

Abstract
The green tea component epigallocatechin-3-gallate (EGCG) may be beneficial in autoimmune diseases; however, the underlying mechanisms are not well understood. In this study, we determined the effect of EGCG on the development of experimental autoimmune encephalomyelitis, an animal model for human multiple sclerosis, and the underlying mechanisms. Female C57BL/6 mice were fed EGCG (0%, 0.15%, 0.3%, and 0.6% in diet) for 30 days and then immunized with specific antigen myelin oligodendrocyte glycoprotein 35-55. EGCG dose dependently attenuated clinical symptoms and pathological features (leukocyte infiltration and demyelination) in the central nervous system and inhibited antigen-specific T-cell proliferation and delayed-type hypersensitivity skin response. We further showed that EGCG reduced production of interferon-γ, IL-17, IL-6, IL-1β, and tumor necrosis factor-α; decreased types 1 and 17 helper T cells (Th1 and Th17, respectively); and increased regulatory T-cell populations in lymph nodes, the spleen, and the central nervous system. Moreover, EGCG inhibited expression of transcription factors T-box expressed in T cells and retinoid-related orphan receptor–γt, the specific transcription factor for Th1 and Th17 differentiation, respectively; the plasma levels of intercellular adhesion molecule 1; and CCR6 expression in CD4+ T cells. These results indicate that EGCG may attenuate experimental autoimmune encephalomyelitis autoimmune response by inhibiting immune cell infiltration and modulating the balance among pro- and anti-autoimmune CD4+ T-cell subsets. Thus, we identified a novel mechanism that underlies EGCG's beneficial effect in autoimmune disease.
Multiple sclerosis (MS) is a T-cell–mediated inflammatory autoimmune disease of the central nervous system (CNS). Experimental autoimmune encephalomyelitis (EAE) is a well-established animal model for human MS, owing to the similarities in clinical, immunological, and neuropathological features between MS and EAE.1 The EAE model, which has been widely used to study MS, is also one of the most favored tools for the study of T-cell–mediated autoimmunity. The key pathogenesis of MS involves a process in which myelin-reactive CD4+ T cells enter the CNS and orchestrate their effector function on the myelin sheath, resulting in tissue destruction and consequent loss of function.2, 3, 4 MS or EAE has long been thought to be a type 1 helper T-cell (Th1)–mediated autoimmune disorder in which myelin-specific, IL-12–induced, interferon (IFN)-γ–producing CD4+ Th1 cells were considered to be the only driving factor in the induction of the disease. However, compelling evidence, accumulated in the past few years, has developed a new theory indicating the additional involvement of a newly identified population of CD4+ T cells, named Th17 cells. Furthermore, Th17 cells are implicated in this model and in several other autoimmune diseases. Although some autoimmune responses formerly attributed to Th1 cells are now believed to be mediated by Th17 cells, recent studies5, 6, 7, 8, 9 continue to emphasize the importance of Th1 cells in EAE development. In fact, EAE can be induced by transfer of either Th17 or Th1 cells.10 Another important T-cell type involved in regulation of EAE development is regulatory T cells (Tregs), characterized by their CD4+CD25+ phenotype, together with expression of their master transcription factor forkhead box3 (Foxp3). Tregs reduce the inflammatory responses and clinical symptoms in mice with EAE.11, 12, 13
The etiology of most autoimmune diseases, including MS, is not clearly understood. Both genetic predisposition (a key risk factor) and environmental factors are involved. Current therapies, which mainly focus on the use of immune-suppressant drugs, have limited efficacy and various adverse effects. Nutrition represents an alternative and complementary approach that could potentially improve autoimmune disorders. Green tea may be such a nutrition factor. Catechins in green tea are thought to be the major components responsible for the tea's biological effects. These catechins include epicatechin, epigallocatechin, epicatechin-3-gallate, and epigallocatechin-3-gallate (EGCG), among which EGCG is the most biologically active and most abundant (accounting for 50% to 80% of the total tea catechins).14 Although green tea or EGCG can quench several different reactive oxygen species and its health benefits have been partially attributed to its antioxidant properties, our previous study15 indicates that the T-cell–suppressive effect of EGCG is not related to its effect on oxidative stress. Results from a few animal studies suggest that green tea EGCG might be effective in improving the symptoms and pathological conditions associated with autoimmune inflammatory diseases in several animal models.16, 17, 18, 19, 20, 21 Preliminary evidence17, 19, 21 has linked this effect of EGCG to altered T-cell function, including production of cytokines, such as IFN-γ and tumor necrosis factor (TNF)-α. Although these results are encouraging, further studies are needed to address several important issues. First, studies to determine a clear, definitive effect of EGCG on autoimmune diseases are inconsistent. Second, little is known about the dose-response relationship because of a lack of appropriately designed dietary supplementation studies, which would have a great relevance to its use as a food component, supplement, or therapeutic agent. Finally, and most important, the mechanisms for the proposed beneficial effect of EGCG in autoimmunity are not well elucidated. In particular, for the most part, the studies thus far have not considered the previously mentioned, recently developed theory on the immunopathogenesis of autoimmune inflammatory diseases (eg, reciprocal action of Th17 and Treg). To address these issues, we conducted a study using a mouse EAE model to determine how dietary supplementation with EGCG at different doses affects the disease's symptoms and pathological characteristics, as well as the related immune responses as the mechanisms by which EGCG exerts its effect.
Materials and Methods
Animals
Specific pathogen-free C57BL/6 female mice (aged 6 to 8 weeks) were obtained from Charles River (Wilmington, MA). These mice were maintained at a constant temperature and humidity, with a 12-hour light-dark cycle. Water and a nutritionally adequate, nonpurified mouse diet (Teklad 7012; Harlan Teklad, Madison, WI) were provided ad libitum. In the feeding study, mice were pair fed the experimental diets, as described later. All mice were observed daily for general health and clinical signs of disease. At the end of the study, mice were euthanized by CO2 asphyxiation, followed by exsanguination, and tissues were collected postmortem. All conditions and handling of the animals were approved by the Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University (Boston, MA) and conducted according to the NIH Guidelines for the Care and Use of Laboratory Animals.
Dietary EGCG Supplementation
In the experiment to determine effect of EGCG at different doses on EAE development, mice were randomly divided into four groups and pair fed the AIN 93M diet (Research Diet) supplemented with 0%, 0.15%, 0.3%, or 0.6% EGCG (w/w) for 30 days. EGCG (TEAVIGO, containing 95% EGCG) was provided by DSM Nutritional Products (Kaiseraugst, Switzerland). After 30 days of feeding, mice were immunized to induce EAE, as described later, while they continued to receive their experimental diets. In the experiment to determine whether EGCG is still effective at treating EAE after disease initiation, mice were divided into three groups and fed the control diet for 1 week before being immunized to induce EAE. While continuing to feed the control group the control diet throughout the entire study, we switched one group at day 7 and the other group at day 12 after immunization to the diet containing 0.6% EGCG. Mice were maintained on these diets until day 30 after immunization.
Induction and Evaluation of EAE
Mice were immunized s.c. in the flanks with 200 μg of myelin oligodendrocyte glycoprotein (MOG)35-55 peptide (synthesized by the Tufts University Core Facility Laboratory) in a 200-μL emulsion of complete freund's adjuvant (CFA) containing 5 mg/mL heat-killed Mycobacterium tuberculosis H37Ra extract (Sigma-Aldrich, St. Louis, MO) on day 0 and i.p. injected with 200 ng per mouse of pertussis toxin (List Biological Laboratories, Campbell, CA) on days 0 and 2. The clinical symptoms were scored daily from day 0 to 30 after immunization, as follows: 0, no signs; 0.5, partial tail paralysis; 1, limp tail or tail paralysis; 2, complete loss of tail tonicity or abnormal gait; 3, partial hind limb paralysis; 4, complete hind limb paralysis; and 5, moribund.
Histological and IHC Data
Mice were euthanized on day 30 after immunization and perfused by intracardiac infusion with 10% paraformaldehyde. The brain and spinal cord were removed and fixed in 10% paraformaldehyde. Fixed samples were embedded in paraffin, and cross sections were stained with H&E or Luxol fast blue for evaluation of inflammation or demyelination, respectively. For immunohistochemistry (IHC), the sections were deparaffinized with xylene, rehydrated through a series with ethanol, and incubated in 0.3% hydrogen peroxide for 10 minutes to quench endogenous peroxidase activity. Immunostaining was conducted using the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). Nonspecific binding was blocked by incubation with normal rat serum for 30 minutes. The sections were then incubated with the following primary antibodies at indicated dilutions at 4°C overnight: anti-CD3 (1:100) and anti-F4/80 (1:50) from AbD SeroTec (Oxford, UK) and anti-CD45R/B220 (1:25) and anti-Ly-6G and Ly-6C (1:25) from BD Pharmingen (San Diego, CA). Sections were then incubated with a biotinylated secondary antibody (anti-rat IgG) for 30 minutes, washed, and incubated for another 30 minutes with ABC (avidin and biotinylated enzyme complex) reagent. Color was developed by adding peroxidase substrate diaminobenzidine. Sections were counterstained with Mayer's hematoxylin (Sigma-Aldrich) and, finally, mounting solution and coverslips were added.
MOG-Specific DTH
On day 27 after immunization, mice were injected with MOG35-55 peptide in the right footpad and saline in the left footpad as control. The footpad thickness was measured 24, 48, and 72 hours after the injection using a micrometer (QUICKmini; Mitutoyo, Kawasaki, Japan). Specific responses were calculated by subtracting the thickness of the left footpad (saline) from that of the right footpad (MOG).
T-Cell Proliferation
After mice were euthanized, the draining lymph nodes (LNs) were aseptically removed and single-cell suspensions were prepared. LN cells were placed in sterile RPMI 1640 medium (Biowhittaker, Walkerville, MD), supplemented with 25 mmol/L HEPES, 2 mmol/L glutamine, 100 kU/L penicillin, and 100 mg/L streptomycin (all from Invitrogen, Carlsbad, CA). Cells suspended in RPMI 1640 medium containing 5% heat-inactivated fetal bovine serum (Invitrogen) were plated in triplicate in 96-well round-bottom cell culture plates and stimulated with 0, 1, 10, or 100 μg/mL MOG35-55 peptide for 72 hours. Cultures were pulsed with 1 μCi/well [3H]-thymidine (Perkin Elmer; Life Sciences, Boston, MA) during the final 4 hours of incubation. The cells were harvested onto glass-fiber filter mats (Wallac, Gaithersburg, MD) by a Tomtec harvester (Wallac), and cell proliferation was quantified as the amount of [3H]-thymidine incorporation into DNA, as determined by liquid scintillation counting in a 1205 Betaplate counter (Wallac). Results are expressed as cpm.
Cytokine Production
The LNs and spleen were collected, and single-cell suspensions in RPMI 1640 medium and 5% fetal bovine serum were prepared. The LNs and spleen cells were separately cultured in 24-well culture plates in the presence of 100 μg/mL MOG35-55 peptide for 72 hours. Supernatants were then collected and measured with an enzyme-linked immunosorbent assay (ELISA) for IL-17 (kit from eBiosciences, San Diego, CA), transforming growth factor (TGF)-β (kit from R&D Systems, Minneapolis, MN), and IFN-γ, IL-6, and TNF-α (all from BD Pharmingen). Intracellular cytokine levels were determined using a flow cytometry, as described later.
Plasma IL-1β, IL-12/IL-23 p40, and ICAM-1
Heparinized plasma samples were collected and centrifuged to obtain plasma. Plasma concentrations of intercellular adhesion molecule 1 (ICAM-1) and IL-1β were measured using the Duoset ELISA kit (R&D Systems), and IL-12/IL-23 p40 was measured using the ELISA Ready-SET-Go! kit (eBiosciences), following the manufacturer's instructions.
Isolation of Mononuclear Cells in CNS
Mice were euthanized on day 12 after immunization and perfused through the left ventricle with cold PBS. Brain and spinal cord were removed, and the tissues from two mice were pooled into one CNS sample. Mononuclear cells were isolated from the CNS samples using the Neural Tissue Dissociation Kits (Mitenyi Biotec, Auburn, CA). The isolated cells were either directly analyzed for surface markers or stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) and 500 ng/mL ionomycin (both from Sigma-Aldrich) in the presence of monensin (Golgi Stop; BD Pharmingen) for 4 hours for measurement of intracellular markers. The expression of cell surface and intracellular markers was determined by flow cytometry, as described later.
Flow Cytometry
To determine the cell phenotype and expression of CCRs, cells were surface stained by appropriate antibodies labeled with fluorochromes. For intracellular cytokine measurements, cells were restimulated with 50 ng/mL PMA and 500 ng/mL ionomycin (both from Sigma-Aldrich) in the presence of monensin (BD Pharmingen) for 4 hours. Cells were blocked by anti-CD16/32 (Fc block; BD Pharmingen), fixed and permeabilized with the Cytofix/Cytoperm kit (BD Pharmingen), and stained with fluorochrome-labeled antibodies for each cytokine tested. The expression of transcription factors was analyzed after intracellular staining using a similar procedure. Foxp3 staining was performed using the Mouse Foxp3 Buffer Set (BD Pharmingen). The antibodies used for flow cytometry were as follows: CD3 (145-2C11), anti-CD4 (GK1.5), anti-IFN-γ (XMG1.2), and anti-IL-10 (JES5-16E3) were from eBiosciences; anti-transcription factors T-box expressed in T cells (T-bet; O4-46), anti-Foxp3 (MF23), anti-IL-4 (11B11), and anti-IL-17 (TC11-18H10) were from BD Pharmingen; and anti-CCR6 (140706) and anti-retinoid-related orphan receptor (ROR)–γt (600380) were from R&D Systems. All flow cytometric measurements were conducted using an Accuri C6 flow cytometer (BD Accuri Cytometers, Ann Arbor, MI), and acquired data were analyzed with FlowJo 7.6 software (Treestar Inc., Ashland, OR).
Real-Time RT-PCR
To determine mRNA levels of selected analytes, total RNA was extracted from brain, spinal cord, and splenocyte samples using TRIzol reagent (Invitrogen) and was then reverse transcribed into cDNA using a SuperScript VILO cDNA synthesis kit (Invitrogen), following the manufacturer's instructions. Real-time PCR was performed in triplicate using SYBR Green Master Mix (Qiagen, Valencia, CA) in an ABI 7300 real-time PCR system (Applied Biosystems, Foster City, CA). The levels of tested genes were normalized to β-actin gene as an internal control.
The sequences of the primers used are as follows: CCL20, 5′-CGACTGTTGCCTCTCGTACA-3′ (forward) and 5′-AGCCCTTTTCACCCAGTTCT-3′ (reverse); T-bet, 5′-GCCAGGGAACCGCTTATATGTC-3′ (forward) and 5′-CTGTGAGATCATATCCTTGGGCTG-3′ (reverse); RORγt, 5′-TGCAAGACTCATCGACAAGG-3′ (forward) and 5′-AGGGGATTCAACATCAGTGC-3′ (reverse); IL-23p19, 5′-GACTCAGCCAACTCCTCCAG-3′ (forward) and 5′-GGCACTAAGGGCTCAGTCAG-3′ (reverse); IL-12p40, 5′-AGGTCACACTGGACCAAAGG-3′ (forward) and 5′-TGGTTTGATGATGTCCCTGA-3′ (reverse); IL-12p35, 5′-CATCGATGAGCTGATGCAGT-3′ (forward) and 5′-CAGATAGCCCATCACCCTGT-3′ (reverse); IL-27p28, 5′-CTCTGCTTCCTCGCTACCAC-3′ (forward) and 5′-GGGGCAGCTTCTTTTCTTCT-3′ (reverse); EBi3, 5′-CGGTGCCCTACATGCTAAAT-3′ (forward) and 5′-GCGGAGTCGGTACTTGAGAG-3′ (reverse); and β-actin, 5′-TGTTACCAACTGGGACGACA-3′ (forward) and 5′-GGGGTGTTGAAGGTCTCAAA-3′ (reverse).
Statistical Analysis
All results are expressed as mean ± SEM. Statistical analysis was conducted using Systat 12 statistical software (Systat Software, Chicago, IL). Differences were determined using one-way analysis of variance, followed by Tukey's honestly significant difference post hoc test for multiple comparisons, or a nonpaired Student's t-test. Significance was set at P < 0.05.
Results
Dietary EGCG Supplementation Ameliorates Clinical Symptoms of EAE
Mice were fed different doses of EGCG for 30 days and then immunized with MOG35-55 peptide to induce EAE while continuing their EGCG consumption. Almost all of the mice developed EAE after immunization, but the time of onset varied to some extent among individual mice. Nevertheless, compared with the control group, mice fed 0.6% EGCG had a delayed average time of onset (14 ± 0.7 versus 11.5 ± 0.8 days; P < 0.05), whereas no significant difference was observed in the mice fed 0.15% or 0.3% EGCG (Figure 1 and Table 1). Furthermore, the cumulative disease index (mean sum of clinical scores over the entire observation period) showed a dose-dependent decrease in EGCG-fed mice, which lasted until the completion of the study (ie, 30 days after immunization) (Figure 1 and Table 1). The improvement in symptoms was not statistically significant in the 0.15% EGCG group.
Figure 1.
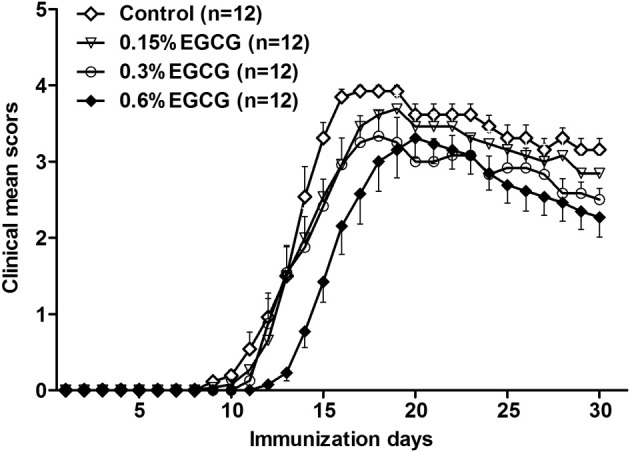
Dietary EGCG dose dependently ameliorates clinical symptoms of EAE. Female C57BL/6 mice were fed diets containing 0%, 0.15%, 0.3%, and 0.6% EGCG for 30 days and then immunized with MOG35-55/CFA. Clinical symptoms of EAE were scored daily while mice were fed the same experimental diets for another 30 days. Values are mean ± SEM (n = 12).
Table 1.
Effect of Dietary EGCG on EAE Symptoms
| Group | Day of onset | CDI⁎ | Area of curve† |
|---|---|---|---|
| Control | 11.5 ± 0.76 | 62.18 ± 1.10 | 60.50 (100) |
| EGCG (%) | |||
| 0.15 | 12.0 ± 0.80 | 54.82 ± 1.91 | 54.31 (89.8) |
| 0.3 | 12.5 ± 0.65 | 49.68 ± 4.88‡ | 49.41 (81.7) |
| 0.6 | 14.0 ± 0.70‡ | 45.41 ± 3.84§ | 42.78 (70.7) |
Values are given as the mean ± SEM (n = 12/group) unless otherwise indicated.
CDI, cumulative disease index.
Sum of clinical scores over the entire observation period.
Values are given as number (percentage) of 60.50.
P < 0.05 versus the control.
P < 0.01 versus the control.
EGCG Inhibits Autoantigen MOG35-55-Induced Specific T-Cell Response Both ex Vivo and in Vivo
T cells play a key role in the development of EAE through their antigen (Ag)–specific effector response. Clonal expansion of Ag-specific T cells on antigen encounter is a prerequisite for the initiation and development of T-cell–mediated immunopathological characteristics. We, thus, determined whether EGCG attenuates EAE development via its impact on Ag-specific T-cell response. We used two approaches to address this question: an ex vivo proliferation assay to determine Ag-specific proliferation and an in vivo delayed-type hypersensitivity (DTH) assay to assess T-cell–mediated inflammatory response after rechallenge with the immunization autoantigen. In the assay of ex vivo T-cell proliferation, we rechallenged LN cells with MOG35-55 peptide or ovalbumin as a control after these cells were isolated from mice with EAE fed different doses of EGCG. The addition of MOG35-55 (1 to 100 μg/mL) stimulated T-cell proliferation in a concentration-dependent manner, which was dose dependently inhibited by dietary EGCG supplementation (Figure 2A). Nonspecific Ag ovalbumin did not induce proliferative response (data not shown). In concordance with the results for symptoms, this effect was not significant in the 0.15% EGCG group. Consistent with the results of an ex vivo T-cell proliferation assay, EGCG also dose dependently reduced the DTH response, as determined by the Ag rechallenge-induced footpad thickness, and a significant reduction was observed even in the 0.15% EGCG group (Figure 2B).
Figure 2.
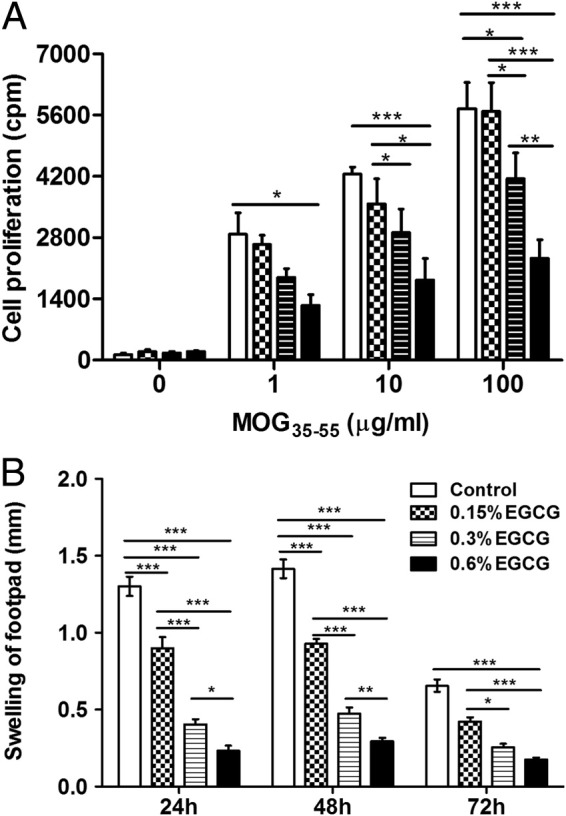
Dietary EGCG dose dependently inhibits autoantigen-specific T-cell proliferation and DTH skin response. A: At day 60, after being fed and immunized with MOG35-55/CFA, as described in the legend to Figure 1, mice were euthanized and cells were isolated from the draining LNs. LN cells were restimulated with 0, 1, 10, or 100 μg/mL MOG35-55 peptide for 72 hours, and proliferation was determined by [3H]-thymidine incorporation. B: At day 57 (ie, 3 days before being euthanized), mice were rechallenged by injecting 20 μL of MOG35-55 peptide (10 μg/mL) into footpads, and DTH was measured as the thickness of footpads was recorded at 24, 48, and 72 hours after injection. Values are mean ± SEM (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001, as determined by one-way analysis of variance, followed by Tukey's honestly significant difference post hoc test for multiple comparisons.
Because mice fed 0.6% EGCG had the greatest protective effect, and they tolerated this level well, in the subsequent experiments we used this dose to further determine the working mechanisms of EGCG and to compare its efficacy when administered at different times during EAE development.
EGCG Reduces Inflammation and Demyelination in the CNS of Mice with EAE
In the pathogenesis of EAE, autoreactive T cells and other associated immune cells cross the blood-brain barrier (BBB), infiltrating the CNS, where these cells and their products orchestrate inflammatory cascades, ultimately causing damage to the myelin sheath. Because we found that EGCG ameliorated EAE symptoms and inhibited the Ag-specific T-cell response, we intended to learn if this protective effect of EGCG would be reflected in the corresponding changes in tissue inflammation and damage. Figure 3A shows the pathological characteristics of spinal cord samples collected from mice 30 days after immunization. Mice with EAE fed the control diet showed extensive inflammatory cell infiltration into the white matter of spinal cords, and this change was greatly reduced by EGCG treatment. We further found that EAE-induced tissue inflammation and the protective effect of EGCG were well reflected in the levels of tissue destruction (ie, demyelination) (Figure 3B).
Figure 3.

Dietary EGCG reduces the neuropathological features associated with EAE. After being fed and immunized with MOG35-55/CFA, as described in the legend to Figure 1, mice fed 0.6% EGCG or a control diet were euthanized and spinal cords were collected. Sections of the fixed samples were obtained and stained with H&E (A) or Luxol fast blue (LFB) (B) for assessment of inflammation or demyelination, respectively. Each section is the representative of six mice in each diet group. On day 12 after immunization, mice were euthanized and brains and spinal cords were collected for IHC and profiling inflammatory infiltration. C: IHC for neutrophils (Gr-1), B cells (CD45R), macrophage (MΦ) (F4/80), and T cells (CD3). Each section is the representative of six mice in each diet group. D: Isolated cells were counted under a microscope for total number of infiltrated cells and stained with appropriate fluorescence-conjugated antibody, followed by flow cytometry to determine the cellular composition. Each dot represents one sample pooled from two mice. Significant differences were determined by the nonpaired Student's t-test. *P < 0.05 compared with the control.
Profile of Immune Cell Infiltration in the CNS and Its Alteration by Dietary EGCG
To identify the populations of immune cells that infiltrated into the CNS of mice with EAE and to determine how they are affected by EGCG supplementation, we conducted IHC and found that EGCG treatment reduced the frequency of neutrophils (Gr-1), MΦ (F4/80), and T cells (CD3), but not B cells (CD45R, B220), in the spinal cords on day 12 after immunization (estimated peak time for inflammatory infiltration) (Figure 3C). We also isolated mononuclear cells from spinal cords and brains on day 12 after immunization and conducted a quantitative analysis using flow cytometry. In agreement with the IHC results, EGCG treatment reduced total infiltrating cells, neutrophils, MΦ, total T cells, and CD4+ T cells; there was also a trend for reduced B cells (P = 0.09) (Figure 3D). However, EGCG treatment did not significantly change the composition of infiltrating cells, except for a reduced percentage of neutrophils (data not shown). These results indicate that EGCG reduces infiltration of most cell types to a similar extent, without substantial preference.
EGCG Inhibits Th1/Th17 and Enhances Treg but Does Not Affect Th2 Response
In EAE pathogenesis, a key step after effective autoantigen challenge is T-cell differentiation into different subtypes, as characterized by predominant production of the corresponding cytokines by each population. Therefore, determining how EGCG affects the profile of T-cell subsets would provide a mechanistic insight toward helping us understand their protective effect in EAE and other autoimmune diseases. We, thus, measured the hallmark cytokines IFN-γ, IL-4, and IL-17, for Th1, Th2, and Th17, respectively, and transcription factor Foxp3, specific for Treg in the CNS (brain and spinal cord) and peripheral lymphoid organs (spleen and draining LNs), which were collected on day 12 after immunization. Compared with the control mice, those fed EGCG had significantly smaller populations of Th1 and Th17 cells and a larger population of Tregs in the CNS (Figure 4). Intracellular IL-4 and IL-10 were not detectable under our experimental conditions; thus, we do not know if EGCG affects the Th2 population or IL-10 production in infiltrated T cells in the CNS.
Figure 4.
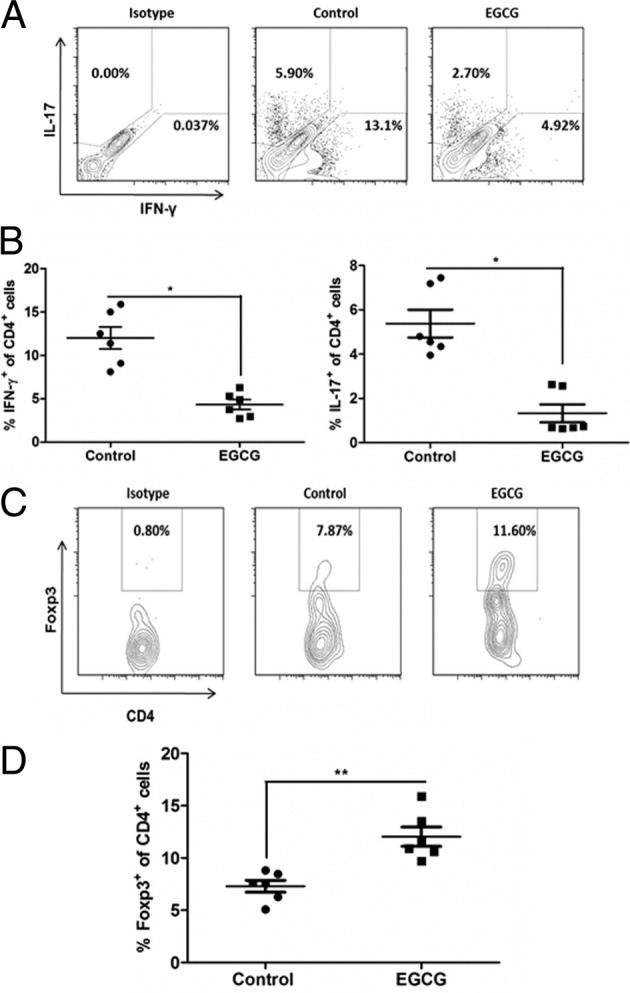
Dietary EGCG down-regulates Th1 and Th17 and up-regulates Treg response in the CNS. On day 12 after immunization, mice were euthanized and brains and spinal cords were collected. The isolated mononuclear cells from the CNS were stimulated with PMA plus ionomycin in the presence of Golgi Stop for 4 hours. After that, appropriate surface and intracellular stainings were performed and cell type and subpopulation were determined using flow cytometry. A: Representatives of flow cytometry results for Th1 (IFN-γ–producing CD4+ T cells) and Th17 (IL-17–producing CD4+ T cells) populations. B: Summary of Th1 and Th17 populations. Each dot represents one sample pooled from two mice. C: Representatives of flow cytometry results for Treg (CD4+Foxp3+) T cells. D: Summary of the Treg population. Each dot represents one sample pooled from two mice. All values in this figure are mean ± SEM (n = 6/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.01, **P < 0.001 compared with the control.
To determine whether EGCG affects priming/expansion of pathogenic Th1 and Th17 responses in peripheral lymphoid tissues, we measured production of IFN-γ and IL-17 by spleen and LN cells after Ag rechallenge with MOG35-55 peptide. EGCG supplementation significantly reduced IFN-γ and IL-17 production in the cultures, as measured by ELISA (Figure 5A). These results were further confirmed by analysis of the intracellular levels of these cytokines, as demonstrated by a reduction in both IFN-γ–secreting (Th1) and IL-17–secreting (Th17) CD4+ cells and in a subpopulation of Th17 CD4+ T cells that concomitantly express IL-17 and IFN-γ (Figure 5, B and C). Consistent with the findings in the CNS, EGCG supplementation resulted in increased proportions of Tregs in both spleen and LN CD4+ T cells (Figure 6A); furthermore, we obtained detectable levels of intracellular IL-4 and IL-10, which were not different between the control and EGCG-fed mice (Figure 6, B and C). Because cytokines IL-12, IL-23, and TGF-β are key factors in driving Th1, Th17, and Treg differentiation and maintaining their expansion, respectively,22, 23 whereas IL-27 suppresses the generation of encephalitogenic Th17 cells and the effector phase of EAE,24 we determined the effect of EGCG treatment on theses cytokines to gain a further mechanistic insight. Results showed that EGCG supplementation reduced plasma levels of IL-12/IL-23 total p40 (Figure 5D) and mRNA expression of IL-12p40 and IL-23p19 but did not affect mRNA expression for IL-12p35 (Figure 5D) and IL-27 (IL-27p28 and IL-27EBi3) (data not shown) and TGF-β production in splenocytes (Figure 6D).
Figure 5.
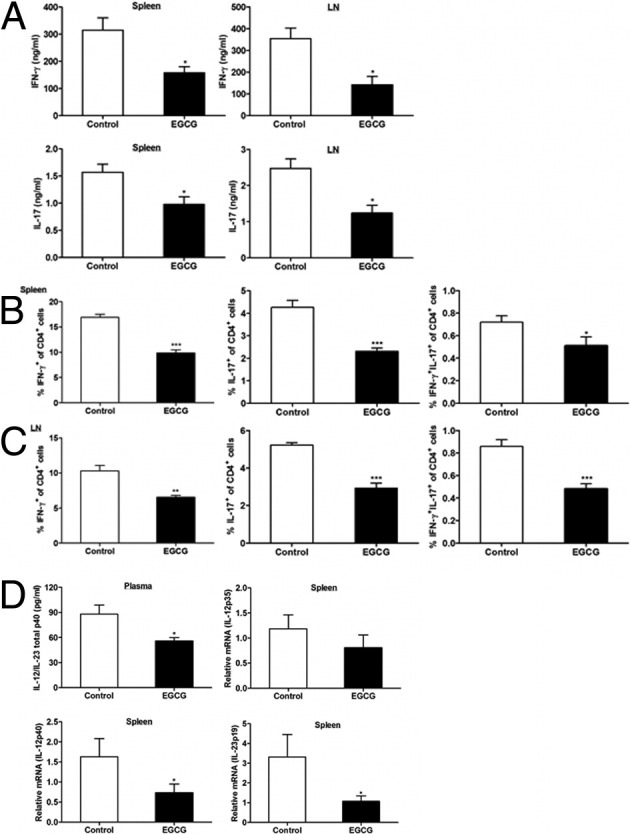
Dietary EGCG inhibits Th1 and Th17 response in peripheral lymphoid organs of mice with EAE. On day 12 after immunization, mice were euthanized and spleen and draining LNs were collected. Spleen and LN cells were stimulated in vitro with 100 μg/mL MOG35-55 peptide for 72 hours to determine secretion of IFN-γ and IL-17 using ELISA (A). In addition, these cells were restimulated with PMA and ionomycin in the presence of Golgi Stop for an additional 4 hours for analysis of intracellular levels of IFN-γ and IL-17 to identify Th1 and Th17 populations in CD4+ T cells of spleen (B) and LNs (C). D: IL-12/IL-23 total p40 levels in plasma samples were determined by ELISA. Total RNA extracted from the splenocytes was used to determine IL-12p35, IL-12p40, and IL-23p19 mRNA expression levels using real-time RT-PCR. All values in this figure are mean ± SEM (n = 8/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control.
Figure 6.
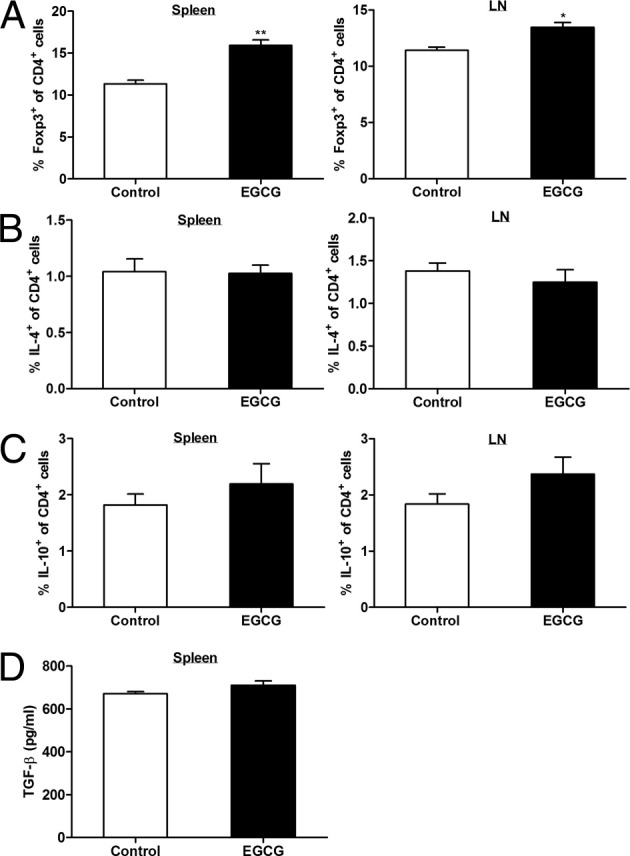
Dietary EGCG up-regulates Treg but does not affect Th2 response in peripheral lymphoid organs of mice with EAE. On day 12 after immunization, mice were euthanized and spleen and draining LNs were collected. A: Spleen and LN cells were stained with fluorescence-conjugated antibody for CD4 and Foxp3 in the Mouse Foxp3 buffer and analyzed by a flow cytometer. B and C: To determine the intracellular IL-4 and IL-10 levels in CD4+ T cells from spleen and LN, spleen and LN cells were stimulated in vitro with 100 μg/mL MOG35-55 peptide for 72 hours, followed by restimulation with PMA and ionomycin in the presence of Golgi Stop for an additional 4 hours. Intracellular levels of IL-4 (B) and IL-10 (C) in CD4+ T cells were analyzed by a flow cytometer. D: Splenocytes were stimulated in vitro with 100 μg/mL MOG35-55 peptide for 72 hours, and supernatant was analyzed for TGF-β production using ELISA. All values in this figure are mean ± SEM (n = 8/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.01, **P < 0.001 compared with the control.
Together, these results suggest a novel mechanism: EGCG's protective ability to impede EAE development may be mediated by reducing the pro-autoimmune, inflammatory Th1 and Th17 cells while increasing the protolerance, anti-inflammatory Tregs.
EGCG Suppresses the Expression of Transcription Factors T-bet and RORγt
Transcription factors T-bet and RORγt are master regulators that direct the differentiation of Th1 and Th17 cells, respectively. Therefore, after observing the EGCG-induced reduction in Th1 and Th17 populations, we wanted to learn if these two transcription factors are involved in the suppressive effect of EGCG on Th1 and Th17 development. Spleens were collected on day 12 after immunization, and isolated cells were used to conduct intracellular staining for T-bet and RORγt expression analysis. In accordance with the findings of EGCG's effect on Th1 and Th17, EGCG supplementation resulted in reduced expression of both T-bet and RORγt (Figure 7A). These results were further supported by the finding that EGCG-fed mice had significantly lower expressions of T-bet and RORγt in the CNS (Figure 7B). Thus, the reduced Th1 and Th17 populations in mice with EAE after EGCG supplementation were probably mediated by down-regulation of their respective master transcription factors.
Figure 7.
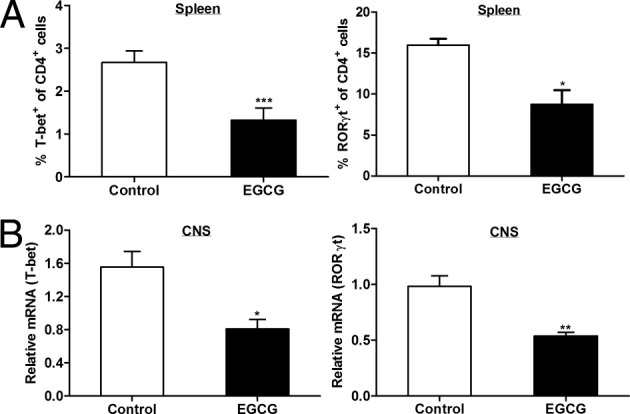
Dietary EGCG suppresses T-bet and RORγt expression. On day 12 after immunization, mice were euthanized and spleens, brains, and spinal cords were collected. A: Isolated spleen cells were used to conduct intracellular staining for T-bet and RORγt in CD4+ T cells and analyzed by flow cytometry. B: Total RNA extracted from the CNS tissue (brains and spinal cords) was used to determine T-bet and RORγt mRNA expression using real-time RT-PCR. All values in this figure are mean ± SEM (n = 6/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control.
EGCG Decreases Pro-Inflammatory Cytokine Production
Pro-inflammatory cytokines IL-6, IL-1β, and TNF-α have been important in the development of inflammation and neurological damage in EAE and in the pathogenesis of other autoimmune diseases. IL-6 is a decisive factor in switching differentiation toward Th17 cells away from Treg conversion.25, 26, 27 IL-1β and IL-628 or IL-23 and IL-1β29 are essential for Th17 development; TNF-α, a Th1 cytokine, promotes EAE symptoms and pathological characteristics.30, 31, 32, 33 All these cytokines are up-regulated in human MS and murine EAE.28, 29, 34, 35 Spleen and LN cells isolated on day 12 after immunization were restimulated with MOG35-55 peptide, and the collected supernatants were analyzed by ELISA for cytokine production. The cells from both spleen and LN produced less IL-6 and TNF-α in EGCG-fed mice compared with the control mice; furthermore, EGCG-fed mice had lower plasma levels of IL-1β than control mice (Figure 8). These results suggest that EGCG-induced improvement in EAE may be partly mediated by its effect on these inflammatory cytokines.
Figure 8.
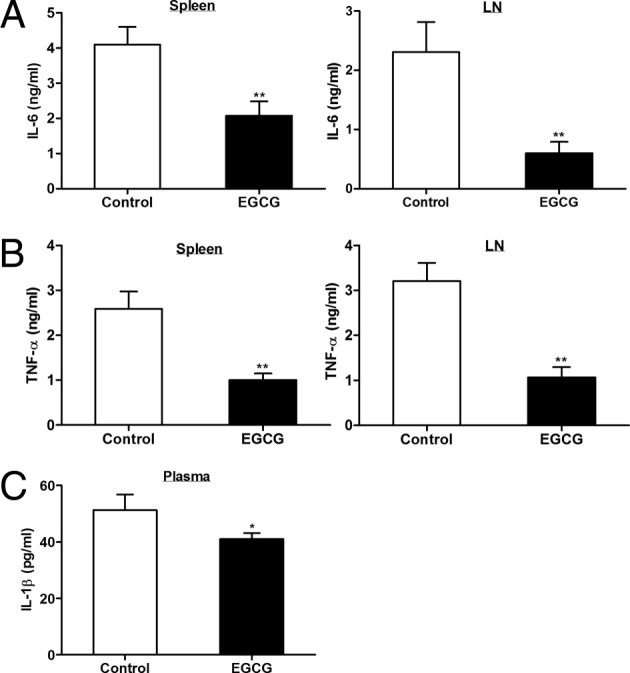
Dietary EGCG inhibits production of pro-inflammatory cytokines. On day 12 after immunization, mice were euthanized and spleens and LNs were collected. Spleen and LN cells were restimulated with MOG35-55 peptide for 72 hours, and supernatants were used to determine production of IL-6 (A) and TNF-α (B). Plasma samples were used to determine IL-1β concentration (C). All cytokines were determined using ELISA. All values in this figure are mean ± SEM (n = 8/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.05, **P < 0.01 compared with the control.
EGCG Reduces Circulating ICAM-1 and Down-Regulates CCR6 Expression in CD4+ T Cells
In the EAE model, after immunization, the myelin Ag–primed and subsequently expanded autoreactive effector T cells in peripheral lymphoid tissues need to enter the CNS to initiate tissue inflammation. Adhesion molecules play an important role in the migration of immune cells into the CNS. Patients with MS have increased serum levels of soluble ICAM-1 (sICAM-1),36, 37 and recent studies9, 38 have shown that ICAM-1 is more important than other adhesion molecules in promoting pathological cell infiltration into the CNS and the development of EAE. To determine whether this molecule is involved in the EGCG-induced reduction of inflammatory infiltration, we measured the plasma sICAM-1 level and found that it was lower in mice fed EGCG compared with those fed the control diet (Figure 9A). CCR6 has been essential for the first wave of autoreactive Th17 migrating into the uninflamed CNS by interacting with its only ligand, CCL20, which is constitutively expressed in the epithelial cells of the choroid plexus.39 Because we observed that inflammatory infiltration in the CNS of mice with EAE was greatly reduced by dietary EGCG supplementation, we wanted to know if this effect of EGCG affects T-cell migration across the BBB in addition to the reduced expansion of autoreactive T cells. In our first attempt to address this issue, we determined CCR6 expression on spleen CD4+ T cells harvested on day 12 after immunization. We found that both the number of CD4+ T cells expressing CCR6 (Figure 9B) and the amount of CCR6 expressed per cell (Figure 9C) were significantly lower in EGCG-fed mice compared with control mice. We further measured CCL20 mRNA expression in the CNS samples and found no difference between EGCG-fed mice and control mice (Figure 9D).
Figure 9.
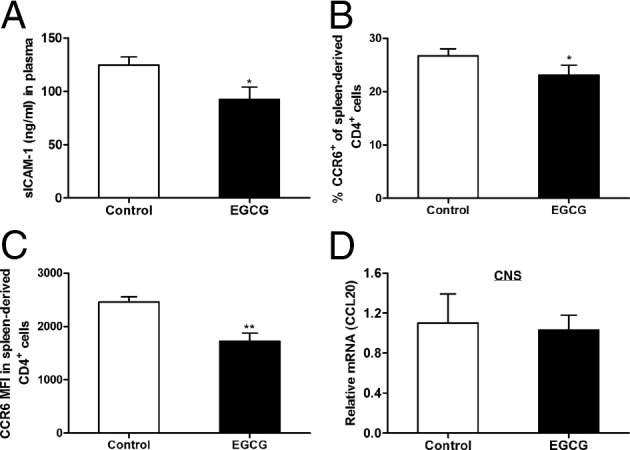
Dietary EGCG supplementation reduces circulating ICAM-1 and down-regulates CCR6 expression in CD4+ T cells but does not affect CCL20 expression in the CNS. On day 12 after immunization, mice were euthanized and blood, spleens, brains, and spinal cords were collected. A: Blood was centrifuged to obtain plasma, and sICAM-1 levels in the plasma samples were determined by ELISA. B and C: Spleen cells were isolated and CCR6 expression in CD4+ T cells was determined by flow cytometry. These data are expressed as percentage of CCR6+ CD4+ T cells (B) and CCR6 expression level per cell, as indicated by mean fluorescence intensity (MFI; C). D: Total RNA extracted from the CNS tissue (brains and spinal cords) was used to determine CCL20 mRNA expression using real-time RT-PCR. All values in this figure are mean ± SEM (n = 6/group). Significant differences were determined by the nonpaired Student's t-test. *P < 0.05, **P < 0.01 compared with the control.
EGCG Is Effective in Treating EAE When Administered during the Induction and Effector Phase of the Disease
In the experiments previously described, EGCG supplementation started 30 days before immunization and continued throughout the study. As such, we could not determine whether EGCG supplementation could still provide a benefit during the induction and after the onset of disease. To answer this question, we started EGCG supplementation on days 7 and 12 after immunization and then observed its effect on the severity of EAE symptoms. These time points were selected because, in a typical EAE model, day 7 is the time for full induction of autoimmune response (induction phase) and day 12 is the time when the symptoms have already developed for most animals (effector phase). We found that EGCG administration started at day 7 delayed disease onset and attenuated the symptoms. When EGCG was started at day 12, it attenuated the symptoms at a comparable magnitude but did not delay disease onset (Figure 10 and Table 2). These results indicate a therapeutic effect of EGCG on EAE.
Figure 10.
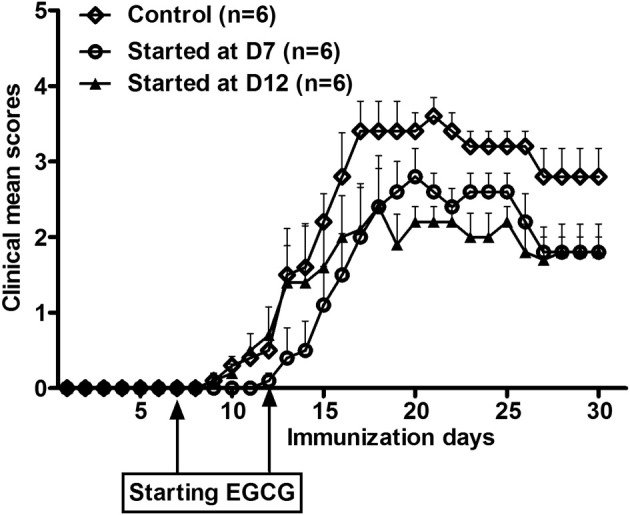
Dietary EGCG supplementation started after EAE initiation effectively ameliorated clinical symptoms of EAE. Three groups of mice fed the control diet were immunized with MOG35-55/CFA. Although one group continued receiving the control diet throughout the study, the remaining two groups were fed a 0.6% EGCG diet from days 7 (D7) and 12 (D12) after immunization on. Clinical symptoms of EAE were scored daily until day 30 after immunization. Values are mean ± SEM (n = 6).
Table 2.
Effect of EGCG Administered after Immunization on EAE Symptoms
| Group | Day of onset | CDI⁎ | Area of curve† |
|---|---|---|---|
| Control | 11.0 ± 0.65 | 54.00 ± 4.72 | 52.60 (100) |
| EGCG | |||
| At day 7 | 14.5 ± 0.94‡ | 35.60 ± 3.27‡ | 34.70 (66.0) |
| At day 12 | 10.5 ± 0.65 | 36.40 ± 5.50‡ | 35.10 (66.7) |
Values are given as the mean ± SEM (n = 6/group) unless otherwise indicated.
CDI, cumulative disease index.
Sum of clinical scores over the entire observation period.
Values are given as number (percentage) of 52.60.
P < 0.05 versus the control.
Discussion
Altered T-cell responses play a key role in autoimmune pathological conditions, as typically manifested in MS and rheumatoid arthritis. Attenuation of T-cell expansion and function is the most common therapeutic approach for these diseases. The theories for the immunopathogenesis of autoimmune diseases have significantly evolved over the past few years. The most pronounced progress is that T-cell–mediated autoimmune disorders are no longer discussed within the context of the Th1 and Th2 paradigm; rather, Th17 cells and Tregs are currently the main focus of research. MS and its rodent model, EAE, are the typical examples of such cases. This new information, therefore, needs to be fully considered in investigations designed to identify the bioactive food components with preventive and/or therapeutic potential against autoimmune diseases and to illuminate their mechanisms of action.
Most evidence suggesting an anti-inflammation property of EGCG is from cell-based studies in which EGCG is added at much higher than physiologically relevant concentrations. A few studies in animal studies using oral administration of EGCG or green tea polyphenols have been reported on some autoimmune diseases, such as rheumatoid arthritis,19 Sjögren's syndrome,40 colitis,41 and encephalomyelitis.17 These studies have shown, to a varied extent, EGCG's ability to improve symptoms, pathological characteristics, and the cytokine profile. In the only study17 to investigate EGCG's effect on EAE, Atkas et al showed that administration of EGCG by gavage (300 μg/mouse twice daily from the day of immunization on) to mice with EAE reduced clinical symptoms, brain inflammation and neuronal damage, proliferation, and TNF-α production of LN cells. The authors also reported a lack of effect of EGCG on Tregs, defined as CD4+CD25+ cells. However, given the fact that activated CD4+ T cells also express CD25 (IL-2Rα) and that the authors did not use the specific marker Foxp3 to identify Tregs, it is likely that the change in activated T cells might have masked real changes in the Treg population. This study, for the first time to our knowledge, demonstrated the efficacy of EGCG in improving EAE symptoms and provided evidence suggesting that the effect of EGCG was mediated through suppression of T-cell activity. Published before the introduction of Th17/Treg concept into immunopathogenesis, the study understandably provided limited mechanistic information. Taking advantage of the recent advancements in the understanding of pathogenesis of autoimmune immunity and using a multidose, dietary supplementation design, we revisited this issue. Our results confirmed and expanded the previous findings by showing that EGCG dose dependently ameliorated clinical symptoms and delayed disease onset in mice with EAE, and this EGCG-induced change was well associated with reduced inflammatory infiltration and demyelination damage in the CNS. Furthermore, EGCG supplementation suppressed expansion of the autoreactive pathological T cells and their effector function after rechallenge with the autoantigen. More important, we demonstrated a novel mechanism for the EGCG-induced effect (ie, altered regulation in CD4+ T-cell subsets, reducing Th1 and Th17 populations while promoting the Treg population). This effect is most likely to be mediated through the corresponding changes in the transcription factors that control their differentiation. Moreover, we showed that reduced expression of CCR6 on CD4+ T cells in EGCG-fed mice may contribute to the decreased cell infiltration seen in the CNS of these mice. Together, this study has provided compelling evidence to support the therapeutic efficacy of EGCG in EAE and, thus, a promising potential treatment for human MS and possibly other autoimmune diseases as well.
In the development of a T-cell–mediated autoimmune disease, the autoantigen-driven T-cell expansion needs to reach an adequate level to exert its effector functions, leading to tissue damage. In EAE, administered myelin antigen is first absorbed on site (skin) and processed by antigen-presenting cells (APCs) and then presented to T cells in peripheral lymphoid tissue. Primed T cells undergo proliferation and differentiation to expand the antigen-specific colony while polarizing into different effector subsets. The infiltration of these activated cells into the CNS from the periphery and the induction of an inflammatory response thereafter are key to the initiation of disease. In this study, we found that T cells from EGCG-fed mice had lower levels of proliferation on ex vivo rechallenge with the autoantigen MOG, which was further supported by a reduction in MOG-induced DTH skin response in EGCG-fed mice. Because the magnitude of an antigen-driven T-cell expansion depends on T cell and APC function, it is unclear whether these results represent a direct effect of EGCG on T cells, an indirect effect by acting on APCs, or both. We have previously shown that both in vitro and in vivo EGCG supplementation inhibits T-cell proliferation.15, 42 In a co-culture study,42 separately treating either APCs (during the antigen ovalbumin pulse) or T cells (ovalbumin specific) with EGCG-reduced T-cell proliferation, however, the direct effect of EGCG on T cells was predominant. Together with the previous studies showing that in vitro supplementation of EGCG suppressed maturation of mouse bone marrow–derived43 and human monocyte–derived44 dendritic cells, we believe that, although T cells are the main target of EGCG effect, contribution of APCs to EGCG-induced inhibition of autoreactive T-cell proliferation should be considered. More directed, definitive work is needed to accurately elucidate this question.
In the active EAE model, the autoreactive myelin-specific effector T cells, primed and expanded in peripheral lymphoid tissues, crossed the BBB and infiltrated the CNS, where these pathological cells exert their effector functions, leading to tissue damage and resulting disease symptoms. We found that EGCG inhibited autoreactive T-cell proliferation in the periphery and greatly reduced the inflammatory infiltration in the CNS, which is likely to contribute to the attenuated clinical symptoms and demyelination damage in the mice with EAE fed EGCG. Although unable to define the exact mechanisms by which EGCG reduces leukocyte infiltration, we propose some attributable factors. First, reduced pathological cells in the periphery are assumed to reduce the frequency of these cells coming across the CNS. Second, EGCG reduces the production or expression of the molecules known to facilitate leukocyte transendothelial migration, such as chemokines IL-8 and monocyte chemoattractant protein-145, 46, 47 and adhesion molecules48, 49 and to inhibit migration of neutrophils,50, 51 CD8+ T cells,49 and B cells.52 Third, the observed EGCG-induced reduction in IFN-γ and IL-17 production and the proportion of IL-17–IFN-γ double-positive CD4+ T cells may contribute to the reduced leukocyte infiltration because, as reported, IFN-γ facilitates Th17 migration across the BBB by enhancing endothelial expression of adhesion molecule ICAM-1, and IL-17 is capable of disrupting the BBB; IL-17–IFN-γ double-positive cells have a higher capacity for crossing the BBB than either single-positive cells.9 The observed reduction in plasma levels of sICAM-1 in EGCG-fed mice further supports this speculation. Last, we found that CD4+ T cells isolated from EGCG-fed mice had lower levels of expression for CCR6, a CCR primarily expressed in Th17 cells. A recent study39 showed that the binding of CCR6 expressed by Th17 cells to its ligand CCL20 in the epithelial cells of the choroid plexus is essential for triggering the entry of autoreactive Th17 cells into the CNS, leading to EAE. However, we did not see a significant effect of EGCG on CCL20 expression in the CNS. These results partly explain the EGCG-induced reduction in CD4+ T-cell infiltration and open a new direction for future studies on the working mechanisms of EGCG.
CD4+ T cells are classified into at least four functionally distinct subsets (namely, Th1, Th2, Th17, and Treg cells).25, 53, 54, 55, 56, 57, 58 Differentiation of naïve CD4+ T cells into these different effector cells is initiated by T-cell receptor engagement and costimulation in the presence of specific cytokines produced by the innate immune system on encounter with particular pathogens or antigens. Hallmark cytokines for Th1, Th2, and Th17 are IFN-γ, IL-4, and IL-17, respectively, and Tregs are often identified as CD4+CD25+Foxp3+ cells. The recently identified Th17 cells are believed to play a critical role in the clearance of extracellular bacteria and fungi, particularly those not well defended by Th1 or Th2 immunity.59, 60, 61 Tregs mainly function to maintain self-tolerance and regulate immune responses.62 Although these responses are necessary for a normal immune response, their dysregulation can be harmful, as evidenced in the case of autoimmune diseases. Up-regulated activation of Th1 and Th17 and down-regulated Treg development are viewed as characteristic components in the immunopathological features of EAE and some other organ-specific autoimmune diseases. Although studies have shown a protective effect of EGCG or green tea polyphenols in several autoimmune animal models, it is not known if this effect of EGCG is related to an altered modulation of polarization and function of these CD4+ T-cell subsets. Previous studies19 showed that green tea polyphenols in water given to mice with collagen-induced arthritis resulted in a lower expression of IFN-γ and TNF-α in the arthritic joints; EGCG gavage in mice with EAE reduced ex vivo TNF-α production in LN cells,17 and in mice with concanavalin A–induced hepatitis, it reduced levels of plasma IFN-γ and TNF-α and their mRNA expression in the liver.63 Although these results appear to imply an involvement of Th1 response in EGCG's protective effect on autoimmunity, in the current study, we provided direct evidence showing that EGCG inhibits Th1 and Th17 response in both the CNS and peripheral lymphoid tissue of mice with EAE. Transcription factors T-bet and RORγt are the master regulators in directing differentiation of naïve CD4+ T cells to Th1 and Th17, respectively. Thus, our further observation that EGCG inhibited expression of transcription factors T-bet and RORγt in CD4+ T cells of both the CNS and peripheral lymphoid tissue strongly suggests that EGCG may inhibit CD4+ T-cell differentiation into Th1 and Th17 by acting on their respective transcription controls. On the other hand, we found that EGCG increased the percentages of Tregs in CD4+ T cells of both the CNS and spleen. Although this result may represent an additional mechanism for the protective effect of EGCG in EAE, it is difficult to dissect the effect of EGCG on one subset, independent of its interplay with the other, because there is a well-recognized reciprocal regulation between Th17 and Treg development. Th17 differentiation is initiated by TGF-β and IL-6 and amplified by IL-21. Th17 cell proliferation and stabilization are supported by IL-23.22 TGF-β is important for natural Treg development64 and plays a key role in induced Treg differentiation.65 Given the duplicity of TGF-β in driving naïve CD4+ T cells into both Th17 and Treg, the presence of IL-6 appears to be the decisive factor.26 Compelling evidence has indicated that IL-6 is an essential differentiation factor for Th17 cells, and its presence redirects TGF-β–induced Treg differentiation toward Th17 cells. Because we found that EGCG inhibited IL-6 production, the observed up-regulation of Th17 and down-regulation of Treg by EGCG may be mediated, in part, through altered IL-6 production. IL-4 has been implicated as a suppressor cytokine in EAE, and IL-4–producing Th2 cells have helped limit the development of EAE.34, 35, 66, 67 By using intracellular staining and flow cytometry, we were unable to detect IL-4 and IL-10 produced by infiltrated CD4+ T cells in the CNS but found that their production in LN and spleen CD4+ T cells was not affected by EGCG. These results indicate that EGCG may not affect Th2 response; thus, the protective effect of EGCG in mice with EAE does not seem to be related to Th2 cells. Furthermore, although Tregs are the main producers of IL-10 and IL-10 contributes to Treg's function, EGCG can still promote Treg development without necessarily affecting IL-10 production.
In this study, we used two supplementation protocols to test the efficacy of EGCG in ameliorating EAE. We found that EGCG treatment started either 30 days before immunization or 7 or 12 days after immunization, effectively alleviating EAE. EGCG administration for 30 days before induction of EAE did not reduce disease incidence, although it slightly delayed disease onset; however, starting EGCG treatment 7 days after immunization also similarly delayed disease onset. Therefore, we believe that EGCG's effect is largely therapeutic, rather than preventive, at the doses used. Based on our findings, taking EGCG may benefit those who have T-cell–mediated autoimmune disorders. One limitation of this study, which needs to be further determined, is the relevance of effective doses for the treatment of disease in humans. The lowest effective dose (0.3% in diet) in this study is equivalent to 360 mg/kg body weight/day for a mouse of 25 g, based on the average diet consumption of 3 g/day. When this dose is converted from mice consuming 12 kJ/day to humans consuming 2000 kJ/day by using isocaloric calculation, it is equivalent to 26 mg/kg body weight/day in humans, or 1820 mg EGCG/day consumed by a 70-kg person. This dose can be achieved by drinking large quantities (at least 10 cups or 2 L/day) of tea or by taking supplements. In previous animal studies,42 mice fed 1% EGCG for a longer time than that used in the current study did not show any adverse effect, as assessed by histological characteristics, weight, and general condition; in addition, the doses used in our studies are within the safe range, as defined by the comprehensive EGCG toxicology studies in animals.68, 69, 70 Although we believe that equivalent EGCG doses for humans would be safe, nevertheless, this needs to be confirmed.
In summary, we showed that dietary EGCG supplementation is effective in ameliorating the symptoms and pathological changes in EAE animals. This effect of EGCG is associated with the reduced inflammatory infiltration in the CNS and the reduced proliferation of autoreactive T cells and their differentiation into different subsets with corresponding effector functions (namely, down-regulated Th1 and Th17 cells and up-regulated Tregs). These results strongly suggest a promising potential for using EGCG as a therapeutic agent in MS and possibly in other T-cell–mediated autoimmune diseases as well. This study has also revealed a novel mechanism to help us understand this beneficial effect of EGCG. Future studies are needed to determine the efficacy of EGCG in patients with MS and to demonstrate whether the mechanism for EAE in this study can also be applied to other T-cell–mediated autoimmune diseases.
Acknowledgments
We thank Stephanie Marco for help in the preparation of the manuscript.
Footnotes
Supported by a contract from the US Department of Agriculture (USDA) Agriculture Research Service (58-1950-7-707 to S.N.M.) and a grant from the USDA National Institute of Food and Agriculture (2010-65200-20360 to D.W.).
References
- 1.Gold R., Hartung H.P., Toyka K.V. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- 2.Ando D.G., Clayton J., Kono D., Urban J.L., Sercarz E.E. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 3.Kuchroo V.K., Anderson A.C., Waldner H., Munder M., Bettelli E., Nicholson L.B. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 4.Merrill J.E., Kono D.H., Clayton J., Ando D.G., Hinton D.R., Hofman F.M. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci U S A. 1992;89:574–578. doi: 10.1073/pnas.89.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmachari S., Pahan K. Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol. 2007;179:275–283. doi: 10.4049/jimmunol.179.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyake M., Sasaki K., Ide K., Matsukura Y., Shijima K., Fujiwara D. Highly oligomeric procyanidins ameliorate experimental autoimmune encephalomyelitis via suppression of Th1 immunity. J Immunol. 2006;176:5797–5804. doi: 10.4049/jimmunol.176.10.5797. [DOI] [PubMed] [Google Scholar]
- 7.O'Connor R.A., Prendergast C.T., Sabatos C.A., Lau C.W., Leech M.D., Wraith D.C., Anderton S.M. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Y., Lu L., Guo T.B., Qiu J., Yang Y., Liu A., Zhang J.Z. Novel immunomodulatory properties of berbamine through selective down-regulation of STAT4 and action of IFN-gamma in experimental autoimmune encephalomyelitis. J Immunol. 2008;181:1491–1498. doi: 10.4049/jimmunol.181.2.1491. [DOI] [PubMed] [Google Scholar]
- 9.Kebir H., Ifergan I., Alvarez J.I., Bernard M., Poirier J., Arbour N., Duquette P., Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 10.Luger D., Silver P.B., Tang J., Cua D., Chen Z., Iwakura Y., Bowman E.P., Sgambellone N.M., Chan C.C., Caspi R.R. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy J., Illes Z., Zhang X., Encinas J., Pyrdol J., Nicholson L., Sobel R.A., Wucherpfennig K.W., Kuchroo V.K. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vila J., Isaacs J.D., Anderson A.E. Regulatory T cells and autoimmunity. Curr Opin Hematol. 2009;16:274–279. doi: 10.1097/MOH.0b013e32832a9a01. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X., Koldzic D.N., Izikson L., Reddy J., Nazareno R.F., Sakaguchi S., Kuchroo V.K., Weiner H.L. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 14.Yang C.S., Maliakal P., Meng X. Inhibition of carcinogenesis by tea. Annu Rev Pharmacol Toxicol. 2002;42:25–54. doi: 10.1146/annurev.pharmtox.42.082101.154309. [DOI] [PubMed] [Google Scholar]
- 15.Wu D., Guo Z., Ren Z., Guo W., Meydani S.N. Green tea EGCG suppresses T cell proliferation through impairment of IL-2/IL-2 receptor signaling. Free Radic Biol Med. 2009;47:636–643. doi: 10.1016/j.freeradbiomed.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Abboud P.A., Hake P.W., Burroughs T.J., Odoms K., O'Connor M., Mangeshkar P., Wong H.R., Zingarelli B. Therapeutic effect of epigallocatechin-3-gallate in a mouse model of colitis. Eur J Pharmacol. 2008;579:411–417. doi: 10.1016/j.ejphar.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 17.Aktas O., Prozorovski T., Smorodchenko A., Savaskan N.E., Lauster R., Kloetzel P.M., Infante-Duarte C., Brocke S., Zipp F. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol. 2004;173:5794–5800. doi: 10.4049/jimmunol.173.9.5794. [DOI] [PubMed] [Google Scholar]
- 18.Gillespie K., Kodani I., Dickinson D.P., Ogbureke K.U., Camba A.M., Wu M., Looney S., Chu T.C., Qin H., Bisch F., Sharawy M., Schuster G.S., Hsu S.D. Effects of oral consumption of the green tea polyphenol EGCG in a murine model for human Sjogren's syndrome, an autoimmune disease. Life Sci. 2008;83:581–588. doi: 10.1016/j.lfs.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haqqi T.M., Anthony D.D., Gupta S., Ahmad N., Lee M.S., Kumar G.K., Mukhtar H. Prevention of collagen-induced arthritis in mice by a polyphenolic fraction from green tea. Proc Natl Acad Sci U S A. 1999;96:4524–4529. doi: 10.1073/pnas.96.8.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morinobu A., Biao W., Tanaka S., Horiuchi M., Jun L., Tsuji G., Sakai Y., Kurosaka M., Kumagai S. (-)-Epigallocatechin-3-gallate suppresses osteoclast differentiation and ameliorates experimental arthritis in mice. Arthritis Rheum. 2008;58:2012–2018. doi: 10.1002/art.23594. [DOI] [PubMed] [Google Scholar]
- 21.Ran Z.H., Chen C., Xiao S.D. Epigallocatechin-3-gallate ameliorates rats colitis induced by acetic acid. Biomed Pharmacother. 2008;62:189–196. doi: 10.1016/j.biopha.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J., Paul W.E. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J., Paul W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald D.C., Ciric B., Touil T., Harle H., Grammatikopolou J., Sarma J.D., Gran B., Zhang G.-X., Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- 25.Bettelli E., Carrier Y., Gao W., Korn T., Strom T.B., Oukka M., Weiner H.L., Kuchroo V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 26.Bettelli E., Oukka M., Kuchroo V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 27.Korn T., Mitsdoerffer M., Croxford A.L., Awasthi A., Dardalhon V.A., Galileos G., Vollmar P., Stritesky G.L., Kaplan M.H., Waisman A., Kuchroo V.K., Oukka M. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acosta-Rodriguez E.V., Napolitani G., Lanzavecchia A., Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 29.Wilson N.J., Boniface K., Chan J.R., McKenzie B.S., Blumenschein W.M., Mattson J.D., Basham B., Smith K., Chen T., Morel F., Lecron J.C., Kastelein R.A., Cua D.J., McClanahan T.K., Bowman E.P., de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 30.Begolka W.S., Vanderlugt C.L., Rahbe S.M., Miller S.D. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998;161:4437–4446. [PubMed] [Google Scholar]
- 31.Issazadeh S., Ljungdahl A., Hojeberg B., Mustafa M., Olsson T. Cytokine production in the central nervous system of Lewis rats with experimental autoimmune encephalomyelitis: dynamics of mRNA expression for interleukin-10, interleukin-12, cytolysin, tumor necrosis factor alpha and tumor necrosis factor beta. J Neuroimmunol. 1995;61:205–212. doi: 10.1016/0165-5728(95)00100-g. [DOI] [PubMed] [Google Scholar]
- 32.Issazadeh S., Navikas V., Schaub M., Sayegh M., Khoury S. Kinetics of expression of costimulatory molecules and their ligands in murine relapsing experimental autoimmune encephalomyelitis in vivo. J Immunol. 1998;161:1104–1112. [PubMed] [Google Scholar]
- 33.Kuroda Y., Shimamoto Y. Human tumor necrosis factor-alpha augments experimental allergic encephalomyelitis in rats. J Neuroimmunol. 1991;34:159–164. doi: 10.1016/0165-5728(91)90125-q. [DOI] [PubMed] [Google Scholar]
- 34.Imitola J., Chitnis T., Khoury S.J. Cytokines in multiple sclerosis: from bench to bedside. Pharmacol Ther. 2005;106:163–177. doi: 10.1016/j.pharmthera.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Sospedra M., Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 36.Rieckmann P., Nunke K., Burchhardt M., Albrecht M., Wiltfang J., Ulrich M., Felgenhauer K. Soluble intercellular adhesion molecule-1 in cerebrospinal fluid: an indicator for the inflammatory impairment of the blood-cerebrospinal fluid barrier. J Neuroimmunol. 1993;47:133–140. doi: 10.1016/0165-5728(93)90023-r. [DOI] [PubMed] [Google Scholar]
- 37.Sharief M.K., Noori M.A., Ciardi M., Cirelli A., Thompson E.J. Increased levels of circulating ICAM-1 in serum and cerebrospinal fluid of patients with active multiple sclerosis: correlation with TNF-alpha and blood-brain barrier damage. J Neuroimmunol. 1993;43:15–21. doi: 10.1016/0165-5728(93)90070-f. [DOI] [PubMed] [Google Scholar]
- 38.Bullard D.C., Hu X., Schoeb T.R., Collins R.G., Beaudet A.L., Barnum S.R. Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:851–857. doi: 10.4049/jimmunol.178.2.851. [DOI] [PubMed] [Google Scholar]
- 39.Reboldi A., Coisne C., Baumjohann D., Benvenuto F., Bottinelli D., Lira S., Uccelli A., Lanzavecchia A., Engelhardt B., Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 40.Hsu S.D., Dickinson D.P., Qin H., Borke J., Ogbureke K.U., Winger J.N., Camba A.M., Bollag W.B., Stoppler H.J., Sharawy M.M., Schuster G.S. Green tea polyphenols reduce autoimmune symptoms in a murine model for human Sjogren's syndrome and protect human salivary acinar cells from TNF-alpha-induced cytotoxicity. Autoimmunity. 2007;40:138–147. doi: 10.1080/08916930601167343. [DOI] [PubMed] [Google Scholar]
- 41.Varilek G.W., Yang F., Lee E.Y., deVilliers W.J., Zhong J., Oz H.S., Westberry K.F., McClain C.J. Green tea polyphenol extract attenuates inflammation in interleukin-2-deficient mice, a model of autoimmunity. J Nutr. 2001;131:2034–2039. doi: 10.1093/jn/131.7.2034. [DOI] [PubMed] [Google Scholar]
- 42.Pae M., Ren Z., Meydani M., Shang F., Meydani S.N., Wu D. Epigallocatechin-3-gallate directly suppresses T cell proliferation through impaired IL-2 utilization and cell cycle progression. J Nutr. 2010;140:1509–1515. doi: 10.3945/jn.110.124743. [DOI] [PubMed] [Google Scholar]
- 43.Ahn S.C., Kim G.Y., Kim J.H., Baik S.W., Han M.K., Lee H.J., Moon D.O., Lee C.M., Kang J.H., Kim B.H., Oh Y.H., Park Y.M. Epigallocatechin-3-gallate, constituent of green tea, suppresses the LPS-induced phenotypic and functional maturation of murine dendritic cells through inhibition of mitogen-activated protein kinases and NF-kappaB. Biochem Biophys Res Commun. 2004;313:148–155. doi: 10.1016/j.bbrc.2003.11.108. [DOI] [PubMed] [Google Scholar]
- 44.Yoneyama S., Kawai K., Tsuno N.H., Okaji Y., Asakage M., Tsuchiya T., Yamada J., Sunami E., Osada T., Kitayama J., Takahashi K., Nagawa H. Epigallocatechin gallate affects human dendritic cell differentiation and maturation. J Allergy Clin Immunol. 2008;121:209–214. doi: 10.1016/j.jaci.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 45.Ahn H.Y., Xu Y., Davidge S.T. Epigallocatechin-3-O-gallate inhibits TNFalpha-induced monocyte chemotactic protein-1 production from vascular endothelial cells. Life Sci. 2008;82:964–968. doi: 10.1016/j.lfs.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 46.Shin H.Y., Kim S.H., Jeong H.J., Kim S.Y., Shin T.Y., Um J.Y., Hong S.H., Kim H.M. Epigallocatechin-3-gallate inhibits secretion of TNF-alpha, IL-6 and IL-8 through the attenuation of ERK and NF-kappaB in HMC-1 cells. Int Arch Allergy Immunol. 2007;142:335–344. doi: 10.1159/000097503. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Y., Toborek M., Hennig B. Epigallocatechin gallate-mediated protection against tumor necrosis factor-alpha-induced monocyte chemoattractant protein-1 expression is heme oxygenase-1 dependent. Metabolism. 2010;59:1528–1535. doi: 10.1016/j.metabol.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 48.Chae Y.J., Kim C.H., Ha T.S., Hescheler J., Ahn H.Y., Sachinidis A. Epigallocatechin-3-O-gallate inhibits the angiotensin II-induced adhesion molecule expression in human umbilical vein endothelial cell via inhibition of MAPK pathways. Cell Physiol Biochem. 2007;20:859–866. doi: 10.1159/000110446. [DOI] [PubMed] [Google Scholar]
- 49.Kawai K., Tsuno N.H., Kitayama J., Okaji Y., Yazawa K., Asakage M., Hori N., Watanabe T., Takahashi K., Nagawa H. Epigallocatechin gallate attenuates adhesion and migration of CD8+ T cells by binding to CD11b. J Allergy Clin Immunol. 2004;113:1211–1217. doi: 10.1016/j.jaci.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 50.Dona M., Dell'Aica I., Calabrese F., Benelli R., Morini M., Albini A., Garbisa S. Neutrophil restraint by green tea: inhibition of inflammation, associated angiogenesis, and pulmonary fibrosis. J Immunol. 2003;170:4335–4341. doi: 10.4049/jimmunol.170.8.4335. [DOI] [PubMed] [Google Scholar]
- 51.Takano K., Nakaima K., Nitta M., Shibata F., Nakagawa H. Inhibitory effect of (-)-epigallocatechin 3-gallate, a polyphenol of green tea, on neutrophil chemotaxis in vitro and in vivo. J Agric Food Chem. 2004;52:4571–4576. doi: 10.1021/jf0355194. [DOI] [PubMed] [Google Scholar]
- 52.Kawai K., Tsuno N.H., Kitayama J., Sunami E., Takahashi K., Nagawa H. Catechin inhibits adhesion and migration of peripheral blood B cells by blocking CD11b. Immunopharmacol Immunotoxicol. 2011;33:391–397. doi: 10.3109/08923973.2010.522195. [DOI] [PubMed] [Google Scholar]
- 53.Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 54.Mangan P.R., Harrington L.E., O'Quinn D.B., Helms W.S., Bullard D.C., Elson C.O., Hatton R.D., Wahl S.M., Schoeb T.R., Weaver C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 55.Mosmann T.R., Cherwinski H., Bond M.W., Giedlin M.A., Coffman R.L. Two types of murine helper T cell clone, I: definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 56.Mosmann T.R., Coffman R.L. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 57.Street N.E., Mosmann T.R. Functional diversity of T lymphocytes due to secretion of different cytokine patterns. FASEB J. 1991;5:171–177. doi: 10.1096/fasebj.5.2.1825981. [DOI] [PubMed] [Google Scholar]
- 58.Veldhoen M., Hocking R.J., Atkins C.J., Locksley R.M., Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 60.Weaver C.T., Harrington L.E., Mangan P.R., Gavrieli M., Murphy K.M. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 61.Weaver C.T., Hatton R.D., Mangan P.R., Harrington L.E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 62.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y., Mei Y., Feng D., Xu L. (-)-Epigallocatechin-3-gallate protects mice from concanavalin A-induced hepatitis through suppressing immune-mediated liver injury. Clin Exp Immunol. 2006;145:485–492. doi: 10.1111/j.1365-2249.2006.03137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y., Zhang P., Li J., Kulkarni A.B., Perruche S., Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 65.Chen W., Jin W., Hardegen N., Lei K.J., Li L., Marinos N., McGrady G., Wahl S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Falcone M., Bloom B.R. A T helper cell 2 (Th2) immune response against non-self antigens modifies the cytokine profile of autoimmune T cells and protects against experimental allergic encephalomyelitis. J Exp Med. 1997;185:901–907. doi: 10.1084/jem.185.5.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen-specific T-cell autoimmunity in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 68.Isbrucker R.A., Bausch J., Edwards J.A., Wolz E. Safety studies on epigallocatechin gallate (EGCG) preparations, part 1: genotoxicity. Food Chem Toxicol. 2006;44:626–635. doi: 10.1016/j.fct.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 69.Isbrucker R.A., Edwards J.A., Wolz E., Davidovich A., Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations, part 3: teratogenicity and reproductive toxicity studies in rats. Food Chem Toxicol. 2006;44:651–661. doi: 10.1016/j.fct.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Isbrucker R.A., Edwards J.A., Wolz E., Davidovich A., Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations, part 2: dermal, acute and short-term toxicity studies. Food Chem Toxicol. 2006;44:636–650. doi: 10.1016/j.fct.2005.11.003. [DOI] [PubMed] [Google Scholar]