olfactory neuroepithelium is principally composed of olfactory neurons that have odorant receptors, the sustentacular cells that have supportive and xenobiotic metabolizing function, and basal cells. The surface is coated by mucous, which is carried through the epithelium by excretory ducts originating in the Bowman's glands of the lamina propria. In our study, PD caused acute injury to the olfactory neuroepithelium, with foci of invagination centered at the excretory ducts of the Bowman's glands, the loss of Bowman's glands, and an increase in PAS-positive material within the lumen. That pattern of damage suggests that PD is selectively more toxic to the part of the neuroepithelium containing the ducts of the Bowman's glands. The epithelial cells lining these ducts selectively express several enzymes important for xenobiotic metabolism, including aldehyde oxidase 1 and cytochrome P450 2A5.59, 60 Another xenobiotic metabolizing enzyme, cytochrome P450 4B1, is believed to be secreted into the lumen of the Bowman's glands.61 Indeed, a recent study62 suggests that, as a group, the xenobiotic metabolizing enzymes of the murine neuroepithelium are generally expressed in the Bowman's glands and/or ducts and the sustentacular cells. Some of these enzymes, including cytochrome P450 2A4, glutathione-S-transferase mu2, and carbonyl reductase 2, are also expressed in the respiratory epithelium.62 Furthermore, our dual-label immunofluorescence for OMP and activated caspase 3 suggests that foci of damage centered at ducts of Bowman's glands and that apoptosis of olfactory neurons was less frequent than apoptosis of sustentacular cells. Each of these observations suggests that the remarkable airway epithelial toxicity of PD may have a selective component. In particular, because xenobiotic metabolism in general is frequently localized to the ducts of the Bowman's glands, the sustentacular cells, and the respiratory epithelium,59, 60, 61, 62 metabolism or another process localized to the sites of metabolism could play a role in the toxicity of PD. Conversely, xenobiotic metabolism may be localized to sustentacular cells and ducts of the Bowman's glands to metabolize xenobiotics to less toxic compounds before they reach the delicate olfactory neurons.
Indeed, existing studies suggest that neurons should be particularly sensitive to the cytotoxic effects of the α-diketones. As a class, the α-dicarbonyl compounds cause cytotoxicity through three described mechanisms: i) modification of essential proteins63, 64, 65, 66, 67, 68; ii) interactions with DNA, including the formation of 2-deoxyguanosine adducts69; and iii) cell injury by reactive oxygen species.70, 71, 72 Much less is known about factors, such as metabolism, that may influence sites of cytotoxicity in vivo. Studies of the role of metabolism in α-dicarbonyl compound cytotoxicity are largely focused on metabolism of methylglyoxal, a three-carbon α-dicarbonyl compound with one ketone group and one aldehyde group. Methylglyoxal can be metabolized by reduction and oxidation reactions, but is principally metabolized and detoxified by a conjugation reaction catalyzed by glyoxalase 1, followed by a hydrolysis reaction catalyzed by glyoxalase 2.73, 74, 75 Consistent with a detoxifying role, enhanced activity of glyoxalase 1 decreases methylglyoxal-associated reactive oxygen species, protein modification, and cytotoxicity.73, 76 However, α-dicarbonyl compounds, such as PD and diacetyl, have two adjacent ketone groups. These α-dicarbonyl compounds are known as α-diketones and are principally metabolized by reduction reactions that are catalyzed by DCXR, with NADPH as a cofactor.46, 77 DCXR is believed to be important in detoxifying α-diketones.46 The relationship between DCXR and PD toxicity is particularly important because DCXR has a somewhat higher affinity and a greater kcat/Km for PD than for diacetyl.46 However, DCXR is not always detoxifying, because increased DCXR expression actually enhanced 9,10-phenanthrenequinone-induced cytotoxicity in vitro.70 What our in vivo molecular pathology study shows is that it is possible for DCXR to be both detoxifying and to increase cytotoxicity in cells that have DCXR activity. The net effect for the olfactory neurons should be that they receive little diketone exposure in the normal in vivo environment because the adjacent sustentacular cells have abundant DCXR activity in the apical cytoplasm, which is in contact with the air interface.
Our study demonstrates that exposures to PD at ≥320 ppm can alter the organized expression of PD in neuroepithelium so that the extensive expression of DCXR at the air interface is disrupted. In addition, apoptosis was demonstrated by activated caspase 3 staining in sustentacular cells, suggesting further disruption in PD metabolism through loss of sustentacular cells. In the disordered PD-exposed neuroepithelium, olfactory neurons have more cytoplasm in close proximity to the dorsal air stream in the nose, where the cellular dose of volatile α-diketones will correspondingly increase in subsequent exposures. Our study demonstrates that neurons have less DCXR and should, thus, be less capable of metabolizing PD than sustentacular cells. Furthermore, in vitro data from other laboratories suggest that neurons may be defective in antioxidant responses needed to respond to dicarbonyl-induced cytotoxicity.72 In addition, we noted the aggregation of DCXR in cytoplasmic vacuoles in some sustentacular cells, suggesting degradation of DCXR after high-dose PD exposures. Impairment of detoxification pathways has previously been noted in hippocampal neurons after exposure to methylglyoxal.78 As previously mentioned, the α-dicarbonyl compounds modify proteins, and this is a mechanism for altering enzyme activity.63, 64, 65, 66, 67, 68
Upper airway metabolism is also important in changing respiratory tract dosimetry to the deep lung. The effect of metabolism on PD dosimetry has not been estimated, but dosimetry estimates are available for diacetyl. In the absence of metabolism, it is estimated that inhaling 1-ppm diacetyl would produce a bronchiolar tissue concentration of 0.002 μmol/L in the presence of metabolism but a bronchiolar tissue concentration of 1.0 μmol/L in the absence of metabolism.25 In our study, the lack of demonstrable DCXR in the ducts of the Bowman's glands, where cytotoxicity was greatest, suggests that it may have a protective role, as originally described.46 The role of DCXR in the toxicity of diacetyl and PD, the potential inactivation of DCXR by its substrates, and important consequences for airway dosimetry in long-term studies will need additional investigation. Our study clearly demonstrates the need for considering metabolism and understanding the role of metabolism in adjacent cells when designing in vitro mechanistic studies for evaluating the toxicity of inhaled diketones.
FluoroJade B staining clearly demonstrated widespread olfactory neuronal degeneration exceeding the neuronal injury demonstrated by H&E staining or immunofluorescence for caspase 3 activation. In addition, altered neuronal cytomorphological characteristics, decreased neuronal cells within the neuroepithelium, and detaching neurons were demonstrated in OMP-stained sections. With olfactory neuron damage demonstrated in the olfactory neuroepithelium by activated caspase-3, FluoroJade B, and OMP staining, damage to the axons of those olfactory nerves was expected. Indeed, we selected activation of caspase 3 as a method to evaluate axon apoptosis because it is present in the cytoplasm and, therefore, can be detected in axons by immunofluorescence.79 Because the axons of olfactory nerve synapse within the OB, this technique demonstrates how apoptotic mediators can cross the cribriform plate. Surprisingly, individual olfactory nerve bundles from PD-exposed rats often had activation of caspase 3 in many, or even most, of the axons. Although rare macrophages were seen in those nerve bundles, these were insufficient to phagocytose the apoptotic axons. Indeed, a few of those macrophages actually expressed activated caspase-3. Thus, many of those apoptotic neurons and possibly the apoptotic macrophages will not be phagocytosed, resulting in secondary necrosis.57 The extensive degeneration and disorganization of olfactory neurons suggest that additional necrotic death occurred within olfactory neurons, but necrotic neurons cannot be detected by caspase 3 activation, although their axons also cross the cribriform plate.
The transmission of a death signal across the cribriform plate seems the most likely explanation for the selective elevation of claudin-1 in the OB and STR in the brains of rats exposed to PD. Although claudin-1 is a component of the BBB, other components of the BBB, including claudin-3, claudin-5, and occludin, were not significantly elevated, suggesting that the elevation in claudin-1 may be something other than an effect on the BBB. Because the OB receives the first synapse from the olfactory neurons, the STR is in the synaptic pathway from the OB, and claudin-1 damage was only seen in these two regions, it seems reasonable that the damage signal was transmitted from the olfactory nerve bundles. In contrast, expression of the inflammatory mediators IL-6 and Nos2 was increased in all brain regions tested, suggesting that these changes may reflect vascular injury. Moderate down-regulation of Vegf-A in all sampled regions of PD-exposed brains is more difficult to explain, but the proposed neuroprotective role of Vegf-A suggests that further investigation of this effect may be needed.80, 81, 82, 83, 84, 85
Neuropathologic injury was not observed in histopathological sections of the OB, suggesting that either PD did not produce detectable morphological change in the OB or the injury was so focal it did not appear on the sections. Consistent with highly focal injury, the localization of marked caspase 3 activation in specific olfactory nerve bundles would be expected to produce focal changes in the OB. This may be comparable to the transmission of signals from odorant receptors, in which roughly 1000 different odorant receptors are randomly and widely distributed within one of the four zones of the neuroepithelium, but these widely distributed neurons for a specific odorant receptor synapse at between one and a few glomeruli in each OB.86, 87, 88, 89 Interestingly, high exposures of diacetyl (2500 ppm for 45 minutes) rapidly increase deoxyglucose uptake in two small foci in the OB.90 Although that uptake is increased in the first hour after exposure during a time period when caspase 3 is not yet activated in the olfactory nerve bundles of PD-exposed rats, it suggests the intriguing hypothesis that the localization of injury to selected olfactory nerve bundles could be related to the highly organized topography of axons from neurons with specific odorant receptors.87, 88 Other possible explanations, such as injury caused by secondary necrosis of macrophages migrating into specific olfactory nerve bundles and foci of necrosis affecting specific nerve bundles, are also worthy of investigation. More important, the structurally related β-diketone, 2,4-pentanedione, is also neurotoxic, with the first histopathological change, malacia, seen after the 12th exposure day to 650 ppm.43 Thus, the neurotoxicity of PD and the absence of morphological changes using standard histopathological techniques have some similarities to what has previously been reported for 2,4-pentanedione.
In summary, in this study, we have found respiratory toxicity, olfactory neurotoxicity, and central neurotoxicity for an α-diketone flavoring agent classified as generally recognized as safe under conditions of its normal use when consumed in food.28, 29 This study, as with previous studies with diacetyl, is a reminder that a chemical with a long history of being eaten without any evidence of toxicity, can still be an agent with respiratory toxicity when appropriate studies are conducted. Our study also suggests that shared features of the short-chain diketones may be related to their toxicity when inhaled. The direct effect of the reactive α-diketone group and the ability of the α-diketones to modify proteins and nucleic acids are features consistent with the direct cytotoxicity of diacetyl and PD.11, 12, 13, 14, 15 This study also provides insights into the role of metabolism in the pathogenesis of injury to the olfactory neuroepithelium and brain of PD-exposed rats. Given the selective toxicity of PD to olfactory neurons in the neuroepithelium that express little DCXR, and the aggregation of immunoreactive DCXR in cytoplasm vacuoles of PD-exposed sustentacular cells, our study suggests that DCXR may have a detoxifying role, but may be a target of PD toxicity. Thus, our study suggests several intriguing potential mechanisms for the toxicity of inhaled volatile α-diketones, demonstrates mRNA changes in the brain, documents olfactory neurotoxicity, and clearly demonstrates that the remarkable airway toxicity of diacetyl is shared with its close structural relative, PD.
Footnotes
Supported by National Occupational Research Agenda intramural project 1927ZBEH from the National Institute for Occupational Safety and Health.
The findings and conclusions herein are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health. Mention of brand names does not constitute product endorsement.
Current address of M.G.: Department of Diagnostic Medicine/Pathobiology, College of Veterinary Medicine, Kansas State University, Manhattan, Kansas.
References
- 1.Centers for Disease Control and Prevention Fixed obstructive lung disease in workers at a microwave popcorn factory–Missouri, 2000–2002. MMWR Morb Mortal Wkly Rep. 2002;51:345–347. [PubMed] [Google Scholar]
- 2.Kreiss K., Gomaa A., Kullman G., Fedan K., Simoes E.J., Enright P.L. Clinical bronchiolitis obliterans in workers at a microwave-popcorn plant. N Engl J Med. 2002;347:330–338. doi: 10.1056/NEJMoa020300. [DOI] [PubMed] [Google Scholar]
- 3.Parmet A.J., Von Essen S. Rapidly progressive, fixed airway obstructive disease in popcorn workers: a new occupational pulmonary illness? J Occup Environ Med. 2002;44:216–218. doi: 10.1097/00043764-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Das R., Blanc P.D. Chlorine gas exposure and the lung: a review. Toxicol Ind Health. 1993;9:439–455. doi: 10.1177/074823379300900304. [DOI] [PubMed] [Google Scholar]
- 5.Visscher D.W., Myers J.L. Bronchiolitis: the pathologist's perspective. Proc Am Thorac Soc. 2006;3:41–47. doi: 10.1513/pats.200512-124JH. [DOI] [PubMed] [Google Scholar]
- 6.Ghanei M., Fathi H., Mohammad M.M., Aslani J., Nematizadeh F. Long-term respiratory disorders of claimers with subclinical exposure to chemical warfare agents. Inhal Toxicol. 2004;16:491–495. doi: 10.1080/08958370490442421. [DOI] [PubMed] [Google Scholar]
- 7.Ghanei M., Mokhtari M., Mohammad M.M., Aslani J. Bronchiolitis obliterans following exposure to sulfur mustard: chest high resolution computed tomography. Eur J Radiol. 2004;52:164–169. doi: 10.1016/j.ejrad.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 8.Ghanei M., Tazelaar H.D., Chilosi M., Harandi A.A., Peyman M., Akbari H.M., Shamsaei H., Bahadori M., Aslani J., Mohammadi A. An international collaborative pathologic study of surgical lung biopsies from mustard gas-exposed patients. Respir Med. 2008;102:825–830. doi: 10.1016/j.rmed.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Rowell M., Kehe K., Balszuweit F., Thiermann H. The chronic effects of sulfur mustard exposure. Toxicology. 2009;263:9–11. doi: 10.1016/j.tox.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Dobler D., Ahmed N., Song L., Eboigbodin K.E., Thornalley P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes. 2006;55:1961–1969. doi: 10.2337/db05-1634. [DOI] [PubMed] [Google Scholar]
- 11.Kovacic P., Cooksy A.L. Role of diacetyl metabolite in alcohol toxicity and addiction via electron transfer and oxidative stress. Arch Toxicol. 2005;79:123–128. doi: 10.1007/s00204-004-0602-z. [DOI] [PubMed] [Google Scholar]
- 12.Miller A.G., Gerrard J.A. Assessment of protein function following cross-linking by alpha-dicarbonyls. Ann N Y Acad Sci. 2005;1043:195–200. doi: 10.1196/annals.1333.024. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez Mellado J.M., Ruiz Montoya M. Correlations between chemical reactivity and mutagenic activity against S. typhimurium TA100 for alpha-dicarbonyl compounds as a proof of the mutagenic mechanism. Mutat Res. 1994;304:261–264. doi: 10.1016/0027-5107(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 14.Wondrak G.T., Cervantes-Laurean D., Roberts M.J., Qasem J.G., Kim M., Jacobson E.L., Jacobson M.K. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem Pharmacol. 2002;63:361–373. doi: 10.1016/s0006-2952(01)00915-7. [DOI] [PubMed] [Google Scholar]
- 15.Roberts D.W., York M., Basketter D.A. Structure-activity relationships in the murine local lymph node assay for skin sensitization: alpha, beta-diketones. Contact Dermatitis. 1999;41:14–17. doi: 10.1111/j.1600-0536.1999.tb06201.x. [DOI] [PubMed] [Google Scholar]
- 16.Schachter E.N. Popcorn worker's lung. N Engl J Med. 2002;347:360–361. doi: 10.1056/NEJMe020064. [DOI] [PubMed] [Google Scholar]
- 17.Kanwal R., Kullman G., Piacitelli C., Boylstein R., Sahakian N., Martin S., Fedan K., Kreiss K. Evaluation of flavorings-related lung disease risk at six microwave popcorn plants. J Occup Environ Med. 2006;48:149–157. doi: 10.1097/01.jom.0000194152.48728.fb. [DOI] [PubMed] [Google Scholar]
- 18.Kreiss K. Flavoring-related bronchiolitis obliterans. Curr Opin Allergy Clin Immunol. 2007;7:162–167. doi: 10.1097/ACI.0b013e3280298235. [DOI] [PubMed] [Google Scholar]
- 19.Hubbs A.F., Goldsmith W.T., Kashon M.L., Frazer D., Mercer R.R., Battelli L.A., Kullman G.J., Schwegler-Berry D., Friend S., Castranova V. Respiratory toxicologic pathology of inhaled diacetyl in Sprague-Dawley rats. Toxicol Pathol. 2008;36:330–344. doi: 10.1177/0192623307312694. [DOI] [PubMed] [Google Scholar]
- 20.Hubbs A.F., Battelli L.A., Goldsmith W.T., Porter D.W., Frazer D., Friend S., Schwegler-Berry D., Mercer R.R., Reynolds J.S., Grote A., Castranova V., Kullman G., Fedan J.S., Dowdy J., Jones W.G. Necrosis of nasal and airway epithelium in rats inhaling vapors of artificial butter flavoring. Toxicol Appl Pharmacol. 2002;185:128–135. doi: 10.1006/taap.2002.9525. [DOI] [PubMed] [Google Scholar]
- 21.Morgan D.L., Flake G.P., Kirby P.J., Palmer S.M. Respiratory toxicity of diacetyl in C57BL/6 mice. Toxicol Sci. 2008;103:169–180. doi: 10.1093/toxsci/kfn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King T.E., Jr Bronchiolitis obliterans. Lung. 1989;167:69–93. doi: 10.1007/BF02714935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hubbs A.F., Battelli L.A., Mercer R.R. Inhalation toxicity of the flavoring agent, diacetyl (2,3-butanedione), in the upper respiratory tract of rats. Toxicol Sci. 2004;78:438–439. [Google Scholar]
- 24.Palmer S.M., Flake G.P., Kelly F.L., Zhang H.L., Nugent J.L., Kirby P.J., Foley J.F., Gwinn W.M., Morgan D.L. Severe airway epithelial injury, aberrant repair and bronchiolitis obliterans develops after diacetyl instillation in rats. PLoS One. 2011;6:e17644. doi: 10.1371/journal.pone.0017644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gloede E., Cichocki J.A., Baldino J.B., Morris J.B. A validated hybrid computational fluid dynamics-physiologically based pharmacokinetic model for respiratory tract vapor absorption in the human and rat and its application to inhalation dosimetry of diacetyl. Toxicol Sci. 2011;123:231–246. doi: 10.1093/toxsci/kfr165. [DOI] [PubMed] [Google Scholar]
- 26.Morris J.B., Hubbs A.F. Inhalation dosimetry of diacetyl and butyric acid, two components of butter flavoring vapors. Toxicol Sci. 2009;108:173–183. doi: 10.1093/toxsci/kfn222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Rooy F.G., Rooyackers J.M., Prokop M., Houba R., Smit L.A., Heederik D.J. Bronchiolitis obliterans syndrome in chemical workers producing diacetyl for food flavorings. Am J Respir Crit Care Med. 2007;176:498–504. doi: 10.1164/rccm.200611-1620OC. [DOI] [PubMed] [Google Scholar]
- 28.Hallagan J.B., Hall R.L., Flavor and Extract Manufacturers Association FEMA GRAS: a GRAS assessment program for flavor ingredients. Regul Toxicol Pharmacol. 1995;21:422–430. doi: 10.1006/rtph.1995.1057. [DOI] [PubMed] [Google Scholar]
- 29.Hallagan J.B., Hall R.L. Under the conditions of intended use: new developments in the FEMA GRAS program and the safety assessment of flavor ingredients. Food Chem Toxicol. 2009;47:267–278. doi: 10.1016/j.fct.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Mombaerts P. Better taste through chemistry. Nat Genet. 2000;25:130–132. doi: 10.1038/75952. [DOI] [PubMed] [Google Scholar]
- 31.Saito H., Chi Q., Zhuang H., Matsunami H., Mainland J.D. Odor coding by a mammalian receptor repertoire. Sci Signal. 2009;2:ra9. doi: 10.1126/scisignal.2000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menini A., Picco C., Firestein S. Quantal-like current fluctuations induced by odorants in olfactory receptor cells. Nature. 1995;373:435–437. doi: 10.1038/373435a0. [DOI] [PubMed] [Google Scholar]
- 33.Sullivan S.L., Bohm S., Ressler K.J., Horowitz L.F., Buck L.B. Target-independent pattern specification in the olfactory epithelium. Neuron. 1995;15:779–789. doi: 10.1016/0896-6273(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 34.Gavin C.L., Williams M.C., Hallagan J.B. Flavor and Extract Manufacturers Association of the United States; Washington, D.C.: 2008. 2005 Poundage and Technical Effects Update Survey; p. 187. [Google Scholar]
- 35.Day G., LeBouf R., Grote A., Pendergrass S., Cummings K.J., Kreiss K., Kullman G. Identification and measurement of diacetyl substitutes in dry bakery mix production. J Occ Env Hygiene. 2011;8:93–103. doi: 10.1080/15459624.2011.547148. [DOI] [PubMed] [Google Scholar]
- 36.Chetschik I., Granvogl M., Schieberle P. Quantitation of key peanut aroma compounds in raw peanuts and pan-roasted peanut meal: aroma reconstitution and comparison with commercial peanut products. J Agric Food Chem. 2010;58:11018–11026. doi: 10.1021/jf1026636. [DOI] [PubMed] [Google Scholar]
- 37.Ganeko N., Shoda M., Hirohara I., Bhadra A., Ishida T., Matsuda H., Takamura H., Matoba T. Analysis of volatile flavor compounds of sardine (Sardinops melanostica) by solid phase microextraction. J Food Sci. 2008;73:S83–S88. doi: 10.1111/j.1750-3841.2007.00608.x. [DOI] [PubMed] [Google Scholar]
- 38.Majcher M.A., Jelen H.H. Identification of potent odorants formed during the preparation of extruded potato snacks. J Agric Food Chem. 2005;53:6432–6437. doi: 10.1021/jf050412x. [DOI] [PubMed] [Google Scholar]
- 39.Morales P., Feliu I., Fernandez-Garcia E., Nunez M. Volatile compounds produced in cheese by Enterobacteriaceae strains of dairy origin. J Food Prot. 2004;67:567–573. doi: 10.4315/0362-028x-67.3.567. [DOI] [PubMed] [Google Scholar]
- 40.Ott A., Germond J.E., Baumgartner M., Chaintreau A. Aroma comparisons of traditional and mild yogurts: headspace gas chromatography quantification of volatiles and origin of alpha-diketones. J Agric Food Chem. 1999;47:2379–2385. doi: 10.1021/jf980650a. [DOI] [PubMed] [Google Scholar]
- 41.Food and Drug Administration (21 CFR 172.515 Synthetic flavoring substances and adjuvants) Federal Register 2012, 21 CFR Ch 1
- 42.Dodd D.E., Garman R.H., Pritts I.M., Troup C.M., Snellings W.M., Ballantyne B. 2,4-Pentanedione: 9-day and 14-week vapor inhalation studies in Fischer-344 rats. Fundam Appl Toxicol. 1986;7:329–339. doi: 10.1016/0272-0590(86)90163-6. [DOI] [PubMed] [Google Scholar]
- 43.Garman R.H., Dodd D.E., Ballantyne B. Central neurotoxicity induced by subchronic exposure to 2,4-pentanedione vapour. Hum Exp Toxicol. 1995;14:662–671. doi: 10.1177/096032719501400807. [DOI] [PubMed] [Google Scholar]
- 44.Morgan D.L., Jokinen M.P., Price H.C., Gwinn W.M., Palmer S.M., Flake G.P. Bronchial and bronchiolar fibrosis in rats exposed to 2,3-pentanedione vapors: implications for bronchiolitis obliterans in humans. Toxicol Pathol. 2012;40:448–465. doi: 10.1177/0192623311431946. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa J., Ishikura S., Asami J., Isaji T., Usami N., Hara A., Sakurai T., Tsuritani K., Oda K., Takahashi M., Yoshimoto M., Otsuka N., Kitamura K. Molecular characterization of mammalian dicarbonyl/L-xylulose reductase and its localization in kidney. J Biol Chem. 2002;277:17883–17891. doi: 10.1074/jbc.M110703200. [DOI] [PubMed] [Google Scholar]
- 46.Thomson E.M., Williams A., Yauk C.L., Vincent R. Impact of nose-only exposure system on pulmonary gene expression. Inhal Toxicol. 2009;21(Suppl 1):74–82. doi: 10.1080/08958370902962309. [DOI] [PubMed] [Google Scholar]
- 47.Ghanem M.M., Battelli L.A., Mercer R.R., Scabilloni J.F., Kashon M.L., Ma J.Y., Nath J., Hubbs A.F. Apoptosis and Bax expression are increased by coal dust in the polycyclic aromatic hydrocarbon-exposed lung. Environ Health Perspect. 2006;114:1367–1373. doi: 10.1289/ehp.8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Islam Z., Harkema J.R., Pestka J.J. Satratoxin G from the black mold Stachybotrys chartarum evokes olfactory sensory neuron loss and inflammation in the murine nose and brain. Environ Health Perspect. 2006;114:1099–1107. doi: 10.1289/ehp.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teeguarden J.G., Bogdanffy M.S., Covington T.R., Tan C., Jarabek A.M. A PBPK model for evaluating the impact of aldehyde dehydrogenase polymorphisms on comparative rat and human nasal tissue acetaldehyde dosimetry. Inhal Toxicol. 2008;20:375–390. doi: 10.1080/08958370801903750. [DOI] [PubMed] [Google Scholar]
- 50.Gloede E., Cichocki J.A., Baldino J.B., Morris J.B. A validated hybrid computational fluid dynamics-physiologically based pharmacokinetic model for respiratory tract vapor absorption in the human and rat and its application to inhalation dosimetry of diacetyl. Toxicol Sci. 2011;123:231–246. doi: 10.1093/toxsci/kfr165. [DOI] [PubMed] [Google Scholar]
- 51.Fedan J.S., Thompson J.A., Zaccone E.J., Hubbs A.F. Complex profile of mechanical responses of guinea-pig isolated airways to the popcorn butter flavorings, diacetyl and 2,3-pentanedione. Am J Resp Crit Care Med. 2011;183:A3250. [Google Scholar]
- 52.Corcoran G.B., Fix L., Jones D.P., Moslen M.T., Nicotera P., Oberhammer F.A., Buttyan R. Apoptosis: molecular control point in toxicity. Toxicol Appl Pharmacol. 1994;128:169–181. doi: 10.1006/taap.1994.1195. [DOI] [PubMed] [Google Scholar]
- 53.Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogasawara J., Watanabe-Fukunaga R., Adachi M., Matsuzawa A., Kasugai T., Kitamura Y., Itoh N., Suda T., Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 55.Wang L., Scabilloni J.F., Antonini J.M., Rojanasakul Y., Castranova V., Mercer R.R. Induction of secondary apoptosis, inflammation, and lung fibrosis after intratracheal instillation of apoptotic cells in rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L695–L702. doi: 10.1152/ajplung.00245.2005. [DOI] [PubMed] [Google Scholar]
- 56.Levin S., Bucci T.J., Cohen S.M., Fix A.S., Hardisty J.F., LeGrand E.K., Maronpot R.R., Trump B.F. The nomenclature of cell death: recommendations of an ad hoc Committee of the Society of Toxicologic Pathologists. Toxicol Pathol. 1999;27:484–490. doi: 10.1177/019262339902700419. [DOI] [PubMed] [Google Scholar]
- 57.Silva M.T. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 58.Harkema J.R., Carey S.A., Wagner J.G. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol. 2006;34:252–269. doi: 10.1080/01926230600713475. [DOI] [PubMed] [Google Scholar]
- 59.Kurosaki M., Terao M., Barzago M.M., Bastone A., Bernardinello D., Salmona M., Garattini E. The aldehyde oxidase gene cluster in mice and rats: aldehyde oxidase homologue 3, a novel member of the molybdo-flavoenzyme family with selective expression in the olfactory mucosa. J Biol Chem. 2004;279:50482–50498. doi: 10.1074/jbc.M408734200. [DOI] [PubMed] [Google Scholar]
- 60.Piras E., Franzen A., Fernandez E.L., Bergstrom U., Raffalli-Mathieu F., Lang M., Brittebo E.B. Cell-specific expression of CYP2A5 in the mouse respiratory tract: effects of olfactory toxicants. J Histochem Cytochem. 2003;51:1545–1555. doi: 10.1177/002215540305101114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Genter M.B., Yost G.S., Rettie A.E. Localization of CYP4B1 in the rat nasal cavity and analysis of CYPs as secreted proteins. J Biochem Mol Toxicol. 2006;20:139–141. doi: 10.1002/jbt.20123. [DOI] [PubMed] [Google Scholar]
- 62.Yu T.T., McIntyre J.C., Bose S.C., Hardin D., Owen M.C., McClintock T.S. Differentially expressed transcripts from phenotypically identified olfactory sensory neurons. J Comp Neurol. 2005;483:251–262. doi: 10.1002/cne.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Epperly B.R., Dekker E.E. Inactivation of Escherichia coli L-threonine dehydrogenase by 2,3-butanedione: evidence for a catalytically essential arginine residue. J Biol Chem. 1989;264:18296–18301. [PubMed] [Google Scholar]
- 64.Mathews J.M., Watson S.L., Snyder R.W., Burgess J.P., Morgan D.L. Reaction of the butter flavorant diacetyl (2,3-butanedione) with N-alpha-acetylarginine: a model for epitope formation with pulmonary proteins in the etiology of obliterative bronchiolitis. J Agric Food Chem. 2010;58:12761–12768. doi: 10.1021/jf103251w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyata T., Kurokawa K., Van Ypersele De Strihou C. Advanced glycation and lipoxidation end products: role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J Am Soc Nephrol. 2000;11:1744–1752. doi: 10.1681/ASN.V1191744. [DOI] [PubMed] [Google Scholar]
- 66.Glomb M.A., Monnier V.M. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem. 1995;270:10017–10026. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 67.Riordan J.F. Functional arginyl residues in carboxypeptidase A: modification with butanedione. Biochemistry. 1973;12:3915–3923. doi: 10.1021/bi00744a020. [DOI] [PubMed] [Google Scholar]
- 68.Lo T.W., Westwood M.E., McLellan A.C., Selwood T., Thornalley P.J. Binding and modification of proteins by methylglyoxal under physiological conditions: a kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 69.More S.S., Raza A., Vince R. The butter flavorant, diacetyl, forms a covalent adduct with 2-deoxyguanosine, uncoils DNA, and leads to cell death. J Agric Food Chem. 2012;60:3311–3317. doi: 10.1021/jf300180e. [DOI] [PubMed] [Google Scholar]
- 70.Matsunaga T., Kamiya T., Sumi D., Kumagai Y., Kalyanaraman B., Hara A. L-xylulose reductase is involved in 9,10-phenanthrenequinone-induced apoptosis in human T lymphoma cells. Free Radic Biol Med. 2008;44:1191–1202. doi: 10.1016/j.freeradbiomed.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 71.Brouwers O., Niessen P.M., Haenen G., Miyata T., Brownlee M., Stehouwer C.D., De Mey J.G., Schalkwijk C.G. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia. 2010;53:989–1000. doi: 10.1007/s00125-010-1677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amicarelli F., Colafarina S., Cattani F., Cimini A., Di Ilio C., Ceru M.P., Miranda M. Scavenging system efficiency is crucial for cell resistance to ROS-mediated methylglyoxal injury. Free Radic Biol Med. 2003;35:856–871. doi: 10.1016/s0891-5849(03)00438-6. [DOI] [PubMed] [Google Scholar]
- 73.Berner A.K., Brouwers O., Pringle R., Klaassen I., Colhoun L., McVicar C., Brockbank S., Curry J.W., Miyata T., Brownlee M., Schlingemann R.O., Schalkwijk C., Stitt A.W. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia. 2012;55:845–854. doi: 10.1007/s00125-011-2393-0. [DOI] [PubMed] [Google Scholar]
- 74.Thornalley P.J. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990;269:1–11. doi: 10.1042/bj2690001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vander Jagt D.L., Hunsaker L.A. Methylglyoxal metabolism and diabetic complications: roles of aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and 2-oxoaldehyde dehydrogenase. Chem Biol Interact. 2003 doi: 10.1016/s0009-2797(02)00212-0. [DOI] [PubMed] [Google Scholar]; –144:341–351143
- 76.Kim K.M., Kim Y.S., Jung D.H., Lee J., Kim J.S. Increased glyoxalase I levels inhibit accumulation of oxidative stress and an advanced glycation end product in mouse mesangial cells cultured in high glucose. Exp Cell Res. 2012;318:152–159. doi: 10.1016/j.yexcr.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 77.Ishikura S., Isaji T., Usami N., Kitahara K., Nakagawa J., Hara A. Molecular cloning, expression and tissue distribution of hamster diacetyl reductase: identity with L-xylulose reductase. Chem Biol Interact. 2001 doi: 10.1016/s0009-2797(00)00315-x. [DOI] [PubMed] [Google Scholar]; –132:879–889130
- 78.Di Loreto S., Zimmitti V., Sebastiani P., Cervelli C., Falone S., Amicarelli F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int J Biochem Cell Biol. 2008;40:245–257. doi: 10.1016/j.biocel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 79.Smith B., Galbiati F., Castelvetri L.C., Givogri M.I., Lopez-Rosas A., Bongarzone E.R. Peripheral neuropathy in the Twitcher mouse involves the activation of axonal caspase 3. ASN Neuro. 2011;3 doi: 10.1042/AN20110019. pii:e00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iacobaeus E., Amoudruz P., Strom M., Khademi M., Brundin L., Hillert J., Kockum I., Malmstrom V., Olsson T., Tham E., Piehl F. The expression of VEGF-A is down regulated in peripheral blood mononuclear cells of patients with secondary progressive multiple sclerosis. PLoS One. 2011;6:e19138. doi: 10.1371/journal.pone.0019138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin K.L., Mao X.O., Greenberg D.A. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yasuhara T., Shingo T., Kobayashi K., Takeuchi A., Yano A., Muraoka K., Matsui T., Miyoshi Y., Hamada H., Date I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson's disease. Eur J Neurosci. 2004;19:1494–1504. doi: 10.1111/j.1460-9568.2004.03254.x. [DOI] [PubMed] [Google Scholar]
- 83.Yasuhara T., Shingo T., Muraoka K., Kameda M., Agari T., Wen Ji Y., Hayase H., Hamada H., Borlongan C.V., Date I. Neurorescue effects of VEGF on a rat model of Parkinson's disease. Brain Res. 2005;1053:10–18. doi: 10.1016/j.brainres.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 84.Greenberg D.A., Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- 85.Feng Y., Rhodes P.G., Bhatt A.J. Dexamethasone pre-treatment protects brain against hypoxic-ischemic injury partially through up-regulation of vascular endothelial growth factor A in neonatal rats. Neuroscience. 2011;179:223–232. doi: 10.1016/j.neuroscience.2011.01.050. [DOI] [PubMed] [Google Scholar]
- 86.Ressler K.J., Sullivan S.L., Buck L.B. A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell. 1993;73:597–609. doi: 10.1016/0092-8674(93)90145-g. [DOI] [PubMed] [Google Scholar]
- 87.Ressler K.J., Sullivan S.L., Buck L.B. Information coding in the olfactory system: evidence for a stereotyped and highly organized epitope map in the olfactory bulb. Cell. 1994;79:1245–1255. doi: 10.1016/0092-8674(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 88.Vassar R., Chao S.K., Sitcheran R., Nunez J.M., Vosshall L.B., Axel R. Topographic organization of sensory projections to the olfactory bulb. Cell. 1994;79:981–991. doi: 10.1016/0092-8674(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 89.Vassar R., Ngai J., Axel R. Spatial segregation of odorant receptor expression in the mammalian olfactory epithelium. Cell. 1993;74:309–318. doi: 10.1016/0092-8674(93)90422-m. [DOI] [PubMed] [Google Scholar]
- 90.Johnson B.A., Arguello S., Leon M. Odorants with multiple oxygen-containing functional groups and other odorants with high water solubility preferentially activate posterior olfactory bulb glomeruli. J Comp Neurol. 2007;502:468–482. doi: 10.1002/cne.21322. [DOI] [PMC free article] [PubMed] [Google Scholar]