

ABSTRACT
Providencia rettgeri is emerging as a new opportunistic pathogen with high antibiotic resistance. The need to find alternative methods to control antibiotic-resistant bacteria and the recent advances in phage therapy motivate the search for new phages able to infect Providencia spp. This study describes the isolation and characterization of an obligatory lytic phage, vB_PreS_PR1 (PR1), with therapeutic potential against drug-resistant P. rettgeri. PR1 is a siphovirus. Its virion DNA size (118,537 bp), transcriptional organization, terminal repeats (10,461 bp), and nicks in the 3′-to-5′ strand are similar to those of phage T5. However, sequence similarities of PR1 to phages of the T5virus genus at the DNA and protein levels are limited, suggesting that it belongs to a new species within the Siphoviridae family. PR1 exhibits the ability to kill P. rettgeri antibiotic-resistant strains, is highly specific to the species, and did not present known genomic markers indicating a temperate lifestyle. The lack of homologies between its proteins and proteins of the only other sequenced Providencia prophage, Redjac, suggests that these two phages evolved separately and may target different host proteins.
IMPORTANCE The alarming increase in the number of bacteria resistant to antibiotics has been observed worldwide. This is particularly true for Gram-negative bacteria. For certain of their strains, no effective antibiotics are available. Providencia sp. has been a neglected pathogen but is emerging as a multidrug-resistant bacterium. This has revived interest in bacteriophages as alternative therapeutic agents against this bacterium. We describe the morphological, physiological, and genomic characterization of a novel lytic virus, PR1, which is able to kill drug-resistant P. rettgeri clinical isolates. Genomic and phylogenetic analyses indicate that PR1 is a distant relative of T5virus genus representatives. The lack of known virulence- or temperate lifestyle-associated genes in the genome of PR1 makes this phage a potential candidate for therapeutic use. Analysis of its genome also improves our knowledge of the ecology and diversity of T5-like siphoviruses, providing a new link for evolutionary studies of this phage group.
KEYWORDS: Providencia rettgeri, bacteriophage, T5virus, comparative genomics, proteomic analysis
INTRODUCTION
Providencia spp. are members of the Enterobacteriaceae family. Providencia rettgeri is one of the most common species known to cause urinary tract infections in hospitalized patients or nursing care facilities (1) and is therefore considered a nosocomial pathogen. Nonetheless, Providencia infections have also been associated with gastroenteritis and bacteremia (2, 3) and are considered to be a major cause of travelers' diarrhea (3).
A clinical study conducted between May 2001 and April 2013 at a tertiary care hospital reported 14 cases of Providencia bacteremia that had evolved from urinary tract infections. The overall mortality rate was 29%. Studies have also demonstrated antibiotic resistance to cefepime, imipenem, and piperacillin-tazobactam in 100%, 86%, and 86% of the isolates, respectively (4). Most importantly, notwithstanding the fact that cases of extended-spectrum beta-lactamases (ESBLs) are rarely reported, there has been an increasing prevalence of ESBL phenotypes. Carbapenem-hydrolyzing enzymes, or carbapenemases (VIM-1, NDM-1, and OXA-48-type enzymes), have recently been found in Providencia stuartii and P. rettgeri isolates in Turkey (5), Greece (6), Mexico (7), Italy (8), and Brazil (9). The emergence of carbapenem-resistant Providencia strains is of particular concern given that they are also naturally resistant to polymyxins and tigecycline, making these strains a major threat to public health globally.
Bacteriophages (phages) have emerged as an alternative therapy to combat multidrug-resistant bacteria. Phages are the most abundant biological entities on the planet (10). They outnumber bacteria by an estimated 10-fold and have a population size estimated at 4.8 × 1031 phage particles (11). Over the last decades, more than 3,100 phages infecting different types of bacterial hosts have been isolated and sequenced, which has contributed to a better understanding of their ecology, genetic diversity, and evolutionary relationships (12).
Compared to the existing knowledge on phages infecting well-known species of the Enterobacteriaceae family, such as Escherichia coli and Salmonella enterica (13–16), little is known about phages that infect Providencia species, adding ecological relevance to their analysis. Our current knowledge of phages infecting Providencia dates back 5 decades, when Coetzee and Vieu (1963) studied the distribution of lysogeny among members of this genus (17, 18). Since then, sequencing of novel Providencia strains and use of specialized bioinformatics tools such as PHAST and PHASTER have been limited to the identification and analyses of additional prophages (19, 20). This is the case of the prophage Redjac that was recently found by applying pyrosequencing and whole-genome mapping techniques to a P. stuartii isolate. Redjac is the only completely sequenced Providencia phage deposited in the NCBI database (accession number JX296113) (20).
In this study, we have isolated and characterized a strictly lytic phage (PR1) able to infect and kill antibiotic-resistant P. rettgeri clinical isolates. Comparative analysis of the genome and virion proteome of PR1 revealed that the diversity of T5virus genus phages and their relatives as well as their ability to adapt to different hosts in evolution is wider than previously anticipated.
RESULTS AND DISCUSSION
Antibiotic susceptibility of Providencia strains.
Providencia sp. is a clinically significant, albeit understudied, pathogen. In this study, a total of 28 strains (12 P. rettgeri, 15 P. stuartii, and one Providencia alcalifaciens) were isolated mainly from urine (62.4%) but also from expectorations, bronchus aspirates, and cutaneous exudates of both male and female patients admitted to the Hospital of Braga (Portugal). The presence of the isolates reveals that although the urinary track is the most common site of Providencia infection, this bacterium has a multifaceted niche of infection. It has been reported that Providencia spp. associated with catheter-related urinary infections occur more frequently than E. coli and Proteus mirabilis (21). All tested strains of this study were resistant to ampicillin, amoxicillin-clavulanic acid, and cefuroxime. Cases of extended antibiotic resistance (89.2% to gentamicin, 89.2% to tobramycin, 42.8% to levofloxacin, 35.7% to ciprofloxacin, and 14.3% to trimethoprim-sulfamethoxazole [co-trimoxazole]) have also been observed, reflecting the alarming problem of drug resistance and the importance of Providencia sp. members as emerging pathogens (22).
Isolation of PR1, a P. rettgeri-infecting virus.
We have isolated and characterized a phage named PR1 (vB_PreS_PR1) from sewage enriched with Providencia strains from different human clinical specimens. PR1 produces very small plaques on a layer of all sensitive bacteria (0.1-mm diameter). Based on transmission electron microscopy (TEM) analysis, PR1 has a 75-nm head with an icosahedral symmetry and a long noncontractile tail (160 nm by 10 nm), indicating that it belongs to the family Siphoviridae (Fig. 1A). Its morphology closely resembles that of T5-like phages (e.g., T5, AKFV33, and SPC35) known to infect E. coli and Salmonella enterica hosts (23–25). The host range analysis indicates that PR1 successfully infects most P. rettgeri strains used in this study, with a high efficiency of plating (EOP), while not being able to lyse certain other closely related bacteria of the tribe Proteeae and the Enterobacteriaceae family, such as P. stuartii, P. alcalifaciens, Citrobacter freundii, Morganella morganii, P. mirabilis, or Proteus vulgaris (Table 1). Compared to other characterized phages of T5virus genus (T5 and AKFV33), PR1 presents a short latent period of 25 min and burst size of 32 (23, 26).
FIG 1.

Morphological and genomic analysis of PR1. (A) Transmission electron micrographs of Providencia rettgeri-infecting phage negatively stained with 2% uranyl acetate. (B) Genome map with predicted 157 ORFs numbered and colored (yellow, green, blue, and gray) according to their predicted functions. Long terminal repeats (LTRs) and major transcriptional units are indicated. The nucleotide positions are given (in kilobases). (C) The major morphological and genomic features have been summarized. Locations and nucleotide sequences of nick regions in PR1 DNA can be seen in detail in Table S4 in the supplemental material. CDS, coding sequences.
TABLE 1.
Lytic spectra and efficiency of plating of the Providencia rettgeri phage PR1
| Species | Strain | Sourcea | Patient gender | Antibiotic resistance profileb | PR1 infectivity | PR1 EOPc |
|---|---|---|---|---|---|---|
| P. rettgeri | PR 1 | Expectoration | Male | AM, AMC CXM, FM | + | High |
| PR 2 | Urine | Male | AM, AMC CXM, FM, GM, NN, SXT | − | ||
| PR 3 | Urine | Male | AM, AMC CXM, FM, GM, NN | + | High | |
| PR 4 | Urine | Male | AM, AMC CXM, FM, GM, NN | + | High | |
| PR 5 | Urine | Male | AM, AMC CXM, FM | + | LFW | |
| PR 6 | Urine | Female | AM, AMC CXM, FM, GM, NN | + | LFW | |
| PR 7 | Pus | Male | AM, AMC CXM, FM, GM, NN | − | ||
| PR 8 | Urine | Male | AM, AMC CXM, FM, GM, NN | − | ||
| PR 9 | Urine | Male | AM, AMC CXM, FM | + | High | |
| PR 10 | Expectoration | Male | AM, AMC CXM, FM, GM, NN, SXT | + | High | |
| PR 11 | Urine | Male | AM, AMC CXM, FM, GM, NN | + | High | |
| PR 12 | Urine | Male | AM, AMC CXM, FM, GM, NN | + | Low | |
| P. stuartii | PS 1 | Urine | Male | AM, AMC CXM, FM, GM, NN, LVX | − | |
| PS 2 | Bronchus aspirate | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP, SXT | − | ||
| PS 3 | Expectoration | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 4 | Cutaneous exudate | Female | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 5 | Urine | Male | AM, AMC CXM, FM, GM, NN, LVX, SXT | − | ||
| PS 6 | Urine | Unknown | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 7 | Urine | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 8 | Urine | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 9 | Unknown | Unknown | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 10 | Urine | Female | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 11 | Urine | Male | AM, AMC CXM, FM, GM, NN | − | ||
| PS 12 | Expectoration | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP, SXT | − | ||
| PS 13 | Cutaneous exudate | Male | AM, AMC CXM, FM, GM, NN, LVX, CIP | − | ||
| PS 14 | Urine | Male | AM, AMC CXM, FM, GM, NN | − | ||
| PS 15 | Bronchial aspirate | Male | AM, AMC CXM, FM, GM, NN | − | ||
| P. alcalifaciens | PA 1 | Urine | Male | AM, AMC CXM, FM, GM, NN | − | |
| C. freundii | CF 1 | Salmonella Genetic Stock Centre (EC592) | − | |||
| M. morganii | MM | Salmonella Genetic Stock Centre (CDC 4195-69) | − | |||
| P. mirabilis | PM | ATCC (CECT 4101) | − | |||
| P. vulgaris | PV | ATCC (CECT 174) | − | |||
| E. coli | EC | ATCC (CECT 432) | − |
ATCC, American Type Culture Collection.
AM, ampicillin; AMC, amoxicillin-clavulanic acid; CXM, cefuroxime; FM, framycetin; GM, gentamicin; NN, tobramycin; LVX, levofloxacin; CIP, ciprofloxacin; SXT, co-trimoxazole.
The EOP was recorded as high or low, representing EOPs higher or lower than 1%, respectively. LFW, lysis-from-without events.
PR1 genomic features.
The PR1 virion contains a linear 118,537-bp double-stranded DNA (dsDNA) molecule (Fig. 1B and C) with 10,461-bp long terminal repeats (LTRs) (see Fig. S1 in the supplemental material). The nonredundant part of the phage PR1 genome is organized into three major transcriptional units, while the LTR regions span two minor transcriptional units (Fig. 1B). This resembles the transcriptional organization of the preearly (i.e., left LTR), early (i.e., transcriptional units I and II), and late (i.e., transcriptional unit III) genes encoding the lytic functions, DNA replication and metabolism, and morphogenetic functions, respectively, which are found in phages of the T5virus genus (27). Of the 157 predicted PR1-encoded proteins, only 50 can be functionally assigned. Where sequence similarity exists, the level of identity is low (48% average amino acid identity), indicating the lack of a close relationship between PR1 and any phage currently available in the databases (Table S1). There are 65 proteins encoded by PR1 conserved among phages of the T5virus genus (Escherichia phages T5, DT57C, AKFV33, EPS7, slur09, and vB_EcoS_FFH1; Salmonella phages SPC35, Stitch, and Shivani; Yersinia phage ϕR201; Vibrio phage ϕ3; and Pectobacterium phage My1). Additionally, the genes encoding homologous proteins in PR1 and related phages of the T5virus genus are mostly colinear in the genomes. Taken together, these observations suggest a common ancestry of PR1 and T5virus genus phages (Table S1). None of the assigned genes code for virulence factors or features characteristic of temperate phages (e.g., an integrase gene and genes for the maintenance of lysogeny), suggesting an obligatory lytic lifestyle. Additionally, we did not find any homologies between PR1 proteins and temperate P. stuartii phage Redjac proteins. Conceivably, host proteins or protein regions that are targeted by these phages at infection and during phage development differ significantly.
Similar to T5, PR1 encodes 22 tRNAs, whose genes are located between ORF57 and ORF86 (Table S2). Overall, PR1 appears to encode tRNAs for highly used codons in its genome. These tRNAs often match to less frequent codons of the host. This would allow increased (or modulated) synthesis of phage proteins (28).
The regulation of PR1 gene transcription is complex, as indicated by the presence in the genome of at least 12 putative host-dependent promoters [identified by searching for TTGACA(N17)TATAAT, allowing for a 1-bp mismatch] and 25 rho-independent terminators. As in T5, the host-dependent promoters are in the regions of early as well as late genes (Fig. 1).
Identification and analysis of localized single-stranded DNA (ssDNA) nicks in the PR1 genome.
In addition to the ends of virion DNA, the assembly pattern of the PR1 library sequence reads revealed nine regions represented by several fragments of identical start sites and sequences indicative of single-stranded nicks in the 3′-to-5′ strand of virion DNA (Table S3). Their number compared to the overall coverage of the relevant regions indicates five minor (Fig. 1B, nicks 2, 3, 5, 7, and 8) and four major (nicks 1, 4, 6, and 9) nick sites, which start from the same sequence (5′-GCGC) in the 3′-to-5′ strand. Sanger sequencing of two exemplary nick regions in PR1 DNA confirmed their presence (Fig. 1; see also Fig. S2). The GCGC sequences in the nick regions are nested in 9-nucleotide (nt) sequences that differ slightly from each other but can be found only in the regions of nicks (Table S3). Single-stranded nicks of conserved 5′ end sequence in one DNA strand of virion DNA are also a characteristic feature of T5 phage (31, 32), its close relative BF23 (31), ϕKMV-like viruses (33), and Pseudomonas putida phage PpG1, an evolutionarily divergent member of the LUZ24-like phage group (34). The deduced consensus sequence of nick sites in PR1 DNA [5′-(R/G)CGCRNDR] resembles that of major nick sites in T5 DNA [5′-(R/G)CGCRGG]. As in T5, the PR1 nick sites are in both noncoding and coding regions, and one of the nicks is in the template strand of ORF120 (primase), indicating that the nicks do not interfere with either phage DNA replication or transcription. Conceivably, the nicks are repaired promptly after the DNA injection to the infected cell and restored in newly replicated phage DNA late in lytic development, e.g., during DNA packaging, as in the case of ϕkF77 (33). The physiological function of nick sites in unclear even though their presence in DNA of distantly related phages implies a critical role. One of the major PR1 nicks is in the large terminase subunit gene, which might suggest involvement in the initiation of DNA packaging. Alternatively, the 3′ ends of nicked strands may serve to initiate recombinational replication or repair. The lack of obvious difference between the development of T5 nick-free mutants and wild-type phages in a laboratory (24, 32) suggests that there may be an alternative pathway for the process in which the nicks are involved or that the nicks are important under conditions that have not yet been studied.
Comparative genome analysis.
Initial blastn analysis demonstrated that, strikingly, the PR1 genome shares less than 1% of overall nucleotide identity with other phages, including prophage Redjac, the only Providencia-infecting virus sequence available in the NCBI database. The PR1 core set of gene products was then compared with representatives of all Siphoviridae genera currently recognized by the International Committee on Taxonomy of Viruses (ICTV) using the CoreGenes tool. Notably, only a relatively significant identity (43.2%) to the T5 proteome was found. Making a further comparison of PR1 to all phages classified as T5virus, a similar value is observed against Escherichia phages AKFV33 (45.0%), DT57C (43.3%), EPS7 (42.0%), and FFH1 (42.7%) and Salmonella phages Shivani (42.7%) SPC35 (42.7%), and Stitch (43.3%). These CoreGenes values lie in close proximity to the 40% threshold that was previously used to taxonomically separate or group species (35). To further assess the phage relatedness, orthologous genes between PR1 and all T5virus members were analyzed through the OrthVenn server (36). The resulting Venn diagram with PR1 shows a total of 173 clusters, but only 60 are shared among all six species (Fig. S3). An important fact is also the identification in PR1 of 91 singletons, which means that more than half of the PR1 proteome (containing 157 open reading frames [ORFs]) is unrelated to T5virus.
Taking a closer look at the pairwise alignments of the closest PR1 relatives, weak homologies of PR1 to the Escherichia phage AKFV33 (phage sharing higher proteome homology) and the Escherichia phage T5 (representative phage genus) were observed (Fig. 2). Notably, the PR1 proteome is strikingly different in the LTR and transcriptional unit I and in several other smaller regions that encode proteins with little or no homology in transcriptional units II and III.
FIG 2.

Divergence of the core gene organization of PR1 from that of the closest phages. The PR1 genome was compared to that of Escherichia virus AKFV33 and Enterobacter virus T5 using tBLASTx within Easyfig. Arrows indicating ORFs are drawn to scale and colored in the reference genome according to their predicted functions. Gene similarity profiles between phages are indicated in grayscale (and percentages). Crisscrossed lines observed in the extremities are a result of the homology between the terminal repeats.
(i) LTR.
In AKFV33 and T5, the long terminal repeat (LTR) region is known to carry 16 preearly genes responsible for two-step DNA injection (23, 27). In T5, the first DNA injection step transfer starts with gp1 up to gp16, including the deoxyribonucleotide 5′ monophosphatase (dmp), A1, and A2 genes, which are separated by a large 1,778-bp noncoding region from the second DNA injection step transfer process attributed to gp17. This injection system seems to be conserved among all T5virus members but is different in PR1 (Table S4) (37). From the first 16 putative genes in PR1, only the A1-A2-dmp complex has homologies to other T5-like phage proteins. Mutants of T5 have shown that A2-A1-dpm are expressed first and that A2 and A1 are essential genes (23, 37). We hypothesize that PR1 DNA injection maintained this core set of genes but has evolved to a specific system as an adaptation to the Providencia host cell wall.
(ii) Transcriptional unit I.
The PR1 transcriptional unit I encodes tRNAs, proteins of unknown function, the lytic system, and some DNA metabolism-associated proteins and is organized in a fashion similar to that of the relevant regions of the AKFV33 and T5 genomes (27). Almost all proteins are unique to PR1. This is also the case for PR1 lysis genes that have either limited or undetectable homology. PR1 encodes a canonically ordered holin type T (gp46) and an endolysin (gp45) with d-alanyl–d-alanine carboxypeptidase (NCBI accession number PF02557; VanY) that has relatively low similarity (<50% amino acid identity) to proteins of non-T5virus members, namely, Klebsiella myovirus KP27 and KP15. Based on genome synteny with the T5 genome (gp40 and gp41), PR1 gp49 and gp50 appear to be the o- and i-spanins with features of amino acid sequence characteristic of prototypical i- and o-spanins of lambda phage (27, 38).
(iii) Transcriptional unit II.
Transcriptional unit II contains mostly the replication module genes located downstream from the predicted phoH gene and includes genes for two DNA ligases (gp116-gp117), two helicases (gp119 and gp125), one DNA primase (gp120), and two DNA polymerase exons (gp121 and gp123) with a conserved DNA_pol_A (PF00476) domain. This replication cluster seems to be highly conserved not only in AKFV33 and T5 but also in all other T5-like phages (>92% amino acid identity) (23). The homing endonucleases are exceptions. None of the PR1, AKFV33, and T5 endonucleases share significant identity. The case of the PR1 LAGLIDADG-type homing endonuclease, which has low identity to other proteins in Domibacillus and Desulfotomaculum strains (<35% amino acid identity), has also been reported in the genomes of two Staphylococcus phages (Romulus and Remus), where the homing endonuclease genes interrupt DNA repair protein genes (62). Another difference between PR1 and T5-like phages is the RNase H-type RNase (gp96), also part of the replisome. It shares only 44% amino acid identity with an RNase of a Cupriavidus bacterium and may be involved in the regulation of PR1 gene expression by processing certain transcripts.
(iv) Transcriptional unit III.
Transcriptional unit III encodes morphogenetic functions arranged together, similarly to AKFV33 and T5 and other siphoviruses (27, 38). Mass spectrometry enabled the identification of 26 proteins representing products of this module (with a sequence coverage ranging from 7.1% to 70.2%) (Fig. 3 and Table 2). Among these, the functions of 17 proteins could be predicted based on structure analysis and protein homology searches using HHpred. Structural functions of two unique identified proteins encoded by this module, gp149 and gp154, have also been predicted. PR1 ORF149 is located between the genes encoding homologs of the T5 prohead protease and portal protein and possibly encodes a new capsid decoration protein, based on genome synteny between PR1 and T5 in the relevant regions. Capsid decoration proteins are dispensable for phage morphogenesis and typically vary even between related phages. PR1 ORF154 is most likely a receptor binding protein (RBP) owing to its homology to known or putative RBPs of T5virus genus phages that infect enteric bacteria (after five PSI-BLAST iterations).
FIG 3.
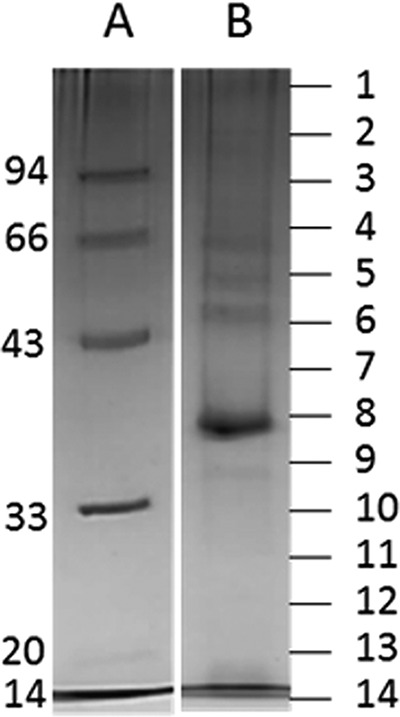
SDS-PAGE analysis of the purified structural phage proteins (lane B) on a 12% SDS-PAGE separation gel alongside a PageRuler prestained protein ladder (lane A). The entire lane was cut into 14 distinct slices corresponding to the molecular mass as indicated (kDa) to the left of the gels.
TABLE 2.
Phage PR1 structural proteins identified by nanoLC-ESI-MS/MSa
| Protein | Identified function | Band no. (most abundant)b | Protein mass (Da) | No. of unique peptidesc | Sequence coverage (%) | HHpred significant match (phage) | E-valued | Identity (%)e |
|---|---|---|---|---|---|---|---|---|
| gp4 | DNA transfer protein | 1–7 (4) | 65,166 | 12 | 23.7 | DNA transfer protein (enterobacterial phage DT57C) | 2e−164 | 47 |
| gp8 | Hypothetical protein | 1–11 (1) | 40,622 | 8 | 39.0 | Hypothetical protein My1_019 (Pectobacterium phage My1) | 7e−19 | 39 |
| gp18 | No hit | 2, 7 | 34,021 | 3 | 11.6 | |||
| gp46 | Holin | 11 | 25,959 | 4 | 22.6 | Putative holin (Pectobacterium phage My1) | 3e−69 | 48 |
| gp48 | Deoxynucleoside monophosphate kinase | 11 | 26,205 | 3 | 16.7 | Deoxynucleoside-5′-monophosphate kinase (Escherichia phage Akfv33) | 1e−24 | 36 |
| gp82 | Hypothetical protein | 14 | 22,008 | 5 | 20.7 | Hypothetical protein Ea357_046 (Erwinia phage Ea35-70) | 9e−63 | 55 |
| gp101 | Ribonucleoside-diphosphate reductase | 1 | 92,766 | 3 | 5.2 | Aerobic ribonucleoside diphosphate reductase, large subunit (Escherichia phage vb_ecos_FFH1) | 0.0 | 58 |
| gp102 | Starvation-inducible protein | 1 | 29,514 | 2 | 8.8 | Phosphate starvation-inducible protein PhoH, predicted ATPase (Yersinia phage ϕR201) | 2e−101 | 60 |
| gp121 | DNA polymerase | 1 | 69,415 | 2 | 4.1 | DNA polymerase (Escherichia phage vB_EcoS_FFH1) | 0.0 | 68 |
| gp128 | Hypothetical protein | 7 | 31,602 | 6 | 29.9 | D11 protein (enterobacterial phage DT57C) | 2e−39 | 36 |
| gp134 | Hypothetical protein | 1–10 (4) | 81,973 | 24 | 41.5 | Hypothetical protein OO7_12349 (Providencia sneebia DSM 19967) | 1e−71 | 47 |
| gp135 | Tail protein | 6 | 15,421 | 1 | 7.1 | Tail protein (Salmonella phage Stitch) | 2e−27 | 42 |
| gp136 | Tail protein | 1–9 (1, 2) | 110,749 | 27 | 38.6 | Tail protein Pb4 (Salmonella phage Spc35) | 1e−173 | 45 |
| gp137 | Baseplate hub protein | 1–9 (2) | 106,764 | 23 | 31.0 | Tail protein Pb3 (Escherichia phage Akfv33) | 0.0 | 57 |
| gp138 | Distal tail protein | 12, 14 | 22,870 | 5 | 40 | Hypothetical protein SPC35_0127 (Salmonella phage Spc35) | 1e−72 | 57 |
| gp139 | Tape measure protein | 1–7 (2) | 134,015 | 46 | 48.6 | Tape measure protein (Salmonella phage Stitch) | 0.0 | 36 |
| gp142 | Minor tail protein | 9–11 (10) | 33,805 | 7 | 34.0 | Minor tail protein (Escherichia phage Akfv33) | 6e−56 | 42 |
| gp143 | Major tail protein | 1–11 (6) | 41,715 | 10 | 41.2 | Major tail protein (Salmonella phage Stitch) | 9e−165 | 60 |
| gp144 | Tail tube terminator protein | 14 | 20,192 | 2 | 8.0 | Tail terminator protein (enterobacterial phage DT57C) | 2e−36 | 41 |
| gp145 | Hypothetical protein | 10 | 28,397 | 5 | 22.7 | Hypothetical protein AGC_0158 (Escherichia phage Eps7) | 3e−88 | 55 |
| gp146 | Hypothetical protein | 13–14 (14) | 19,794 | 7 | 53.5 | Hypothetical protein BN79_156 (Yersinia phage ϕR201) | 9e−17 | 30 |
| gp147 | Major capsid protein | 1–14 (9) | 52,374 | 21 | 52.6 | Phage capsid family protein (Escherichia phage slur09) | 8e−153 | 56 |
| gp148 | Prohead protease | 1–14 (13) | 22,091 | 6 | 40.6 | Putative prohead protease (Escherichia phage vb_ecos_FFH1) | 8e−74 | 62 |
| gp149 | No hit | 1–14 (5) | 44,254 | 26 | 70.2 | |||
| gp150 | Portal protein | 1–7 (6, 7) | 46,128 | 16 | 51.1 | Portal protein (Yersinia phage ϕR201) | 0.0 | 63 |
| gp154 | No hit | 1–5 (4) | 68,027 | 9 | 18.7 |
It should be noted that identifications with a low abundance of peptides (<4) could be the result of protein impurities remaining in the samples.
SDS-PAGE band.
Only phage proteins with a minimum number of two unique peptides were considered (except gp135 based on a reliable function).
HHpred E-value of the most significant matching protein model.
HHpred percent sequence identity for a pairwise amino acid alignment with the template protein.
Phylogenetic analysis.
Siphoviridae are by far the family of phages with the most undefined genera and subfamilies. Therefore, a recent reassessment was attempted to refine taxonomy by taking a holistic approach (39). Combining DNA and protein comparisons, taking their physiological and morphological traits into account, the authors attempted to define new genera or add new members to genera already ratified by the ICTV. While several genera have been proposed, no members have currently been included in the existing T5virus genus. Therefore, to validate the taxonomic positioning of PR1, we have compared it against all ICTV-ratified T5virus species and isolates using four distinct phylogenetic trees based on the amino acid sequences of the large terminase subunit, major capsid, or the tape measure of PR1 and all T5virus members, consistent with previous studies (Fig. S4) (40). From all resulting trees, it is clear that two distinct branches can be observed. One includes all members of the ICTV-recognized T5virus genus, and the other is a clearly separated branch containing PR1.
Conclusions and perspectives.
In conclusion, the PR1 bacteriophage that is described in this work appears to be a lytic phage with therapeutic potential to treat infections with antibiotic-resistant P. rettgeri strains. Based on the genome analysis results, we suggest that PR1 possibly shared a common ancestor with T5virus, retaining its genome size and number of ORFs and tRNAs, conserving genes encoding certain functions, and having nicks in one DNA strand, while exchanging during evolution genes involved in DNA injection, metabolism, and cell lysis systems and several others with unknown function. However, PR1 has a low overall genome and proteome relatedness and is phylogenetically distant from all T5virus representatives. Therefore, PR1 is eligible as a candidate for the creation of a new genus within the Siphoviridae family, named “PR1virus.” This also implies that a new subfamily should be created to group both PR1 and T5 genera.
MATERIALS AND METHODS
Bacterial strains, antibiotic resistance profiles, and growth media.
A total of 28 clinical Providencia isolates from different human clinical specimens were kindly provided by the Hospital of Braga (Table 1). The panel includes 12 isolates of P. rettgeri, 15 isolates of P. stuartii, and a single isolate of Providencia alcalifaciens. Strains were isolated using CLED (cysteine, lactose, and electrolyte-deficient) selective medium. Typing and antimicrobial susceptibility testing were performed using a Vitek2 (bioMérieux) or WalkAway (Beckman Coulter) instrument, according to CLSI guidelines (M100-S24) (41). For a broader comparison, additional strains of closely related species (Citrobacter freundii, Morganella morganii, Proteus mirabilis, Proteus vulgaris, and Escherichia coli) obtained from the Salmonella Genetic Stock Centre or the American Type Culture Collection were also used. All strains were grown at 37°C in Trypticase soy agar (TSA; Oxoid) or Trypticase soy broth (TSB; Oxoid) medium.
Phage isolation, amplification, and purification.
Providencia phages were isolated from raw sewage collected at a wastewater treatment plant (WWTP) (Frossos, Braga, Portugal) by enrichment (42). Briefly, the raw inlet sewage water was centrifuged (10 min at 10,000 × g) and filtered using a 0.22-μm-pore-size cellulose acetate membrane (GE Healthcare) to remove solid particles and bacterial debris. The resulting suspension was mixed with an equal volume of double-strength TSB, enriched with specific Providencia strains, and incubated overnight at 37°C and 120 rpm (Biosan ES-20/60 orbital shaker). The presence of phages was evaluated by spotting 10 μl of filtered supernatants onto Providencia lawns and by visualizing inhibition halos after an overnight incubation at 37°C.
A high-titer suspension of phages was produced through phage infection and multiplication using a fresh cell culture. After visible bacterial lysis, the solution was centrifuged (11,000 × g, 10 min, 4°C), incubated with 1 M NaCl at 37°C for 1 h, and later incubated with polyethylene glycol (PEG) 6000 (10%, wt/vol) overnight at 4 °C. Samples were centrifuged (11,000 × g, 10 min, 4°C), suspended in SM buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl, pH 7.5), and incubated with chloroform (1:4, vol/vol). The phage titer was assessed using a standard double-layer agar method. Finally, supernatants were stored at 4°C until needed.
Lytic spectrum.
The phage lytic spectrum was assessed using the spot-on-lawn method with phage titers of 108 PFU/ml. Briefly, bacterial lawns were made using the strains listed in Table 1 by spreading an inoculum of ≈108 CFU/ml in TSA plates using a soft-agar overlay. A 10-μl drop of phage stock was spotted and allowed to dry. Plates were incubated aerobically at 37°C for 16 h. Assays of the efficiency of plating (EOP) were performed to discriminate low- and high-infecting rates and lysis-from-without events, as described elsewhere (42).
One-step growth curve.
One-step growth curves were determined as previously described (42). A mid-exponential-phase culture (5 ml) was incubated with an equal volume of appropriately diluted phage suspension to a desired multiplicity of infection (MOI) of 0.001. After a short 5-min incubation, the mixture was centrifuged and suspended in 10 ml of fresh TSB medium, and samples were collected for PFU counts at 5, 10, 15, 20, 25, 30, 40, 50, and 60 min. Averages ± standard deviations for all experiments are given for three replicates.
TEM.
The morphology of phages was analyzed by uranyl acetate staining, as previously described (42). A centrifuged (1 h, 25,000 × g, 4°C) phage suspension was washed twice with tap water and deposited on copper grids with a carbon-coated Formvar carbon film on a 200-square-mesh nickel grid. Phages were stained with 2% uranyl acetate (pH 4.0) and visualized under a Jeol JEM 1400 transmission electron microscope (TEM).
DNA isolation and genome sequencing.
Phage genomic DNA was purified from high-titer lysates (≥1010 PFU/ml) using the phenol-chloroform-isoamyl alcohol extraction method (43) and sequenced using the next-generation sequencing (NGS) Illumina MiSeq platform (VIB Nucleomics Core, Belgium). Individual libraries were created by mixing equimolar proportions of two nonhomologous phages and processed with a custom NEBNext Ultra DNA kit to generate 500-bp fragments with individual barcodes. After a quality check by Agilent Bioanalyzer and Qubit measurements, the libraries were pooled and sequenced on the MiSeq System using 2- by 150-bp paired-end reads. Sequenced reads were demultiplexed, preprocessed to remove low-quality reads, and de novo assembled into one contig with average coverage of 574-fold using CLC Bio Genomics Workbench, version 7.0 (Aarhus, Denmark).
In silico genome analysis.
The Providencia phage genome was annotated using the Glimmer algorithm plug-in in Geneious software (44) and manually inspected for alternative start codons. For each coding DNA sequence (ORF), function and motifs were analyzed by Web-based search engines, namely, blastp, HHMER (45), RAST (46), and HHpred (47) (E value of ≤10−5). Additional tRNA-encoding genes were identified using tRNAscan-SE (48) and ARAGORN (49). Phobius (50), TMHMM (51), and HMMTOP (52) servers were used to predict transmembrane domains, and SignalP (53) was used to identify possible signal peptide cleavage sites. Lipo1.0 (29) was used to identify putative lipoproteins. Promoters (100 bp upstream of each ORF) were predicted with MEME (30), and terminators were predicted using ARNold (54) with Mfold (55).
For comparative analysis, whole-genome comparisons between phages were made at the nucleotide level with blastn and EMBOSS Stretcher (56) and at the proteomic level by CoreGenes (57) and OrthoVenn (36). Pairwise comparisons were also made using the tBLASTX plug-in in Easyfig (58) to reveal intercluster relationships.
Phylogeny analysis.
The phylogenetic trees of the terminase large subunit, major capsid, and tape measure proteins of phage PR1 and related phages were generated in one-click mode using Phylogeny.fr (http://www.phylogeny.fr/). Protein alignments were performed using MUSCLE, version 3.8.31, and refined by Gblocks, version 0.91b. Phylogeny was analyzed and drawn by PhyML and TreeDyn, respectively (59). The trees were exported in Newick format and visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Determination of the structure of PR1 virion DNA termini and internal nick sites.
PR1 virion DNA ends and internal nick sites in PR1 DNA were identified using the SeqMan program of the Lasergene, version 9.1, package (DNAStar, Madison, WI, USA), based on the analysis of the assembly patterns of subpopulations of random sequence reads representing the genome fragments of PR1 (100- to 200-fold coverage). The validation of the long terminal repeat (LTR) end prediction was performed by Sanger sequencing using PR1 virion DNA as a template and primers 5′-GGCTGACTTGTTTTACTGGAATGT and 5′-TAGCGGCTGATGCCATTACA. The validation of nick site predictions was verified for two selected sites based on Sanger sequencing results using PR1 DNA as a template and the following primers: 5′-TGCTGGTGGTTGGTTTTAT-3′ (OMLO812), 5′-AACCGCATATTGTTCTAAGTCTA-3′ (OMLO813), 5′-CTAGTTGCGTCGGGAGTAT-3′ (OMLO814), and 5′-CTTGGCAAGAATAGTAATAGACTTG-3′ (OMLO815). The PR1 virion DNA for restriction digestions and Sanger sequencing was isolated with a Qiagen Lambda Midi kit according to the manufacturer's instructions, except that the PEG and NaCl precipitation was performed overnight instead of in 1 h. Sanger sequencing was done using a BigDye Terminator, version 3.1, chemistry kit and ABI3730xl Genetic Analyser (Life Technologies/Thermo Fisher Scientific).
Structural proteome analysis by LC-ESI-MS/MS.
Phage proteins were precipitated by the addition of four volumes of ice-cold acetone and centrifuged (20 min, 1,600 × g, 4 °C). The pellet was air dried and suspended in loading buffer (1% [wt/vol] sodium dodecyl sulfate [SDS], 6% [wt/vol] sucrose, 100 mM 1,4-dithiothreitol, 10 mM Tris-HCl [pH 6.8], 0.0625% [wt/vol] bromophenol blue) before being loaded on a 12% SDS-PAGE gel. Protein bands were separated for 2 h at 200 V, stained with Simply Blue SafeStain (Invitrogen), and trypsinized according to the protocol of Shevchenko et al. (60). Resulting peptides were analyzed using nano-liquid chromatography-electrospray ionization tandem mass spectrometry (nanoLC-ESI MS/MS) and identified using the search engines SEQUEST, version 1.4 (ThermoFinnigan, San Jose, CA) and Mascot, version 2.2 (Matrix Sciences), in combination with a database of all possible phage ORFs, as described previously (61).
Accession number(s).
The PR1 genome has been deposited in the NCBI GenBank database under accession number KY363465.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UID/BIO/04469/2013 unit, COMPETE 2020 (POCI-01-0145-FEDER-006684), and the Project PTDC/BBB-BSS/6471/2014 (POCI-01-0145-FEDER-016678). The Azeredo and Lavigne labs are members of the Phagebiotics research community, supported by the FWO Vlaanderen. H.O. acknowledges the FCT grant SFRH/BPD/111653/2015.
H.O. isolated, sequenced, annotated, and analyzed the two genomes and drafted the manuscript. G.P. helped with the isolation and revised the manuscript. M.Ł. and J.G. helped with the phage annotation, analysis of the genome ends and nick sites, and revised the manuscript. A.M.K. helped with the phage taxonomy analysis. J.-P.N., H.H., and R.L. performed, analyzed, and wrote the proteomics-related work. J.A. devised the study, assisted with experimental design. and edited the manuscript.
We declare that we have no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01567-17.
REFERENCES
- 1.Wie SH. 2015. Clinical significance of Providencia bacteremia or bacteriuria. Korean J Intern Med 30:167–169. doi: 10.3904/kjim.2015.30.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armbruster CE, Smith SN, Yep A, Mobley HL. 2014. Increased incidence of urolithiasis and bacteremia during Proteus mirabilis and Providencia stuartii coinfection due to synergistic induction of urease activity. J Infect Dis 209:1524–1532. doi: 10.1093/infdis/jit663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoh M, Matsuyama J, Ohnishi M, Takagi K, Miyagi H, Mori K, Park KS, Ono T, Honda T. 2005. Importance of Providencia species as a major cause of travellers' diarrhoea. J Med Microbiol 54:1077–1082. doi: 10.1099/jmm.0.45846-0. [DOI] [PubMed] [Google Scholar]
- 4.Choi HK, Kim YK, Kim HY, Park JE, Uh Y. 2015. Clinical and microbiological features of Providencia bacteremia: experience at a tertiary care hospital. Korean J Intern Med 30:219–225. doi: 10.3904/kjim.2015.30.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrer A, Poirel L, Yilmaz M, Akan OA, Feriha C, Cuzon G, Matar G, Honderlick P, Nordmann P. 2010. Spread of OXA-48-encoding plasmid in Turkey and beyond. Antimicrob Agents Chemother 54:1369–1373. doi: 10.1128/AAC.01312-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douka E, Perivolioti E, Kraniotaki E, Fountoulis K, Economidou F, Tsakris A, Skoutelis A, Routsi C. 2015. Emergence of a pandrug-resistant VIM-1-producing Providencia stuartii clonal strain causing an outbreak in a Greek intensive care unit. Int J Antimicrob Agents 45:533–536. doi: 10.1016/j.ijantimicag.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 7.Barrios H, Garza-Ramos U, Reyna-Flores F, Sanchez-Perez A, Rojas-Moreno T, Garza-Gonzalez E, Llaca-Diaz JM, Camacho-Ortiz A, Guzman-Lopez S, Silva-Sanchez J. 2013. Isolation of carbapenem-resistant NDM-1-positive Providencia rettgeri in Mexico. J Antimicrob Chemother 68:1934–1936. doi: 10.1093/jac/dkt124. [DOI] [PubMed] [Google Scholar]
- 8.Barbarini D, Russello G, Brovarone F, Capatti C, Colla R, Perilli M, Moro ML, Carretto E. 2015. Evaluation of carbapenem-resistant Enterobacteriaceae in an Italian setting: report from the trench. Infect Genet Evol 30:8–14. doi: 10.1016/j.meegid.2014.11.025. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho-Assef AP, Pereira PS, Albano RM, Beriao GC, Chagas TP, Timm LN, Da Silva RC, Falci DR, Asensi MD. 2013. Isolation of NDM-producing Providencia rettgeri in Brazil. J Antimicrob Chemother 68:2956–2957. doi: 10.1093/jac/dkt298. [DOI] [PubMed] [Google Scholar]
- 10.Rohwer F. 2003. Global phage diversity. Cell 113:141. doi: 10.1016/S0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 11.Guemes AGC, Youle M, Cantu VA, Felts B, Nulton J, Rohwer F. 2016. Viruses as winners in the game of life. Annu Rev Virol 3:197–214. doi: 10.1146/annurev-virology-100114-054952. [DOI] [PubMed] [Google Scholar]
- 12.Ackermann HW. 2012. Bacteriophage electron microscopy. Adv Virus Res 82:1–32. doi: 10.1016/B978-0-12-394621-8.00017-0. [DOI] [PubMed] [Google Scholar]
- 13.Chibani-Chennoufi S, Canchaya C, Bruttin A, Brussow H. 2004. Comparative genomics of the T4-Like Escherichia coli phage JS98: implications for the evolution of T4 phages. J Bacteriol 186:8276–8286. doi: 10.1128/JB.186.24.8276-8286.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. 2003. Bacteriophage T4 genome. Microbiol Mol Biol Rev 67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn JJ, Studier FW. 1983. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol 166:477–535. doi: 10.1016/S0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 16.De Lappe N, Doran G, O'Connor J, O'Hare C, Cormican M. 2009. Characterization of bacteriophages used in the Salmonella enterica serovar Enteritidis phage-typing scheme. J Med Microbiol 58:86–93. doi: 10.1099/jmm.0.000034-0. [DOI] [PubMed] [Google Scholar]
- 17.Coetzee JN. 1963. Lysogeny in Proteus rettgeri and the hostrange of P. rettgeri and P. hauseri bacteriophages. J Gen Microbiol 31:219–229. doi: 10.1099/00221287-31-2-219. [DOI] [PubMed] [Google Scholar]
- 18.Vieu JF. 1963. Distribution of lysogeny among Proteus and Providencia. C R Hebd Seances Acad Sci 256:4317–4319. (In French.) [PubMed] [Google Scholar]
- 19.Krizsanovich K, De Klerk HC, Smit JA. 1969. A transducing bacteriophage for Proteus rettgeri. J Gen Virol 4:437–439. doi: 10.1099/0022-1317-4-3-437. [DOI] [PubMed] [Google Scholar]
- 20.Onmus-Leone F, Hang J, Clifford RJ, Yang Y, Riley MC, Kuschner RA, Waterman PE, Lesho EP. 2013. Enhanced de novo assembly of high throughput pyrosequencing data using whole genome mapping. PLoS One 8:e61762. doi: 10.1371/journal.pone.0061762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dedeic-Ljubovic A, Hukic M. 2009. Catheter-related urinary tract infection in patients suffering from spinal cord injuries. Bosn J Basic Med Sci 9:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu ZQ, Flavin MT, Flavin J. 2014. Combating multidrug-resistant Gram-negative bacterial infections. Expert Opin Investig Drugs 23:163–182. doi: 10.1517/13543784.2014.848853. [DOI] [PubMed] [Google Scholar]
- 23.Niu YD, Stanford K, Kropinski AM, Ackermann HW, Johnson RP, She YM, Ahmed R, Villegas A, McAllister TA. 2012. Genomic, proteomic and physiological characterization of a T5-like bacteriophage for control of Shiga toxin-producing Escherichia coli O157:H7. PLoS One 7:e34585. doi: 10.1371/journal.pone.0034585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zivanovic Y, Confalonieri F, Ponchon L, Lurz R, Chami M, Flayhan A, Renouard M, Huet A, Decottignies P, Davidson AR, Breyton C, Boulanger P. 2014. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J Virol 88:1162–1174. doi: 10.1128/JVI.02262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim M, Ryu S. 2011. Characterization of a T5-like coliphage, SPC35, and differential development of resistance to SPC35 in Salmonella enterica serovar typhimurium and Escherichia coli. Appl Environ Microbiol 77:2042–2050. doi: 10.1128/AEM.02504-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanni YT. 1954. Infection by bacteriophage T5 and its intracellular growth; a study by complement fixation. J Bacteriol 67:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Jiang Y, Vincent M, Sun Y, Yu H, Bao Q, Kong H, Hu S. 2005. Complete genome sequence of bacteriophage T5. Virology 332:45–65. doi: 10.1016/j.virol.2004.10.049. [DOI] [PubMed] [Google Scholar]
- 28.Bailly-Bechet M, Vergassola M, Rocha E. 2007. Causes for the intriguing presence of tRNAs in phages. Genome Res 17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCorquodale DJ, Shaw AR, Shaw PK, Chinnadurai G. 1977. Pre-early polypeptides of bacteriophages T5 and BF23. J Virol 22:480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers SG, Godwin EA, Shinosky ES, Rhoades M. 1979. Interruption-deficient mutants of bacteriophage T5. I. Isolation and general properties. J Virol 29:716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kulakov LA, Ksenzenko VN, Shlyapnikov MG, Kochetkov VV, Del Casale A, Allen CC, Larkin MJ, Ceyssens PJ, Lavigne R. 2009. Genomes of “phiKMV-like viruses” of Pseudomonas aeruginosa contain localized single-strand interruptions. Virology 391:1–4. doi: 10.1016/j.virol.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 34.Glukhov AS, Krutilina AI, Shlyapnikov MG, Severinov K, Lavysh D, Kochetkov VV, McGrath JW, de Leeuwe C, Shaburova OV, Krylov VN, Akulenko NV, Kulakov LA. 2012. Genomic analysis of Pseudomonas putida phage tf with localized single-strand DNA interruptions. PLoS One 7:e51163. doi: 10.1371/journal.pone.0051163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavigne R, Seto D, Mahadevan P, Ackermann HW, Kropinski AM. 2008. Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol 159:406–414. doi: 10.1016/j.resmic.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Coleman-Derr D, Chen G, Gu YQ. 2015. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 43:W78–W84. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davison J. 2015. Pre-early functions of bacteriophage T5 and its relatives. Bacteriophage 5:e1086500. doi: 10.1080/21597081.2015.1086500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grover JM, Luna AJ, Wood TL, Chamakura KR, Kuty Everett GF. 2015. Complete genome of Salmonella enterica serovar Typhimurium T5-like siphophage stitch. Genome Announc 3:e01435-14. doi: 10.1128/genomeA.01435-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adriaenssens EM, Edwards R, Nash JH, Mahadevan P, Seto D, Ackermann HW, Lavigne R, Kropinski AM. 2015. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 477:144–154. doi: 10.1016/j.virol.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 40.Smith KC, Castro-Nallar E, Fisher JN, Breakwell DP, Grose JH, Burnett SH. 2013. Phage cluster relationships identified through single gene analysis. BMC Genomics 14:410. doi: 10.1186/1471-2164-14-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; 24th informational supplement. CLSI document M100-S24. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 42.Oliveira H, Pinto G, Oliveira A, Oliveira C, Faustino MA, Briers Y, Domingues L, Azeredo J. 2016. Characterization and genome sequencing of a Citrobacter freundii phage CfP1 harboring a lysin active against multidrug-resistant isolates. Appl Microbiol Biotechnol 100:10543–10553. doi: 10.1007/s00253-016-7858-0. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 44.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kall L, Krogh A, Sonnhammer EL. 2004. A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 51.Kall L, Sonnhammer EL. 2002. Reliability of transmembrane predictions in whole-genome data. FEBS Lett 532:415–418. doi: 10.1016/S0014-5793(02)03730-4. [DOI] [PubMed] [Google Scholar]
- 52.Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 53.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 54.Naville M, Ghuillot-Gaudeffroy A, Marchais A, Gautheret D. 2011. ARNold: a web tool for the prediction of Rho-independent transcription terminators. RNA Biol 8:11–13. doi: 10.4161/rna.8.1.13346. [DOI] [PubMed] [Google Scholar]
- 55.Zuker M. 2003. Mfold web server for Nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 57.Zafar N, Mazumder R, Seto D. 2002. CoreGenes: a computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinformatics 3:12. doi: 10.1186/1471-2105-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 61.Lavigne R, Noben JP, Hertveldt K, Ceyssens PJ, Briers Y, Dumont D, Roucourt B, Krylov VN, Mesyanzhinov VV, Robben J, Volckaert G. 2006. The structural proteome of Pseudomonas aeruginosa bacteriophage phiKMV. Microbiology 152:529–534. doi: 10.1099/mic.0.28431-0. [DOI] [PubMed] [Google Scholar]
- 62.Vandersteegen K, Kropinski AM, Nash JHE, Noben J-P, Hermans K, Lavigne R. 2013. Romulus and Remus, two phage isolates representing a distinct clade within the Twortlikevirus genus, display suitable properties for phage therapy applications. J Virol 87:3237–3247. doi: 10.1128/JVI.02763-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.