

Abstract
Purpose
Dedicator of cytokinesis 8 (DOCK8) deficiency is an autosomal recessive combined immunodeficiency whose clinical spectra include recurrent infections, autoimmunity, malignancies, elevated serum IgE, eczema and food allergies. Here, we report on patients with loss of function DOCK8 mutations with profound immune dysregulation suggestive of an Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX)-like disorder.
Methods
Immunophenotyping of lymphocyte subpopulations and analysis of DOCK8 protein expression were evaluated by flow cytometry. T regulatory (Treg) cells were isolated by cell sorting, and their suppressive activity was analyzed by flow cytometry. Gene mutational analysis was performed by whole exome and Sanger sequencing.
Results
Patient 1 (P1) presented at 10 months of age with chronic severe diarrhea and active colitis in the absence of an infectious trigger, severe eczema with elevated serum IgE and autoimmune hemolytic anemia, suggestive of an IPEX-related disorder. Whole exome sequencing revealed a homozygous nonsense mutation in DOCK8 at the DOCK- homology region (DHR)-1 (c.1498C>T; p. R500X). Patient P2, a cousin of P1 who carries the same DOCK8 nonsense mutation, presented with eczema and recurrent ear infections in early infancy, and she developed persistent diarrhea by 3 years of age. Patient P3 presented with lymphoproliferation, severe eczema with allergic dysregulation and chronic diarrhea with colitis. She harbored a homozygous loss of function DOCK8 mutation (c.2402 –1G→A). Treg cell function was severely compromised by both DOCK8 mutations.
Conclusion
DOCK8 deficiency may present severe immune dysregulation with features that may overlap with those of IPEX and other IPEX-like disorders.
Keywords: Combined Immunodeficiency, DOCK8, FOXP3, Immune Dysregulation, IPEX, IPEX-like, Regulatory T cells, Treg
Introduction
Dedicator of cytokinesis 8 (DOCK8) is a 190 kDa protein predominantly expressed in hematopoietic cells that belongs to the DOCK180 family of atypical guanine nucleotide exchange factors (GEF) (1). DOCK8 has critical roles in both humoral and cellular immune responses. It mediates cell differentiation, survival, adhesion and migration by coordinating actin cytoskeletal response through cell cycle 42(Cdc42) activation (2–6). DOCK8 is critical to the translocation of cutaneous dendritic cells to lymph nodes and the persistence of memory CD8+ T cells in the epidermis (7). In addition to its regulation of the actin cytoskeleton, DOCK8 regulates the differentiation of T helper (Th) cell subsets (8, 9). DOCK8 promotes TH17 cell differentiation by regulating STAT3 phosphorylation and translocation to the nucleus (8). In B cells, DOCK8 enables a TLR9-PYK2-STAT3 cascade that promotes B cell proliferation and memory B cell formation (10). DOCK8 deficiency gives rise to an autosomal recessive form of the hyper IgE (AR-HIES) syndrome that overlaps in its features with autosomal dominant hyper IgE (AD-HIES) syndrome caused by loss of function mutations in STAT3 (11–13). Patients with DOCK8 deficiency suffer from recurrent infections, including candidiasis, cutaneous viral infections (molluscum contagiosum, herpes simplex viruses, human papilloma viruses), as well as cutaneous and invasive bacterial infections. DOCK8 deficiency results in defective memory T and B cell responses, associated with poor humoral immune responses, and impaired function of CD8 and NK cells that is likely to be implicated in the risk for cutaneous viral infection and virally driven malignancies (4, 14–16).
DOCK8 deficiency has been associated with a number of immune dysregulatory features, including autoimmune disorders such as vasculitis and autoimmune hepatitis, as well as allergic disorders such as eczema, asthma, and food allergy, with high IgE and hypereosinophilia (17, 18). The immune dysregulatory features of DOCK8 deficiency overlap with those of primary disorders of immune dysregulation resulting from gene defects affecting regulatory T (Treg) cell differentiation and function. The most famous of these, Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX), results from loss of function mutations affecting the transcription factor FOXP3. A number of IPEX-like conditions have also been described due to mutations affecting genetic elements critical to Treg cell function (19). DOCK8 itself has emerged to play an important role in T regulatory (Treg) cell homeostasis and function. DOCK8 deficiency is associated with defective Treg cell suppressive function, and patients with DOCK8 deficiency exhibit decreased numbers and depressed function of peripheral blood Treg cells, associated with increased auto-antibody production (20). In this report, we highlight the capacity of DOCK8 deficiency to present as an immune dysregulatory disorder that overlaps in its phenotype with Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX)-like disorders (21), underscoring the vital function of DOCK8 in immunoregulation.
Methods
Patients
The history, clinical and laboratory findings and genetic analysis of DOCK8-deficient patients P1 and P2 are detailed in the Results section, and those of patients P3 and P4 in the Online Supplemental Materials. All study participants were recruited using written informed consent approved by the respective Institutional Review Boards.
Antibodies and flow cytometry
Anti-human monoclonal antibodies (mAbs) to the following antigens were used for staining: CD3 (UCHT1), CD4 (RPA-T4), CD8 (RPA-T8), CD127 (A019D5), CD25 (BC96), CD19 (HIB19), CD27 (IV T-128), IgD (IA6-2), IFN-γ (4S.B3), IL-4 (8D4-8), IL-17 (BL168) (Biolegend), CTLA4 (14D3), FOXP3 (PCH101) and Helios (22F6) (eBioscience) and the appropriate isotype controls. Whole blood was incubated with mAbs against surface markers for 30 min on ice. Intracellular staining with FOXP3, Helios and CTLA4 was performed using eBioscience Fixation/Permeabilization buffer according to the manufacturer’s instructions. Indirect Intracellular staining for DOCK8 was performed for both patients on freshly isolated peripheral blood mononuclear cells (PBMCs) using BD Biosciences Fixation/Permeabilization buffer with polyclonal mouse anti-DOCK8 antibodies (Santa Cruz) or mouse IgG1 isotype control (Biolegend), followed by secondary detection with FITC anti-mouse IgG1 (Biolegend) in P1. For P2, DOCK8 intracellular staining was performed on PBMC using polyclonal rabbit anti-DOCK8 antibody (Sigma-Aldrich, St. Louis, MO) or Rabbit IgG XP (R) isotype control (cell signaling), followed by secondary detection Brilliant Violet 421™ Donkey anti-Rabbit IgG (Biolegend). For intracellular cytokine staining, cell suspensions were incubated with Phorbol myristate acetate (PMA) (Sigma-Aldrich; 50 ng/mL), Ionomycin (Sigma-Aldrich; 500 ng/mL) and GolgiPlug™ (BD Biosciences; According to manufacturers instructions) for 4 h in complete medium. Permeabilization and intracellular IFN-γ, IL- 4 and IL-17 staining was carried out using an eBioscience Fixation/Permeabilization kit as described above. Data were collected with a Fortessa cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
DOCK8 mutation identification and sequencing
Whole exome sequencing was performed as described (22). For Sanger sequencing analysis, DOCK8 sequences were derived from genomic DNA by polymerase chain reaction (PCR) amplification and sequenced bidirectionally using dye-terminator chemistry, as described (11).
Treg cells suppression assay
This assay was performed on isolated PBMCs. T cells were purified by cell sorting for CD4+CD25+CD127low Treg and CD4+CD25− T effector cells (Teff). The suppression assay was done as outlined in our previous reports (20, 22).
Results
The index case (patient P1) was a 3-year-old boy born to consanguineous parents (Fig. 1A) who presented at 10 months of age with persistent diarrhea and poor weight gain starting at one year of age that did not respond to multiple dietary modifications. His physical examination was notable for weight below the 5th percentile for age, and an eczematous rash involving his face (Fig. 1B), trunk and extremities. His eczema started at the age of 3 months, and while complicated by intermittent secondary bacterial infections, it responded well to therapy with topical steroids and emollients. His upper gastroenterology endoscopy was normal while his initial colonoscopy showed non-specific colitis for which he was treated with parenteral nutrition and oral steroid. Despite the initial favorable clinical response, his diarrhea relapsed in the setting of negative infectious studies that included routine stool cultures for enteric bacterial pathogens and frequent stool exams for ova and parasites. Testing for other infectious agents, including rotavirus, adenovirus, Clostridium difficile, giardia and cryptosporidium, was negative on multiple occasions. Subsequently, multiple immunosuppressive medications were tried, including pulse steroid, azathioprine and cyclosporine, without sustained clinical improvement. Repeated colonoscopy demonstrated chronic active colitis with diffuse superficial ulceration (Fig. 1C, D). Colonic tissue histopathology showed a diffuse mixed inflammatory cell infiltrate, including polymorphonuclear leukocytes, lymphocytes and plasma cells associated with bifid crypts and crypt abscesses (Fig. 1D). These findings were concerning for the possibility of evolving ulcerative colitis for which he was managed with azathioprine and mesalamine. His blood counts revealed a mild anemia with peripheral eosinophilia (Table E1). Evaluation of serum immunoglobulins (IgA, IgM, IgG), specific antibody titers and lymphocyte subsets were normal. His IgE levels were elevated (> 5000 Ku/l) and his specific IgE was highly positive for multiple foods including milk, soy, wheat, egg, peanut and tree nuts. Autoimmune workup showed a positive direct Coombs test, consistent with an autoimmune hemolytic anemia, but was negative for anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, anti-thyroid antibodies and celiac disease screening tests. On follow-up he developed a pneumonia complicated by severe acute respiratory distress syndrome leading to his death at the age of 3 years.
Fig. 1. Identification of autosomal recessive DOCK8 mutation in two patients with IPEX-Like phenotype.

(A) Pedigree of the extended family of patients P1 and P2. Squares represent males while circles for females. Filled symbol denote patients, and half-filled symbols denote carriers. The double lines between the parents indicate consanguinity. (B) Representative picture showing facial eczematous lesions in P1 with secondary bacterial infection. (C) Large intestinal mucosal lesions in P1 identified by colonoscopy. (D) Tissue pathology of active colonic lesions in P1. Arrow indicates a crypt abscess. (E) Sanger sequencing fluorograms showing germline homozygous DOCK8 mutation in patients P1 and P2 and its heterozygous carriage in the parents of P2. (F) Flow cytometric analysis of DOCK8 expression in CD3+ T cells of P1 and P2 as compared to those of healthy controls.
The presence of persistent diarrhea with active colitis, eczema and allergic dysregulation and Coombs-positive autoimmune hemolytic anemia led us to investigate genetic lesions associated with IPEX and IPEX-like related disorders (21). Flow cytometry revealed normal expression of FOXP3, CD25 and CTLA4 on Treg cells. Sanger sequencing analysis of the FOXP3 gene was normal. Whole exome sequencing analysis identified 96 homozygous gene variants (Table E2). While many variants associated with missense or in-frame deletions/insertions scored benign by Polyphen and/or SIFT protein function prediction algorithms, the one deleterious gene variant identified that could explain his presentation was in DOCK8. He was found to have a homozygous c.1498 C>T mutation in DOCK8, resulting in a premature stop codon (p. R500X), which was confirmed by Sanger sequencing (Fig. 1E). No other deleterious variants were found in genes associated with early onset inflammatory bowel disease or IPEX-like phenotype, including IL10/IL10R and LRBA. The DOCK8 mutation, which mapped to the DOCK homology domain 1 of the DOCK8 protein, was not found in dbSNP 38 or the 1000 Genomes database. Flow cytometric analysis of his peripheral blood T cells showed complete absence of DOCK8 expression, indicative of a DOCK8 null phenotype (Fig. 1F).
Patient 2 (P2) is a 4-year-old girl, cousin of patient P1, who presented for evaluation following her cousin’s death. She had a generalized eczematous rash since early infancy. Her eczema was managed successfully with topical steroids and emollients with no history of skin super-infections. At 12 months of age, she started to have multiple ear infections that were treated with multiple courses of antibiotic, and which were eventually controlled by the placement of bilateral tympanostomy tubes. At the age of 3 years, she started to have chronic diarrhea, similar to her cousin’s history. On physical exam, the patient had a generalized eczema and bilateral perforated eardrums. Her complete blood counts, immunoglobulins and tetanus IgG antibody titer were normal (Table E1). Her total IgE was 408 IU/ml, and she had positive specific IgE antibodies to milk, soy, peanut, sesame and tree nuts. Sanger sequencing analysis of DOCK8 identified the same homozygous mutation that was described in P1 (c.1498 C>T; p. R500X), also leading to complete absence of DOCK8 expression (Fig. 1E, F). She was started on antimicrobial prophylaxis and monthly IVIG infusion with good clinical improvement. Currently, the patient is being evaluated for hematopoietic stem cell transplantation (HSCT) from her full-matched healthy sibling (23).
We examined the impact of DOCK8 deficiency on Treg cell phenotype and function in our patients. The frequencies of Treg cells in the peripheral blood of patients P1 and P2 were comparable to those of healthy controls (Fig. 2A, B). Analysis of Treg cell markers revealed normal expression of FOXP3, CTLA4 and CD127 in both patients, but decreased expression of CD25 in patient P2 and decreased Helios expression in both patients (Fig. 2C, D). Strikingly, the capacity of patient Treg cells to suppress the in vitro proliferation of the patient T cells in response to anti-CD3 mAb stimulation was absent, thus establishing the presence of a profound functional defect in the patient Treg cells (Fig. 2E, F).
Fig. 2. DOCK8 deficiency leads to profound Treg cell dysfunction.

(A, B) Representative dot plot analysis of FOXP3 expression in CD4+ T cells of P1 (A) and P2 (B) as compared to those of a control subject. (C, D) Representative flow cytometric histogram plots of FOXP3, CD127, CD25, CTLA4 and Helios expression in FOXP3+CD4+ T cells of P1 (C) and P2 (D) (solid lines) versus those of healthy control (HC) subjects (dotted lines). Gray area represents expression in conventional T cells (Tconv) of a HC subject. (E) Flow cytometric analysis of CellTrace Violet proliferation dye dilution in cell-sorted control Teff (CD4+CD25−) cells stimulated with anti-CD2/CD3/CD28 mAb-coated bead in the absence or presence of Treg cells (CD4+CD25+CD127low) from HC subjects or from P2 only. (F) Representative curve of in vitro suppression assays in response to different Treg/Teff ratio in DOCK8-deficient patients P2, P3, and P4 and in control subject. ***p<0.001 by two-way ANOVA.
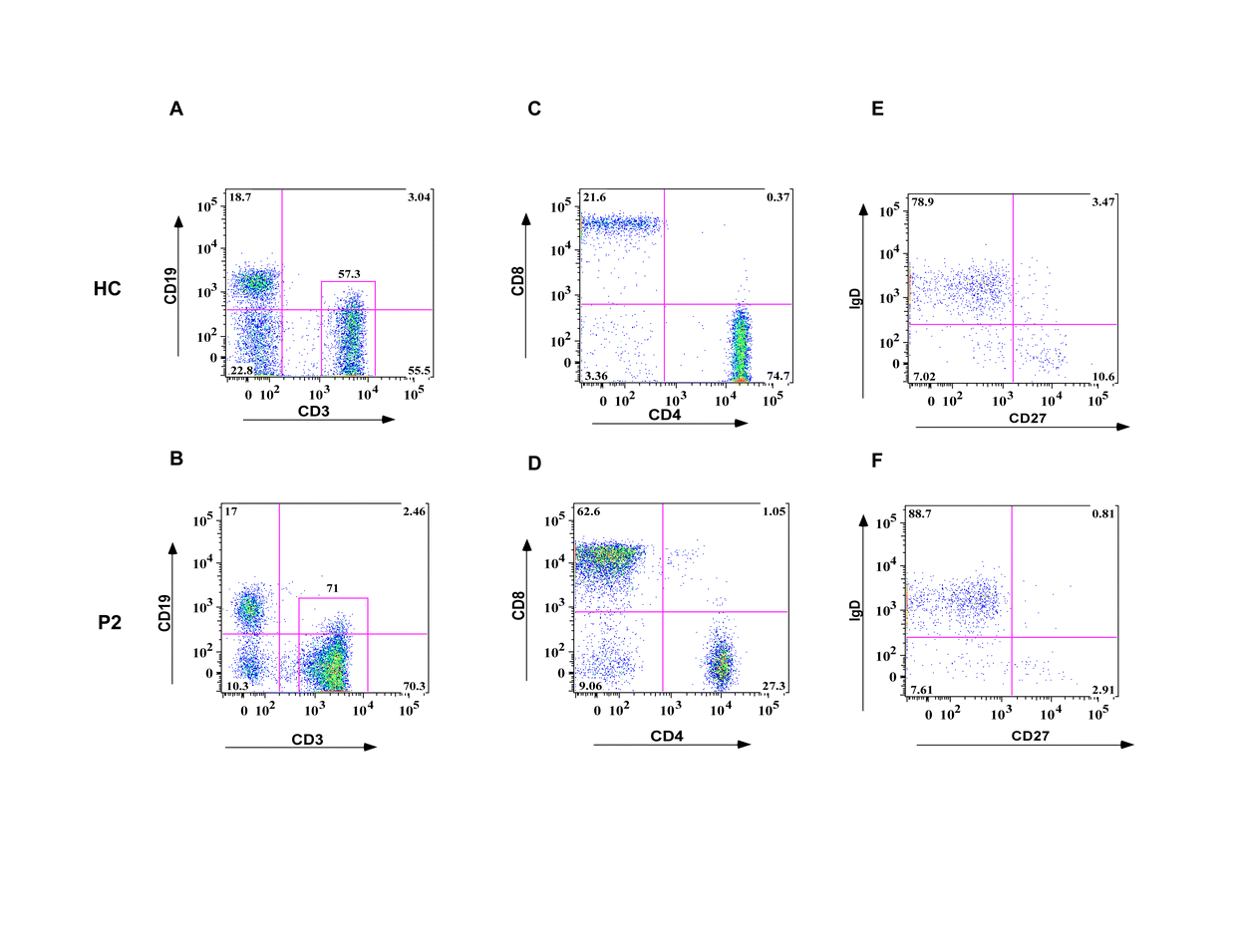
Further analysis of peripheral blood lymphocyte populations on patient P2 revealed findings characteristic of DOCK8-deficient subjects, including an inverted CD4+ to CD8+ T cell ratio, and profound deficiency of CD27+IgD−memory B cells (Fig. E1) (18).
We further analyzed the immunological features of another DOCK8-deficient patient with severe immundysregulation. Patient 3 (P3) is a 6-year-old girl, who has had severe atopic dermatitis since early infancy. She later evolved generalized lymphadenopathy and hepatosplenomegaly, with a Lymph node biopsy showing cortical lymphoid hyperplasia with no dysplastic changes or granulomas. At 5 years of age, she developed chronic diarrhea in the setting of negative infectious studies. Multiple antimicrobial medications were tried without sustained clinical improvement. Her upper and lower gastrointestinal endoscopy showed eosinophilic esophagitis, chronic inactive gastritis and chronic colitis with an eosinophilic infiltrate (Fig. 3A, B). Subsequently, she was started on oral steroid and mast cell stabilizer with resolution of her diarrhea and significant weight gain. Evaluation of serum immunoglobulins, specific antibody titers and lymphocyte subsets were normal apart from high IgE (> 5000 Ku/l) (Table E1). Her specific IgE was highly positive for multiple foods including egg, peanut and tree nuts. Mutational analysis revealed a homozygous mutation in DOCK8 (c.2402 –1G→A). Her brother, patient P4, is a 4-year-old boy who presented with atopic dermatitis. He had no history of recurrent infections, diarrhea or hospitalization. Immunological evaluation showed high IgE with no other abnormalities (Table E1). He was found to have the same mutation as his sister. Both patients had markedly decreased staining of DOCK8 protein in their CD4+ T cells, consistent with the expression of mutant DOCK8 protein (Fig. 3C).
Fig. 3. Pathological and immunological features of patient P3.

(A, B) Representative pictures of esophageal and colonic tissue pathology in patient P3. Representative arrows illustrate eosinophil cell infiltration. (C) Flow cytometric analysis of DOCK8 expression in CD3+ T cells of P3 and P4 as compared to those of a HC subject. (D) Representative dot plot analysis of FOXP3 expression in CD4+ T cells of P3 and P4 as compared to those of a control subject. (E) Representative flow cytometric histogram plots of FOXP3, CD127, CD25, CTLA4 and Helios expression in FOXP3+CD4+ Treg cells of P3 (red tracing) and P4 (green tracing) versus those of a HC subject (blue tracing). Gray area represents expression in Tconv cells of a HC subject.
Analysis of the Treg cell phenotype of patients P3 and P4 revealed normal expression of FOXP3 (Fig. 3D, E). Expression of the Treg cell markers Helios was decreased, similar to the findings in patients P1 and P2 (Fig. 3E). Patient P3 also exhibited decreased CD25 and CTLA4 expression, while CD127 expression was unaffected. Also, and similar to patients P1 and P2, the Treg cell suppression function of patients P3 and P4 were severely compromised (Fig. 2F). Overall, these results confirmed the functional incapacitation of Treg cell function by DOCK8 deficiency.
We further analyzed the frequencies of different T helper (Th) cells in circulation in the surviving patient P2 and in patients P3 and P4 as compared to control subjects. In all three patients, the frequency of circulating IL-4+CD4+ T cells was markedly increased, indicative of Th2 cell skewing (Fig. 4A, B) (8). Circulating IL-17+CD4+ T cells were profoundly deficient, while IFN-γ+CD4+ T cells were normal, consistent with our previous studies on DOCK8 deficient subjects (Fig. 4A, B) (8). Overall, these findings were consistent with the patients suffering immunopathological alterations related to DOCK8 deficiency.
Fig. 4. Exacerbated TH2 and impaired TH17 cell responses in Patient P2.

(A) Flow cytometric analyses of IFN-γ, IL-4 and IL-17 expression by peripheral CD4+ T cells from controls and patient P2. (B) The frequencies of IFN-γ, IL-4 and IL-17 producing CD4+ cells in three healthy controls and three DOCK8-deficient patients. **p<0.01 by unpaired two-tailed Student’s t-test.
Discussion
Whereas DOCK8 deficiency typically manifests as a combined immunodeficiency, with recurrent viral, bacterial and fungal infections, it also presents with several features of immune dysregulation, including allergic dysregulation, lymphoproliferation and autoimmunity, which may dominate the clinical picture (13, 17, 18). The clinical findings in our index case, patient P1, were skewed towards immune dysregulation, with prominence of enteropathy and colitis as clinical features in the absence of an obvious infectious agent, in addition to the other features such allergic dysregulation and autoimmunity (21, 24). The triad of an enteropathy not attributable to an infectious agent, eczema with allergic dysregulation and autoimmunity in a male child raised suspicion of IPEX or an IPEX like disorder, leading to the identification of DOCK8 deficiency as the underlying disorder. Two other patients, P2 and P3, also suffered from enteropathy and allergic dysregulation in the absence of an overt autoimmunity. The intense immune dysregulation suffered by the DOCK8-deficient subjects was reflected by the lack of suppressor function by Treg cell of patients available for testing, namely P2, P3 and P4, a finding in agreement with the profound deficiency in Treg cell suppression previously documented in DOCK8-deficient subjects (20). Overall, the triad of enteropathy/colitis, eczema with allergic dysregulation and autoimmunity manifested by patient P1, and the partial phenotypes of enteropathy and allergic dysregulation manifested by patients P2 and P3, overlaps with the phenotypes of IPEX and IPEX-like disorders. Thus, DOCK8 deficiency should be considered in the differential diagnosis of these disorders.
A number of primary genetic defects impacting Treg cell differentiation and function have been described. The most famous of these, IPEX, is a disorder originally associated with loss of function mutations in FOXP3. While the classical IPEX presentation is a triad of enteropathy, eczema with allergic dysregulation and autoimmunity, including autoimmune endocrinopathies, cytopenias and organ specific autoimmunity (lung, liver, kidney etc) (25–27). It is now recognized that the clinical phenotypes of IPEX may evolve over time, and may also present with partial phenotypes, associated with both classical, fully penetrant FOXP3 mutations as well as hypomorphic ones (24, 28). Furthermore, a number of IPEX-like conditions have been described due to mutations affecting genetic elements critical to Treg cell function (19, 21). These include loss of function mutations in IL2RA (29) CTLA4 (30, 31), LRBA (22, 32), STAT5b (33) and ITCH (34), as well as in some cases of gain of function (GOF) mutations in STAT1 (35) and STAT3 (36, 37). These conditions also present with various combinations of IPEX-related phenotypes as well as distinguishing features of their own. In particular, the intersection between immunodeficiency and immune dysregulation observed in some patients with DOCK8 deficiency is shared by some other IPEX-like disorders, including CTLA4 and LRBA deficiencies as well as in some cases of STAT1 and STAT3 GOF mutations. The myriad clinical phenotypes of these disorders is not surprising, given the broad impact of their underlying gene defects on different arms of the immune system as well as the interaction of these defects with host genetic and exposure factors.
There are several mechanisms by which DOCK8 deficiency may impact Treg cell function (13). DOCK8 regulates the actin cytoskeleton by virtue of its activation of CDC42, which in turn activates Wiskott-Aldrich protein (WASP) (6). WASP, together with WASP interacting protein (WIP) promotes actin polymerization upon T cell activation. DOCK8 physically interacts with the WASP/WIP complex, thus enabling tight spatial regulation of the actin cytoskeleton by the complexed proteins (6). WASP deficiency in Treg cells incapacitates their function, suggesting that DOCK8 deficiency may impair Treg cell function in part by preventing activation of WASP/WIP (38, 39). Another mechanism by which DOCK8 deficiency may act to impair Treg cell function involves STAT3, a transcription factor that plays an essential role in Treg cell function (8). DOCK8 amplifies STAT3 activation by a variety of cytokines, and its deficiency would likely impair STAT3 activation in Treg cells (8). Further studies would be required to dissect the contribution of the respective mechanism to Treg cell dysfunction in DOCK8 deficiency.
Several mechanisms could contribute to the enteropathy in our DOCK8-deficient subjects. While an infectious etiology is always suspect and should be sought out, evidence of a frank infection was lacking. In mice with Foxp3 deficiency, the absence of Treg cell function leads to a breakdown in mucosal tolerance to the gut commensal microbiota (40). This loss of immune regulation gives rise to a gut mucosal inflammatory response driven by the commensal bacteria that is toll-like receptor (TLR)-dependent and which could be abrogated by concurrent deficiency of the TLR adaptor protein MyD88 (40). Both IPEX and DOCK8 deficient patients also suffer from food allergies, which may contribute to the Th2 inflammatory response in the gut (17, 18, 41, 42). Avoidance of the suspected food allergens may contribute to the management of enteropathy in these subjects.
It is well appreciated that the clinical manifestations of patients with the same genetic mutations may vary depending on interactions with other genetic elements and with environmental exposures. In this report, the divergence in the phenotypic presentation of the same DOCK8 gene defect was especially pronounced for the siblings P3 and P4, the former suffering severe disease manifestations while the latter was very mildly affected. In that regard, the recent identification of NEIL3, encoding the base excision repair enzyme Nei endonuclease VIII-like 3 (NEIL3), as a modulator of autoimmunity in LPS-responsive beige-like anchor (LRBA) deficiency, a heritable disor0der of dysregulation and autoimmunity, provides one such example of gene-gene interactions influencing disease phenotype (43). We postulate that similar interaction(s) in our patients may have influenced their disease phenotype in favor of intense immune dysregulation and IPEX-like manifestations.
Finally, and in terms of therapy, the definitive treatment of DOCK8 deficiency is bone marrow transplantation (23). The emergence of immune dysregulation in the context of an immunodeficiency disorder presents challenges in terms of therapy. The benefits of interim focused interventions aimed at restraining potentially life threatening immune dysregulation, including IPEX-like manifestations, should be carefully weighed against the risk of compounding the immune deficiency by such interventions.
Supplementary Material
{kind=link}
PBMC of patient P2 and those of a control subject were analyzed for CD3 and CD19 expression (A, B). CD3+ T cells were further analyzed for CD4 and CD8 expression (C, D). CD19+ B cells were further analyzed for IgD and CD27 expression (E, F).
Hematological and Immunological findings on DOCK8-deficient subjects. The quantitative determination of serum immunoglobulins is reported for the respective patients before starting IVIG therapy.
The 96 candidate variants were homozygous and nonsynonymous in the patient and absent from dbSNP. The gene symbol (DOCK8) for the candidate gene identified on chromosome 9 is in boldface.
Acknowledgments
This work was supported by the National Institutes of Health R01AI085090 to Talal A. Chatila and 4R01AI100315 to Raif S. Geha, and a grant from the Scientific and Technological Research Council of Turkey (1059B191300622) to Sevgi Keles.
Footnotes
Conflict of Interest Statement
All authors have no conflict of interest.
References
- 1.Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends in cell biology. 2007;17(8):383–93. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Disease markers. 2010;29(3–4):151–6. doi: 10.3233/DMA-2010-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119(19):4451–61. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizesko MC, Banerjee PP, Monaco-Shawver L, Mace EM, Bernal WE, Sawalle-Belohradsky J, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2013;131(3):840–8. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ham H, Guerrier S, Kim J, Schoon RA, Anderson EL, Hamann MJ, et al. Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J Immunol. 2013;190(7):3661–9. doi: 10.4049/jimmunol.1202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, et al. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J Clin Invest. 2016;126(10):3837–51. doi: 10.1172/JCI85774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211(13):2549–66. doi: 10.1084/jem.20141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keles S, Charbonnier LM, Kabaleeswaran V, Reisli I, Genel F, Gulez N, et al. Dedicator of cytokinesis 8 regulates signal transducer and activator of transcription 3 activation and promotes TH17 cell differentiation. J Allergy Clin Immunol. 2016;138(5):1384–94. e2. doi: 10.1016/j.jaci.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tangye SG, Pillay B, Randall KL, Avery DT, Phan TG, Gray P, et al. Dedicator of cytokinesis 8-deficient CD4+ T cells are biased to a TH2 effector fate at the expense of TH1 and TH17 cells. J Allergy Clin Immunol. 2017;139(3):933–49. doi: 10.1016/j.jaci.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat Immunol. 2012;13(6):612–20. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124(6):1289–302. e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361(21):2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biggs CM, Keles S, Chatila TA. DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin Immunol. 2017;181:75–82. doi: 10.1016/j.clim.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford G, Enders A, Gileadi U, Stankovic S, Zhang Q, Lambe T, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122(12):2052–61. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208(11):2305–20. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Domaschenz H, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. 2009;10(12):1283–91. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. J Clin Immunol. 2015;35(2):189–98. doi: 10.1007/s10875-014-0126-0. [DOI] [PubMed] [Google Scholar]
- 18.Engelhardt KR, Gertz ME, Keles S, Schaffer AA, Sigmund EC, Glocker C, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2015;136(2):402–12. doi: 10.1016/j.jaci.2014.12.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alroqi FJ, Chatila TA. T Regulatory Cell Biology in Health and Disease. Curr Allergy Asthma Rep. 2016;16(4):27. doi: 10.1007/s11882-016-0606-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen E, Morbach H, Ullas S, Bannock JM, Massad C, Menard L, et al. Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells. J Allergy Clin Immunol. 2014;134(6):1365–74. doi: 10.1016/j.jaci.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verbsky JW, Chatila TA. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Current opinion in pediatrics. 2013;25(6):708–14. doi: 10.1097/MOP.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol. 2015;135(1):217–27. doi: 10.1016/j.jaci.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Herz W, Chu JI, van der Spek J, Raghupathy R, Massaad MJ, Keles S, et al. Hematopoietic stem cell transplantation outcomes for 11 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2016;138(3):852–9. e3. doi: 10.1016/j.jaci.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Frontiers in immunology. 2012;3:211. doi: 10.3389/fimmu.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheikine Y, Woda CB, Lee PY, Chatila TA, Keles S, Charbonnier LM, et al. Renal involvement in the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) disorder. Pediatric nephrology. 2015;30(7):1197–202. doi: 10.1007/s00467-015-3102-x. [DOI] [PubMed] [Google Scholar]
- 26.Baris S, Schulze I, Ozen A, Karakoc Aydiner E, Altuncu E, Karasu GT, et al. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. 2014;34(6):601–6. doi: 10.1007/s10875-014-0059-7. [DOI] [PubMed] [Google Scholar]
- 27.Duclaux-Loras R, Collardeau-Frachon S, Nancey S, Fabien N, Kaiserlian D, Lachaux A. Long-term disease course in a patient with severe neonatal IPEX syndrome. Clin Res Hepatol Gastroenterol. 2015;39(4):e43–7. doi: 10.1016/j.clinre.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Annals of the New York Academy of Sciences. 2016 doi: 10.1111/nyas.13011. [DOI] [PubMed] [Google Scholar]
- 29.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119(2):482–7. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–6. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–7. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–40. doi: 10.1126/science.aaa1663. [DOI] [PubMed] [Google Scholar]
- 33.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177(5):2770–4. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 34.Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. 2010;86(3):447–53. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. 2013;131(6):1611–23. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591–9. doi: 10.1182/blood-2014-09-602763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nature genetics. 2014;46(8):812–4. doi: 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med. 2007;204(2):381–91. doi: 10.1084/jem.20061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, et al. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med. 2007;204(2):369–80. doi: 10.1084/jem.20061334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivas MN, Koh YT, Chen A, Nguyen A, Lee YH, Lawson G, et al. MyD88 is critically involved in immune tolerance breakdown at environmental interfaces of Foxp3-deficient mice. J Clin Invest. 2012;122(5):1933–47. doi: 10.1172/JCI40591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106(12):R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torgerson TR, Linane A, Moes N, Anover S, Mateo V, Rieux-Laucat F, et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132(5):1705–17. doi: 10.1053/j.gastro.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 43.Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E, et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest. 2016;126(11):4219–36. doi: 10.1172/JCI85647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PBMC of patient P2 and those of a control subject were analyzed for CD3 and CD19 expression (A, B). CD3+ T cells were further analyzed for CD4 and CD8 expression (C, D). CD19+ B cells were further analyzed for IgD and CD27 expression (E, F).
Hematological and Immunological findings on DOCK8-deficient subjects. The quantitative determination of serum immunoglobulins is reported for the respective patients before starting IVIG therapy.
The 96 candidate variants were homozygous and nonsynonymous in the patient and absent from dbSNP. The gene symbol (DOCK8) for the candidate gene identified on chromosome 9 is in boldface.