

ABSTRACT
Recombination is a feature of many alphaherpesviruses that infect people and animals. Infectious laryngotracheitis virus (ILTV; Gallid alphaherpesvirus 1) causes respiratory disease in chickens, resulting in significant production losses in poultry industries worldwide. Natural (field) ILTV recombination is widespread, particularly recombination between attenuated ILTV vaccine strains to create virulent viruses. These virulent recombinants have had a major impact on animal health. Recently, the development of a single nucleotide polymorphism (SNP) genotyping assay for ILTV has helped to understand ILTV recombination in laboratory settings. In this study, we applied this SNP genotyping assay to further examine ILTV recombination in the natural host. Following coinoculation of specific-pathogen-free chickens, we examined the resultant progeny for evidence of viral recombination and characterized the diversity of the recombinants over time. The results showed that ILTV replication and recombination are closely related and that the recombinant viral progeny are most diverse 4 days after coinoculation, which is the peak of viral replication. Further, the locations of recombination breakpoints in a selection of the recombinant progeny, and in field isolates of ILTV from different geographical regions, were examined following full-genome sequencing and used to identify recombination hot spots in the ILTV genome.
IMPORTANCE Alphaherpesviruses are common causes of disease in people and animals. Recombination enables genome diversification in many different species of alphaherpesviruses, which can lead to the evolution of higher levels of viral virulence. Using the alphaherpesvirus infectious laryngotracheitis virus (ILTV), we performed coinfections in the natural host (chickens) to demonstrate high levels of virus recombination. Higher levels of diversity in the recombinant progeny coincided with the highest levels of virus replication. In the recombinant progeny, and in field isolates, recombination occurred at greater frequency in recombination hot spot regions of the virus genome. Our results suggest that control measures that aim to limit viral replication could offer the potential to limit virus recombination and thus the evolution of virulence. The development and use of vaccines that are focused on limiting virus replication, rather than vaccines that are focused more on limiting clinical disease, may be indicated in order to better control disease.
KEYWORDS: herpesvirus, recombination, replication, diversity, SNP genotyping assay, vaccine, infectious laryngotracheitis virus (ILTV), recombination hot spot, gallid herpesvirus 1, ILTV
INTRODUCTION
Infectious laryngotracheitis virus (ILTV; Gallid alphaherpesvirus 1) is an alphaherpesvirus that causes mild to severe respiratory disease in chickens. The virus causes major economic losses due to mortality and decreases in weight gain and egg production in poultry industries worldwide (1). Recombination between different strains of ILTV has been recognized as a problem for the poultry industry, particularly because natural recombination between attenuated vaccine strains of ILTV has been shown to generate virulent viruses (2). The importance of recombination as a mechanism of viral genome diversification and evolution in ILTV and other alphaherpesviruses is increasingly being recognized (3). Viruses can acquire genetic changes through several mechanisms, including point mutation and recombination, with the latter particularly important in many alphaherpesviruses (4–6). Viruses that belong to the order Herpesvirales have double-stranded linear DNA genomes and have complex viral DNA replication machinery, comprising a DNA polymerase with a highly efficient proofreading capacity, resulting in very low rates of spontaneous mutation (7–9). Therefore, rather than random changes or point mutations throughout the genome, recombination is considered to be a major evolutionary driving force enabling many alphaherpesviruses to persist, evolve and eventually become more virulent over time (2, 10, 11).
Four types of recombination have been described, based on the structure of the crossover site: site-specific recombination, transposition recombination, nonhomologous or illegitimate recombination, and homologous recombination (12). Homologous and illegitimate recombination, specifically intraspecific recombination, are the most commonly described recombination mechanisms in alphaherpesviruses (12). Intraspecific recombination under in vitro conditions has been extensively described for several alphaherpesvirus species, including Human alphaherpesvirus 1, also known as herpes simplex virus 1 (HSV-1) (13), Bovine alphaherpesvirus 1, also known as bovine herpesvirus 1 (BoHV-1) (14), Human alphaherpesvirus 3, also known as varicella-zoster virus (VZV) (15), Felid alphaherpesvirus 1, also known as felid herpesvirus 1 (FeHV-1) (16) and Suid alphaherpesvirus 1, also known as pseudorabies virus (PRV) (17). Full-genome sequencing and sequence analysis are the most suitable methods for detecting and describing alphaherpesvirus recombination events that occur under natural (field) conditions, but even nowadays these techniques have their limitations, including intensive labor and costs. In contrast, under experimental conditions, other techniques to detect recombination are still more efficient and cost-effective, as well as more suitable for testing large numbers of viruses (14, 18).
We have recently described the development and validation of a TaqMan single nucleotide polymorphism (SNP) genotyping assay to study recombination under experimental conditions in specific-pathogen-free (SPF) chickens (18), the natural host of ILTV. Use of the natural host to study recombination may reveal aspects of herpesvirus biology that may not be apparent in studies that utilize laboratory animal models of infection (19). The aim of this study was to apply this ILTV SNP genotyping assay to examine in vivo recombination between two ILTV strains over time. This was performed in order to describe recombination patterns and to examine genetic diversity among the viral progeny during the course of an ILTV infection. Additionally, we aimed to identify any recombination hot spots in the ILTV genome by performing full-genome sequencing of selected recombinants and analyzing them along with other ILTV genomes that were already available from different geographical regions, including Asia, Australia, Europe, and the United States.
RESULTS
Bird survival rates and virus genome quantification.
Groups of chickens were coinoculated with the V1-99 and CSW-1 strains of ILTV at two different doses or were inoculated with only either V1-99 or CSW-1. The survival rates in groups of birds that were inoculated only with low or high doses of V1-99 ILTV were around 70% and 45% throughout the experiment, respectively (Fig. 1). Groups that received only CSW-1 had 100% survival rates throughout the experiment (Fig. 1). In the coinoculated groups, the survival rates were 40% and 0% at 4 days after coinoculation with low and high doses of virus, respectively (Fig. 1).
FIG 1.
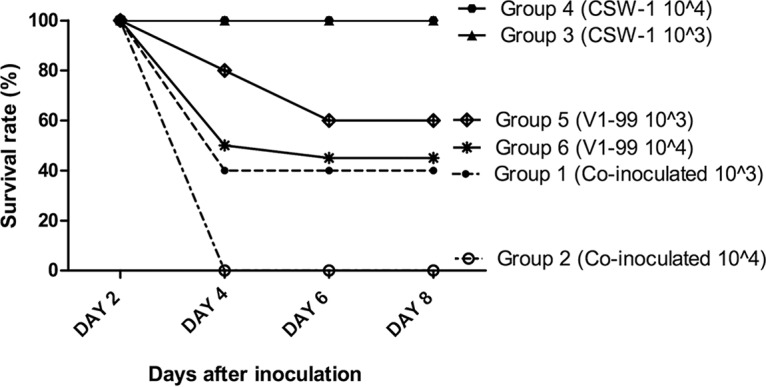
Survival rates within six groups of SPF chickens following intratracheal inoculation with either low (103 PFU) or high (104 PFU) doses of CSW-1 or V1-99 ILTV or coinoculation with the two viruses at the same doses. All birds inoculated only with CSW-1 survived. The group inoculated with a low dose of only V1-99 (group 5) had an 80% survival rate at day 4 and then a survival rate of 60% at days 6 and 8. Meanwhile, in the group inoculated with a high dose of V1-99 (group 6), a survival rate of 45% was seen at day 4, and the survival rate was 40% at days 6 and 8. Group 1 (birds coinoculated with a low dose of both viruses) had a survival rate similar to that of the group that received the single high dose of V1-99. None of the birds in group 2 (coinoculated with a high dose of both viruses) survived beyond day 4.
In the high-dose-coinoculated group, virus could be isolated from only 5 of the 10 birds at both 2 and 4 days after coinoculation (birds A, B, C, D, and E), so tracheal samples from these birds were used to compare virus replication and diversity across days 2 and 4. Significantly higher log10 viral genome copy numbers (means ± standard deviations) were detected at day 4 than at day 2 (4.94 ± 0.648 versus 3.02 ± 1.11, respectively; P = 0.0008, Mann-Whitney test) (Fig. 2E and F). The samples collected from the five birds in this group from which virus could be isolated at only one time point (4 days after infection) were used to verify the performance of SNP genotyping assay, and these results have been previously reported (18). None of the birds survived in the high-dose-coinoculated group after day 4 (Fig. 1), so no further sampling was possible in this group.
FIG 2.

Replication of ILTV in SPF chickens based on genome copy numbers in tracheal swabs measured using qPCR. Medians are indicated by lines for each group. Birds were inoculated with either CSW-1 or V1-99 or coinoculated (co-inoc) with 103 PFU (low dose) (A to D) or with 104 PFU (high dose) (E to H) of the V1-99 and CSW-1 strains of ILTV. (A and B) Low-dose-coinoculated birds F, G, H, I, and J (in the circles) had higher numbers of viral genome copies at day 4 than at day 2 (P < 0.05, Mann-Whitney test). These birds did not survive to day 6. (C and D) Low-dose-coinoculated birds K, L, and M (in the rectangles) did not have significantly different numbers of viral genome copies between day 6 and day 8 (P > 0.05, Mann-Whitney test). (E and F) High-dose-coinoculated birds A, B, C, D, and E (in the triangles) had higher numbers of viral genome copies at day 4 than at day 2 (P < 0.05, Mann-Whitney test). (G and H) High-dose-coinoculated birds did not survive to days 6 and 8.
In the low-dose-coinoculated group, those animals with high viral genome copy numbers at days 2 and 4 (birds F, G, H, I, and J) (Fig. 2A and B) did not survive, so samples could not be obtained at days 6 and 8. Therefore, we compared virus replication and diversity at days 2 and 4 in birds F, G, H, I, and J and then compared virus replication and diversity at days 6 and 8 in the remaining birds (K, L, and M) (Fig. 2C and D). In this group (low-dose-coinoculated group), significantly higher log10 genome copy numbers were detected 4 days after coinoculation than 2 days after coinoculation (3.77 ± 1.91 versus 2.22 ± 1.22, respectively; P = 0.045, Mann-Whitney test). Log10 genome copy numbers did not differ significantly at 6 days and 8 days after coinoculation (3.375 ± 1.097 versus 3.825 ± 1.338, respectively; P = 0.343, Mann-Whitney test).
Characterization of progeny viruses.
Up to 20 progeny viruses were isolated from each coinoculated chicken, and each was plaque purified before viral DNA extraction. Six SNPs across the length of each of the genomes of the progeny viruses were identified as being of either CSW-1 or V1-99 origin using the SNP genotyping assay interface within the Stratagene Mx3000P quantitative PCR (qPCR) software system, as previously described (18). The genotyping assay detected different patterns of recombination at 2 and 4 days after coinoculation with the high dose of viruses (Fig. 3) and also at 2, 4, 6, and 8 days after coinoculation with the low dose of viruses (Fig. 4). In total, 43 (67%) of the 64 possible genotype patterns were detected among the viral progeny of the two coinoculated groups. In the group of birds that were coinoculated with the high dose of viruses, 19 and 24 genotype patterns were detected at 2 and 4 days postinoculation, respectively (Table 1). In the group of birds that were coinoculated with the low dose of viruses, 14, 26, 15, and 9 genotype patterns were detected at 2, 4, 6, and 8 days postinoculation, respectively (Tables 2 and 3). Some of these genotype patterns were detected consistently over time in both low- and high-dose-inoculated groups. In the singly inoculated groups, no recombination was detected within the 10 viruses that were isolated and purified from each infected chicken at day 4 from the groups inoculated with the higher viral dose of 1 × 104 PFU/ml (see Fig. S1 in the supplemental material).
FIG 3.

Recombination patterns, as determined using SNP genotyping, in progeny viruses isolated 2 and 4 days after coinoculation of SPF chickens with 104 PFU of each of CSW-1 and V1-99 ILTV. Each row corresponds to a virus isolate, with the CSW-1 SNPs indicated by gray boxes and the V1-99 SNPs by black boxes. Each distinct recombination pattern was given a unique genotype code (final column).
FIG 4.

Recombination patterns, as determined using SNP genotyping, in progeny viruses isolated 2, 4, 6, and 8 days after coinoculation of SPF chickens with 103 PFU of each of CSW-1 and V1-99. Each row corresponds to a virus isolate, with the CSW-1 SNPs indicated by gray boxes and the V1-99 SNPs by black boxes. Each distinct recombination pattern was given a unique genotype code (final column).
TABLE 1.
Genotypes of viruses detected in high-dose-coinoculated (104 PFU) chickens at 2 and 4 days after coinoculationa
| Day 2 |
Day 4 |
||||
|---|---|---|---|---|---|
| Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates | Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates |
| 4 | A, B, C, D, E | 12 (13.2) | 4 | A, B, D | 3 (3.9) |
| 6 | A, B, C, D, E | 12 (13.2) | 6 | A, B | 6 (7.8) |
| 9 | A, D | 3 (3.3) | 9 | A, D, E | 4 (5.2) |
| 15 | A, B | 4 (4.4) | 15 | E | 2 (2.6) |
| 1 | A, C | 2 (2.2) | 1 | B | 2 (2.6) |
| 20 | A | 1 (1.1) | 20 | A | 2 (2.6) |
| 8 | A | 1 (1.1) | 8 | C | 1 (1.3) |
| 11 | B | 1 (1.1) | 11 | D | 1 (1.3) |
| 2 | D | 1 (1.1) | 2 | A | 2 (2.6) |
| 16 | E | 1 (1.1) | 16 | B, C, E | 4 (5.2) |
| 19 | E | 1 (1.1) | 32 | A | 2 (2.6) |
| 34 | E | 1 (1.1) | 28 | A | 1 (1.3) |
| 37 | E | 1 (1.1) | 38 | A | 1 (1.3) |
| 14 | D | 1 (1.1) | 42 | D | 3 (3.9) |
| 33 | C | 1 (1.1) | 39 | B, E | 5 (6.5) |
| 36 | C | 1 (1.1) | 40 | B, C | 2 (2.6) |
| 35 | C | 1 (1.1) | 43 | B | 1 (1.3) |
| 30 | C | 1 (1.1) | 3 | C | 1 (1.3) |
| 5 | A, C | 3 (3.3) | 27 | C | 1 (1.3) |
| CSW-1 | A, B, C, D, E | 40 (44) | 17 | C, D | 4 (5.2) |
| V1-99 | A, D | 2 (2.2) | 7 | C | 4 (5.2) |
| 41 | C, D | 4 (5.2) | |||
| 4 | A, B, D | 1 (1.3) | |||
| 10 | E | 4 (5.2) | |||
| CSW-1 | A, B, D, E | 14 (18.2) | |||
| V1-99 | D, E | 2 (2.6) | |||
For day 2, there were a total of 42 (46.2%) parental viruses and 49 (53.8%) recombinants, for a total of 91 (100%) viruses; for day 4, there were a total of 16 (20.8%) parental viruses and 61 (79.2%) recombinants, for a total of 77 (100%) viruses.
TABLE 2.
Genotypes of viruses detected in low-dose-coinoculated (103 PFU) chickens at 2 and 4 days after coinoculationa
| Day 2 |
Day 4 |
||||
|---|---|---|---|---|---|
| Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates | Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates |
| 1 | F, G, H, J | 5 (7) | 1 | H | 1 (1.2) |
| 2 | F | 1 (1.4) | 2 | I | 2 (2.4) |
| 4 | G, H, J | 11 (15.5) | 4 | G, H | 3 (3.7) |
| 5 | G, J | 2 (2.8) | 5 | F, J | 3 (3.7) |
| 6 | G, H | 2 (2.8) | 6 | J | 2 (2.4) |
| 7 | G | 1 (1.4) | 7 | F, H, I | 9 (10.9) |
| 9 | H | 2 (2.8) | 9 | G, I | 5 (6.1) |
| 10 | H, J | 2 (2.8) | 10 | F, H | 3 (3.7) |
| 8 | G | 1 (1.4) | 15 | F, G, H | 4 (4.8) |
| 3 | F | 1 (1.4) | 16 | F, J | 3 (3.7) |
| 11 | H | 1 (1.4) | 17 | F, G | 3 (3.7) |
| 12 | H | 1 (1.4) | 18 | F | 1 (1.2) |
| 13 | I | 2 (2.8) | 19 | F | 1 (1.2) |
| 14 | J | 1 (1.4) | 20 | G, H, J | 3 (3.7) |
| CSW-1 | F, G, I, J | 33 (46.5) | 21 | G | 2 (2.4) |
| V1-99 | F, G, I | 5 (7) | 22 | G | 1 (1.2) |
| 23 | G | 1 (1.2) | |||
| 24 | H | 1 (1.2) | |||
| 25 | H | 1 (1.2) | |||
| 26 | J | 1 (1.2) | |||
| 27 | J | 1 (1.2) | |||
| 28 | J | 1 (1.2) | |||
| 29 | J | 1 (1.2) | |||
| CSW-1 | F, G, H, I | 19 (23.2) | |||
| V1-99 | F, G, H, I | 10 (12.1) | |||
For day 2, there were a total of 38 (53.5%) parental viruses and 33 (46.5%) recombinants, for a total of 71 (100%) viruses; for day 4, there were a total of 29 (35.4%) parental viruses and 53 (64.6%) recombinants, for a total of 82 (100%) viruses.
TABLE 3.
Genotypes of viruses detected in low-dose-coinoculated (103 PFU) chickens at 6 and 8 days after coinoculationa
| Day 6 |
Day 8 |
||||
|---|---|---|---|---|---|
| Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates | Genotype pattern code | Chicken(s) from which virus was isolated | No. (%) of isolates |
| 6 | K, L | 2 (4.7) | 6 | L | 3 (5.6) |
| 5 | K | 1 (2.3) | 5 | K | 1 (1.9) |
| 10 | K | 1 (2.3) | 10 | K | 1 (1.9) |
| 9 | K, L | 5 (11.6) | 14 | K, M | 8 (15) |
| 30 | K, L | 5 (11.6) | 4 | K, L | 2 (3.7) |
| 31 | K, L | 3 (7) | 8 | K | 1 (1.9) |
| 1 | K, L | 2 (4.7) | 4 | K, L | 2 (3.7) |
| 32 | K | 1 (2.3) | 16 | L | 3 (5.6) |
| 16 | K | 1 (2.3) | 15 | L | 2 (3.7) |
| 7 | K | 1 (2.3) | CSW-1 | K, L | 17 (31) |
| 11 | K | 1 (2.3) | V1-99 | M | 14 (26) |
| 33 | K | 1 (2.3) | |||
| 34 | L | 2 (4.7) | |||
| 22 | L | 1 (2.3) | |||
| 20 | L | 1 (2.3) | |||
| CSW-1 | K, L | 7 (16.2) | |||
| V1-99 | K, L, M | 8 (19) | |||
For day 6, there were a total of 15 (34.9%) parental viruses and 28 (65.1%) recombinants, for a total of 43 (100%) viruses; for day 4, there were a total of 31 (57.4%) parental viruses and 23 (42.6%) recombinants, for a total of 54 (100%) viruses.
Viral-diversity analysis.
The higher level of viral replication in the low-dose-coinoculated group at day 4 than day 2 (Fig. 2A and B) coincided with a higher level of viral diversity, as assessed using Renyi diversity profiles (Fig. 5A) and as indicated by significantly higher values for diversity as measured by richness (0), Shannon-Weaver (1), 1/Simpson (2), 1/Berger Parker (infinite [Inf]), and Hill (0.25, 0.5, 4, 8, 16, 32, and 64) scales, as well as evenness values at these time points (see Table S1 in the supplemental material). The Renyi diversity profile was higher at day 6 than at day 8 (Fig. 5B); however, the diversity measurements were not significantly different between these days (see Table S2). In the high-dose-coinoculated group, the Renyi diversity profile was higher at day 4 than at day 2 (Fig. 5C), coinciding with the higher levels of viral replication at day 4 than day 2 in this group (Fig. 2E and F). Significant differences were detected only in the Shannon-Weaver (1), 1/Simpson (2), and Hill (4) values for these days (see Table S3).
FIG 5.
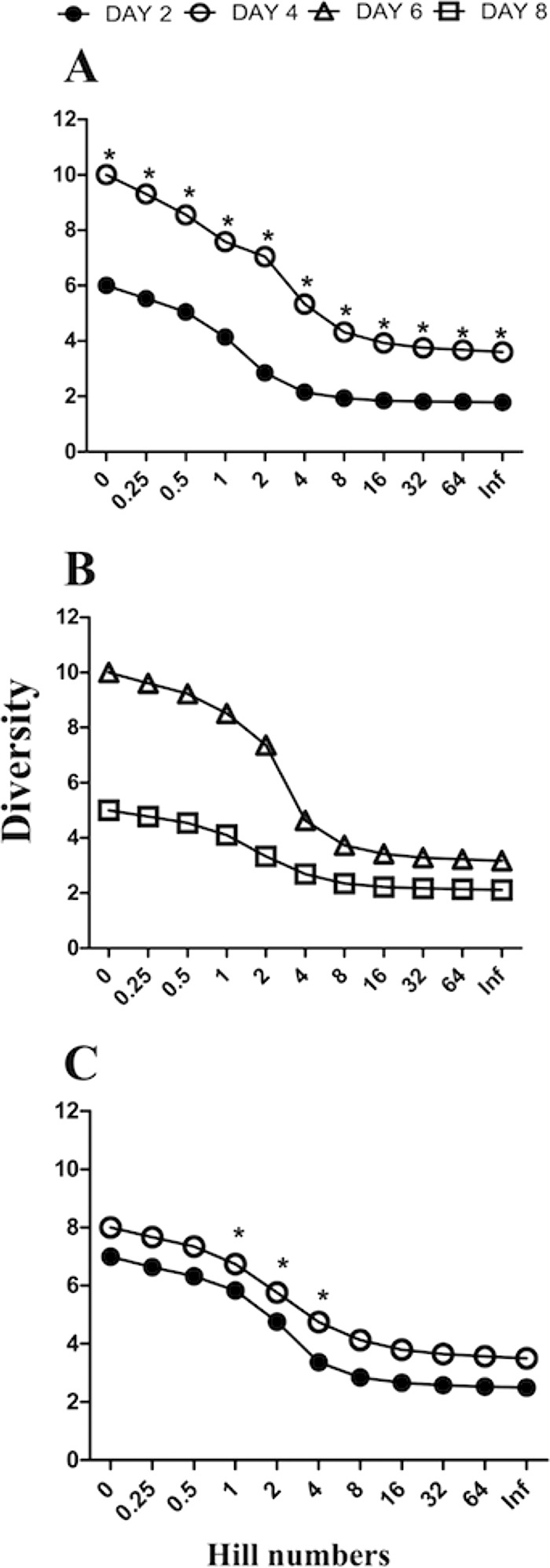
Renyi diversity profiles showing levels of diversity on the y axis and diversity measures on the x axis. Separate plots are shown for recombinants isolated 2 and 4 days after coinoculation with 103 PFU of CSW-1 and V1-99 (A), recombinants isolated 6 and 8 days after coinoculation with 103 PFU of CSW-1 and V1-99 (B), and recombinants isolated 2 and 4 days after coinoculation with 104 PFU of CSW-1 and V1-99 (C). Renyi profiles contain 11 diversity measurements, including richness (x axis value = 0), Hill values (x axis values = 0.25, 0.5, 4, 8, 16, 32, 64), Shannon-Weaver (x axis value = 1), 1/Simpson (x axis value = 2), and 1/Berger Parker (x axis value = Inf) values. One community can be regarded as more diverse than another if all its Renyi diversity measurements are higher (28). Asterisks indicate P values of <0.05 in paired Student's t test analyses on individual diversity measures, which were performed as additional statistical analyses.
Full-genome sequencing and identification of recombination hot spots.
Six progeny viruses were selected for full-genome sequencing and analysis (see Table S4) in order to identify all recombination events and compare these to those detected with the SNP genotyping assay and to examine the distribution of recombination breakpoints along the genome. These recombination patterns were also searched for among 35 other full ILTV genome sequences of isolates from Asia, Australia, Europe, and the United States in order to identify recombination hot spots in ILTV from distinct geographical regions. In addition to the distribution of recombination breakpoints detected by RDP4, reticulate network and phi test analyses were performed as previously described (20) (see Fig. S2).
The six SNPs targeted by the TaqMan SNP genotyping assay were confirmed by full-genome analysis. Additional events were detected outside the genome regions targeted by the TaqMan assay. The full genomes of the two genotype pattern 9 viruses shared 99.9% pairwise sequence identity. Between these two genotype pattern 9 viruses, 11 SNPs were detected within genes UL[−1], US5, US6, US7, US8, and US9, located between bp 110000 and bp 140000 in the aligned sequences. Two SNPs were identified in the protein IF and UL49 genes located between bp 8000 to bp 18000 in the aligned sequences, and two SNPs were identified in the UL43 and UL17 genes between bp 74000 and bp 86000. When CSW-1 was used as the reference strain, a total of 55 polymorphisms were found in the six viruses sequenced in this study (see Table S5 in the supplemental material).
A high number of recombination breakpoints were identified in two locations extending from bp 20000 to bp 50000 and from bp 106000 to bp 137000 in the six genome sequences from viruses isolated in this study (Fig. 6B). When the full-genome sequences from other ILTV isolates from varied geographical regions were examined, the recombination hot spots detected in those genomes were consistent with those found in the six genomes sequenced from the in vivo study (Fig. 6C, D, and E). Recombination events within genes UL[−1] and ICP4 were detected in ILTV sequences from Australia and the United States and in our in vivo recombinants (Table 4). Recombination events among the Asian ILTV strains were detected at sites similar to those detected in the Australian strains, with recombination breakpoints found in genes UL4, UL3.5, UL28, UL29, UL30, UL31, UL32, UL33, and UL34 (Table 4). Only one recombination breakpoint was detected and confirmed by several methods in the ILTV sequences from Europe (Italy) (Table 4). Hence, because of the low number of recombination events in the European group, no recombination hot spots were detected.
FIG 6.
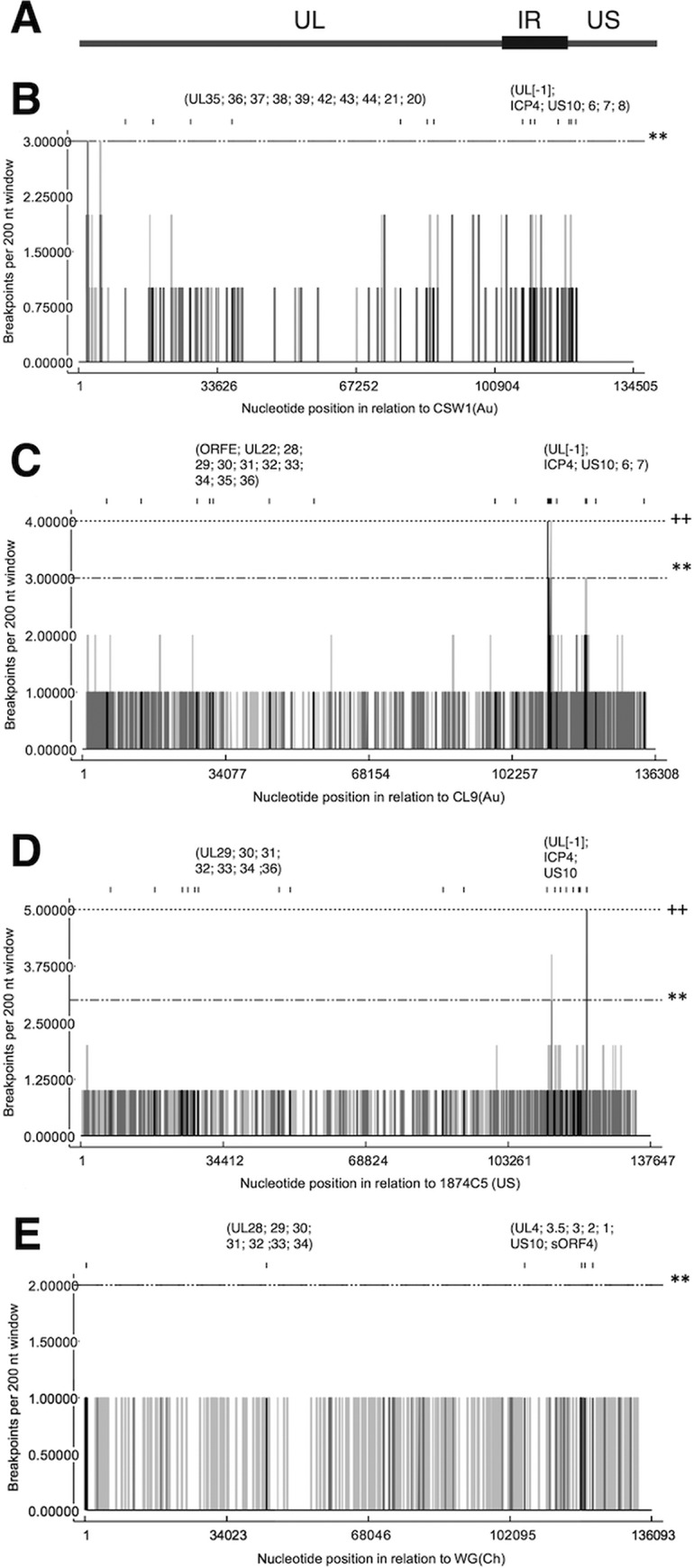
Detection of recombination hot spots in genome sequences. (A) Schematic representation of ILTV genome encompassing unique long (UL), internal repeat (IR), and unique short (US) regions; (B) the 6 ILTV isolates derived from the in vivo study; (C) ILTV isolates originating from Australia (n = 8); (D) ILTV isolates originating from the United States (n = 18); (E) ILTV isolates originating from Asia (n = 3). The vertical lines indicate recombination breakpoints per 200-nucleotide (nt) window in each analyzed sequence, as detected with 95% confidence (gray) or 99% confidence (black). Horizontal lines indicate the limits for global hot spot detection, indicated at a level of 95% confidence (**) or 99% confidence (++).
TABLE 4.
Recombination breakpoint analysis of full ILTV genome sequences and field isolates from Asia, Australia, and the United States and in vivo recombinants
| Origin of isolates | Breakpoint (in alignment), 99% CIa |
Gene(s) at the beginning of the breakpoint locationb | Gene(s) at the end of the breakpoint locationb | Possible viruses involved in recombination eventc | Method(s) by which breakpoint was detected in RDP4 | |
|---|---|---|---|---|---|---|
| Beginning | End | |||||
| Asia (China) | 106423–109054 | 125644–129391 | UL4, UL3.5, UL3, UL2, UL1 | US10, sORF4 | R, WG; M, unknown; m, K317 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ |
| 43560–45099 | 45674–54215 | UL28, UL29 | UL29, UL30, UL31, UL32, UL33, UL34 | R, K317; M, SERVA; m, WG | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| Australia | 27228–28747 | 112173–113630 | ORFB, ORFC | UL[−1] | R, CL9; M, SERVA; m, A20 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ |
| 114752–115584 | 139504–140702 | No gene between UL[−1] and ICP4 | US6, US7 | R, CL10; M, ACC78; m, SA2 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 30261–31965 | 51062–60138 | ORFE, UL22 | UL30, UL31, UL32, UL33, UL34, UL35, UL36 | R, CL10; M, SERVA; m, A20 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 134126–137622 | Undetermined | US6, US7 | Undetermined | R, CL9; M, unknown; m, V1-99 | RDP, GENECONV, MaxChi, Chimaera, SiScan, 3SEQ | |
| 113596–114071 | 120150–124890 | No gene between UL[−1] and ICP4 | US10 | R, CL9; M, unknown; m, CSW-1 | RDP, MaxChi, Chimaera, SiScan, 3SEQ | |
| 111500–113586 | 113598–126007 | No gene between UL[−1] and ICP4 | ICP4, US10 | R, C10; M, CSW-1; m, SA2 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| 30804–33011 | 43052–49251 | ORFE, UL22 | UL28, UL29, UL30 | R, ACC78; M, SERVA; m, SA2 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 99353–101053 | 104765–107587 | UL8 | UL5, UL4, UL3.5 | R, CL10; M, SERVA; m, A20 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 112731–113586 | 114079–114296 | UL[−1] | No gene between UL[−1] and ICP4 | R, V1-99; M, unknown; m, SA2 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 126079–126160 | 126204–126250 | No gene | No gene | R, A20; M, SA2; m, SERVA | GENECONV, 3SEQ | |
| Europe (Italy) | 42226–117783 | 122731–126724 | Entire UL region | Entire US region | R, Nobilis; M, SERVA; m, 757/11 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ |
| United States | 113909–114632 | 122256–124629 | No gene between UL[−1] and ICP4 | No gene | R, 1874c5; M, J2; m, unknown | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ |
| 111754–112964 | 113276–127469 | UL[−1] | ICP4, US10 | R, USDA; M, 81658; m, unknown | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| 115821–119810 | 120055–121242 | ICP4 | No gene | R, BdORFC; M, 6.48.88; m, unknown | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| 117501–119320 | 120028–121127 | ICP4 | No gene | R, 81658; M, unknown; m, 6.48.88 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| 45501–49291 | 50588–59726 | UL29, UL30 | UL30, UL31, UL32, UL33, UL34, UL35, UL36 | R, S2.86; M, unknown; m, USDA | RDP, GENECONV, 3SEQ | |
| 81577–89884 | 90133–95942 | UL19, UL13, UL15, UL17, UL14 | UL13, UL12, UL11, UL10 | R, 6.48.88; M, S2.816; m, J2 | RDP, GENECONV, Bootscan, 3SEQ | |
| 107225–122304 | 122306–128723 | UL 3.5, UL2, UL1, UL0, UL[−1], ICP4 | US10, Sorf4, US2, US3 | R, J2; M, unknown; m, 3.26.90 | GENECONV | |
| 22886–24681 | 24683–26851 | UL46, UL45 | UL45, ORFA, ORFB | R, 6.48.88; M, unknown; m, 14.939 | GENECONV, MaxChi | |
| 14052–16151 | 16153–18973 | UL52 | UL52, UL51, UL50 | R, 6.48.88; M, unknown; m, BdORFC | GENECONV | |
| 1–26560 | 1–26560 | UL56, ORFF, UL54, UL53, UL52, UL50, UL49.5, UL49, UL48, UL46, UL45 | UL56, ORFF, UL54, UL53, UL52, UL50, UL49.5, UL49, UL48, UL46, UL45 | R, USDA; M, 14.939; m, Laryngovac | RDP, MaxChi, SiScan | |
| 42685–50586 | 50591–66866 | UL28, UL29, UL30, UL31 | UL32, UL33, UL34, UL35, UL36, UL37 | R, 1874c5; M, J2; m, unknown | GENECONV, Bootscan | |
| In vivo recombinants | 53623–68876 | 120517–121888 | UL35, UL36, UL37, UL38, UL39 | No gene between UL [−1] and ICP4 | R, sample 23; M, unknown; m, CSW-1 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ |
| 121902–124017 | 134435–137336 | US10, SORF3 | US6, US7, US8 | R, sample 27; M, V1-99; m, unknown | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 71911–78480 | 112397–120377 | UL42, UL43, UL44, UL21, UL20 | ICP4 | R, sample 15; M, V1-99; m, CSW-1 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, SiScan, 3SEQ | |
| 121904–125713 | 129197–137424 | US10, protein SORF3, US2, US3 | US4, US5, US6, US7, US8 | R, sample 15; M, V1-99; m, sample 23 | GENECONV, Bootscan, 3SEQ | |
| 74976–97499 | 111841–114434 | UL44, UL21, UL20, UL19, UL18, UL15, UL17, UL14, UL13, UL12, UL11, UL10, UL9 | No gene Between UL [-1] and ICP4 | R, sample 12; M, sample 1; m, V1-99 | RDP, GENECONV, Bootscan, MaxChi, Chimaera, 3SEQ | |
| 2256–97499 | 106970–137424 | Entire UL region | Entire US region | R, sample 12; M, unknown; m, V1-99 | GENECONV, MaxChi, Chimaera | |
| 1–32193 | 105020–137424 | UL56, protein IF, UL54, UL53, UL52, UL51, UL50, UL49.5, UL49, UL48, UL46, UL45, protein IA-IB-IC-ID-IE, UL22 | Entire US region | R, sample 12; M, unknown; m, V1-99 | GENECONV, MaxChi, Chimaera, 3SEQ | |
| 1–1909 | 5870–17769 | No gene | UL56, protein IF, UL54, UL53, UL52, UL51, UL50 | R, sample 23; M, sample 1; m, sample 12 | SiScan | |
| 1–53621 | 107559–120122 | UL56, protein IF, UL54, UL53, UL52, UL51, UL50, UL49.5, UL49, UL48, UL46, UL45, protein IA-IB-IC-ID-IE, UL22, UL24, UL25, UL26, UL26.5, UL27, UL28, UL29, UL30, UL31, UL32, UL33, UL34 | Entire US region | R, V1-99; M, unknown; m, sample 8 | GENECONV, MaxChi, SiScan, 3SEQ | |
| 1–137424 | 1–137424 | Entire UL region | Entire UL region | R, sample 12; M, CSW-1; m, sample 27 | GENECONV, Bootscan, MaxChi, 3SEQ | |
| 1–113123 | 1–113123 | Entire UL region | Entire UL region | R, sample 27; M, V1-99; m, sample 23 | RDP, GENECONV, MaxChi, Chimaera | |
| 1–17769 | 19248–137424 | UL56, protein IF, UL54, UL53, UL52, UL51, UL50 | UL48, UL46, UL45, protein IA-IB-IC-ID-IE, UL22, UL24, UL25, UL26, UL26.5, UL27, UL28, UL29, UL30, UL31, UL32, UL33, UL34 | R, CSW-1; M, sample 15; m, sample 27 | MaxChi, Chimaera, 3SEQ | |
| 1–137424 | 1–137424 | Entire UL and US regions | Entire UL and US regions | R, sample 8; M, sample 15; m, unknown | GENECONV, MaxChi, 3SEQ | |
| 11775–17921 | 17923–137424 | UL53, UL52, UL51, UL50, UL49.5 | UL49, UL48, UL46, UL45, protein IA-IB-IC-ID-IE, UL22, UL24, UL25, UL26, UL26.5, UL27, UL28, UL29, UL30, UL31, UL32, UL33, UL34 | R, sample 27; M, sample 8; m, unknown | MaxChi, 3SEQ | |
| 1–137424 | 1–137424 | Entire UL and US regions | Entire UL and US regions | R, sample 15; M, sample 1; m, unknown | 3SEQ | |
CI, confidence interval.
UL, unique long; US, unique short.
R, recombinant; M, major parent; m, minor parent.
DISCUSSION
Our results show that higher levels of diversity in the recombinant progeny occurred at the same time as higher levels of virus replication (as measured by viral titers). A direct relationship between viral replication and recombination diversity would have important implications for disease control, as it suggests that measures taken to reduce viral replication, such as administration of vaccines to limit virus replication following challenge (21), may be able to reduce the diversity of recombinant viruses, thus potentially reducing the capacity of ILTV to evolve to higher levels of virulence. Vaccines are used regularly in poultry industries to control ILT. As previously described, most vaccines reduce, but do not prevent, ILTV replication (as measured by viral titers) after challenge, due to cell-mediated immune responses that develop in the host in response to vaccination (21). Many vaccine efficacy studies examine ILTV replication after challenge, but some focus only on clinical signs of disease and mortality (21, 22). Furthermore, studies to meet vaccine registration requirements remain focused mainly on mortality and clinical signs of disease (23). Expanding standard vaccine characterization studies to include examination of a vaccine's capacity to reduce viral replication after subsequent challenge (as measured by viral titers), and potentially viral recombination, would help to further control disease caused by this virus. Similar approaches could be taken for other attenuated live vaccines that are used to control alphaherpesviruses, such as FeHV-1, BoHV-1, and PRV, in veterinary medicine (24, 25) and for varicella-zoster virus (VZV) vaccines in human medicine (26), as these viruses all have capacity for recombination (3).
In this study, a high proportion (67%) of viruses detected following in vivo ILTV coinoculation were recombinants. This is consistent with findings from an HSV-1 study that found that 59% of progeny viruses from the cornea and 74% of progeny viruses from the trigeminal ganglia of mice coinoculated with two strains of HSV-1 were identified as recombinants using restriction fragment length polymorphism (RFLP) analysis (13). Additionally, in vitro studies have also shown abundant recombination following alphaherpesvirus coinoculation, although lower proportions of recombinants are typically detected in cell culture than in in vivo models. For example, approximately 26% of progeny viruses obtained after coinoculation of two HSV-1 strains into cultured cells were identified as recombinants using RFLP analysis (13), and 13% of the viral progeny obtained after coinoculation of two VZV strains into cell culture were identified as recombinants by RFLP analysis (15). Similarly, 10% to 21% of the viral progeny obtained after coinoculation of an FeHV-1 wild-type strain and two vaccine strains were identified as recombinants by restriction endonuclease digestion of PCR products (16). More recently a TaqMan SNP genotyping assay was developed to better detect and describe recombination between two closely related BoHV-1 strains in cell culture. Around 40% of the viral progeny were found to be recombinants using this assay (14), but the assay has not been applied to study recombination between the BoHV-1 strains in vivo. Previous application of the ILTV TaqMan SNP genotyping assay to viruses isolated from SPF chickens 4 days after coinfection with 104 PFU of CSW-1 and V1-99 found that 74% of progeny viruses were recombinants (18). This is consistent with our finding in this study that 79% of the progeny viruses were identified as recombinants 4 days after high-dose coinoculation. In both our present study and the previous in vivo study of ILTV recombination (18), SPF chickens were used as experimental animals. Future studies to examine ILTV recombination in commercial chickens would be advantageous, as differences in responses to infection have been noted between different breeds and lines of poultry (1).
The use of the ILTV SNP genotyping assay to detect recombination following coinoculation of SPF chickens has helped to measure viral diversity over time during the course of acute clinical disease (up to 8 days after coinoculation). Diversity is a product of the number of categories that can be differentiated and the proportions or relative abundances of the objects in each category (27). One community can be regarded as more diverse than another if all Renyi diversity values are higher (28). Our Renyi diversity analyses found that diversity was higher at day 4 than at day 2, regardless of the viral dose used to inoculate the chickens (Fig. 5A and C). However, further analyses of these results found that only those coinoculated with the lower dose had significant differences between day 4 and day 2 in every diversity measure (Fig. 5A; see also Table S1 in the supplemental material). This may indicate that a higher viral coinoculation dose may increase recombinant diversity at day 2 (Fig. 5C) but not necessarily at day 4, which is the peak of viral replication.
In both coinoculated groups some virus genotype pattern codes were detected in multiple birds at both days 2 and 4 after inoculation (Tables 1 and 2), as well as at days 6 and 8 after inoculation (Table 3). The detection of such recombinants in multiple birds over time may indicate that these recombinant viruses have fitness advantages that allow them to persist and be transmitted to other birds. Alternatively, these recombinants may have arisen from multiple independent recombination events, perhaps indicating that the regions of the genomes involved in these events are more prone to recombination. Some data to support the latter possibility were evident in the whole-genome sequence analyses. For example, viruses with genotype pattern codes 2 and 6 had recombination events between the UL46 and UL36 genes, which extend from approximately bp 20000 to 50000. These recombination breakpoints match with a recombination event previously detected in a study of Chinese ILTV strains, in which the K317 vaccine strain was determined to have arisen as a result of recombination between the Serva vaccine strain and virulent WG field strains (29) (Table 4). Recombination in this genome region (Table 4) has also been detected previously in Australian ILTV field isolates (2, 30).
Full-genome analysis of selected recombinants isolated from the in vivo study and ILTV full-genome sequences that were available in GenBank from Asia, Australia, Europe, and the United States (see Table S6) enable better characterization and interrogation of regions in the ILTV genome with a higher frequency of recombination events (recombination hot spots). Two genome regions were identified as hot spots in the recombinants isolated from the in vivo study and in isolates from Australia and the United States. These hot spots were located in the region from bp 20000 to 50000 and the region from bp 120,000 to 140000. Recombination hot spots were not identified in the Asian or the European isolates, although some recombination breakpoints were detected (Table 4). Our results are consistent with those obtained with HSV-1, in which recombination hot spots have been identified within the inverted repeat regions of the genome (31–33). The region from bp 20000 to 50000 has not been found to be a recombination hot spot in alphaherpesviruses in previous studies. Further research is needed in order to fully determine which genes within this region are more likely to be involved in recombination events and the effects on viral phenotypes.
The different numbers of isolates used in our analyses may have affected the detection of recombination hot spots, with more viruses included in the analysis of recombination in U.S. and Australian isolates. Even with the unequal numbers of isolates from the different geographical regions, higher rates of recombination were detected within Australian isolates. Vaccine strains, including the Serva, A20, and SA2 strains, were frequently involved in recombination events among the Australian isolates (2, 34). In contrast, vaccine strains did not typically feature in the recombination events detected in the U.S. strains, although the Laryngovac strain was identified as a minor parent in one recombination event and some field strains, such as 1874c5 and J2, were involved in recombination events (Table 4). In general, the recombination events were detected with more certainty in the Australian, Asian, and European isolates, with most events being detected by five or more of the RDP4 breakpoint detection methods. Some of the recombination events in the U.S. strains, and in the progeny viruses from the in vivo study, were detected by fewer than three of the methods within RDP4 (Table 4). Further work is needed to fully understand the significance of the recombination events described here and to better understand the role of ILTV recombination in the United States.
The precise nature of the relationship between recombination, viral diversity, and evolution of virulence remains to be determined. The results of this study suggest that there may be some correlation between recombination, increased viral diversity, and increased virulence, as the coinfected groups had significantly higher mortality rates than groups that received the same dose of only ILTV V1-99. However, the coinoculated groups did receive two times the total dose of virus compared to the birds that received a single dose of virus, and the larger dose may have resulted in more severe clinical disease. In addition, higher viral genome copy numbers were detected in many of the coinoculated birds than in birds that received just V1-99 ILTV. Future studies to characterize the phenotype of dominant recombinants would be helpful to better understand this relationship. In addition, coinoculation studies that utilize naive, in-contact birds would be useful for understanding how recombinants that arise in individual coinfected chickens may be transmitted to become established in populations of birds. Of great practical importance is an understanding of how all these processes and relationships are affected by vaccines, which could affect recombination events, and therefore have an impact on disease control. This is a complex area of study, but it should be prioritized in order to enhance the control of infectious laryngotracheitis.
MATERIALS AND METHODS
Viruses and cell culture.
The V1-99 and CSW-1 ILTV wild-type strains were used as the parental viruses for in vivo coinoculation. These strains belong to genotype classes 2 and 4, respectively, following the Australian ILTV genotype classification system (35). The V1-99 field strain was isolated from a hen in a commercial layer flock in 1999 and is typical of the predominant field isolates detected during disease outbreaks in Australia up until 2008 (30). The CSW-1 wild type is currently used as a standard laboratory strain in Australia (36, 37) and was originally isolated in 1970 from layer birds (38). The growth characteristics of these viruses have been described previously and compared in vitro (18) and in vivo (39). Both viruses have undergone at least three rounds of plaque purification. For this study, the viruses were propagated and titrated in chicken hepatocellular carcinoma (LMH) cells (40) using a plaque assay, as previously described (41). Virus isolation and purification from clinical material collected during the in vivo experiment were performed in LMH cell monolayers cultured in growth medium (GM) consisting of Dulbecco's minimal essential medium (DMEM) supplemented with amphotericin B (0.005 mg/ml), gentamicin (0.05 mg/ml), and co-trimoxazole (0.01 mg of sulfamethoxazole/ml and 0.002 mg of trimethoprim/ml), 10% (vol/vol) fetal bovine serum (FBS), and 10 mM HEPES (pH 7.7).
In vivo coinoculation experiment.
Five-week-old SPF chickens were obtained from Australian SPF Services Pty. Ltd. and used for the infection experiments. The in vivo experiment extended for 8 days. The birds were maintained in separate groups in isolator units and were managed, monitored, and euthanized following protocols approved by the Animal Ethics Committee (AEC) of the University of Melbourne (AEC number 1413401.1). This committee follows the Australian Code for the Care and Use of Animals for Scientific Purposes (42). Two groups of 10 SPF chickens were coinoculated by the intratracheal route with 300 μl of a 1:1 mixture of the CSW-1 and V1-99 ILTV strains containing either 1 × 103 (low-dose-coinoculated group) or 1 × 104 (high-dose-coinoculated group) PFU of each strain. Four groups containing five SPF birds each were inoculated by the intratracheal route with 300 μl of an inoculum containing 1 × 103 or 1 × 104 PFU of only CSW-1 or only V1-99. The birds were monitored for 8 days. At 2, 4, 6, and 8 days after inoculation, tracheal swabs were collected using standard techniques, placed into 1 ml of viral transport medium (DMEM, 3% [vol/vol] FBS, and 100 μg of ampicillin/ml), transported on ice, and then stored at −80°C until processed for virus isolation and purification. The swabbing technique was not objectively measured, and some apparent variability in viral replication may be attributable to variations in swabbing technique. Prior to storage at −80°C, 200-μl aliquots of tracheal swab suspension were collected and stored separately at −20°C for DNA extraction using a Qiagen DNA robot extractor following the manufacturer's recommendations (QIAxtractor; Qiagen) and quantification of ILTV genome copy number using a qPCR described previously (43).
Virus isolation and purification.
Progeny viruses were isolated and purified as previously described (18). Material from each tracheal swab was serially diluted (10-fold) in GM in order to identify the appropriate dilution for plaque purification. Dilutions were used to inoculate LMH cell monolayers in 6-well plates. After 1 h of incubation at 37°C, the cell monolayer was covered with semisolid methylcellulose overlay medium containing 10% FBS and incubated at 37°C in a humidified atmosphere of 5% (vol/vol) CO2. After incubation for 24 to 48 h, up to 20 plaques were picked from each sample from the coinfected chickens and up to 10 were picked from the samples from the single inoculated birds at day 4 with a micropipette, and then each plaque was propagated individually by the inoculation of LMH cell monolayers in 12-well plates. Three rounds of plaque purification were performed, with one freeze-thaw cycle between each round.
SNP genotyping assay and examination of viral diversity.
To characterize the viral progeny and identify recombinants, DNA from each plaque-purified virus was extracted and used as the template in a TaqMan SNP genotyping assay, as previously described (18). Any samples that were found to contain a mixed population of viruses (i.e., both SNPs present at any of the six locations) were not included in further analyses. To measure diversity, we first identified each recombinant using a unique genotype pattern code. Afterwards, these genotype pattern codes were analyzed in RStudio 0.99.902 using the VeganR (44) and BiodiversityR (27) packages. VeganR calculates the diversity indices used to perform ecological-diversity measurements in communities (45). BiodiversityR was used to generate Renyi profiles. This package provides a graphical user interface (GUI) via R-Commander incorporating functions used by VeganR to analyze measures of diversity such as richness (x axis value = 0), Hill (x axis values = 0.25, 0.5, 4, 8, 16, 32, and 64), Shannon-Weaver (x axis value = 1), 1/Simpson (x axis value = 2), and 1/Berger Parker (x axis value = Inf) values (27).
Whole-genome sequencing, genome assembly, and recombination analysis.
Six progeny viruses that were identified as recombinants using the SNP genotyping assay were selected for whole-genome sequencing (genotype pattern codes/GenBank accession numbers [9A/MF156847, 6/MF156848, 2/MF156849, 9B/MF156850, 4/MF156851, and 40/MF156852]). These viruses included those with genotype patterns that were frequently detected (codes 2, 4, 6, and 9), as well as an infrequently detected genotype pattern (code 40). Two virus isolates with the same genotype pattern (code 9) were included to enable comparisons within a genotype pattern. Each virus was first amplified in LMH cells, and then viral DNA was extracted from purified nucleocapsids as previously described (20) and sequenced using the Illumina MiSeq platform at the Medical Genomic Facility in the Monash Health Translation Precinct (MHTP), Hudson Institute of Medical Research. The genome was assembled by mapping to CSW-1 (GenBank accession number JX646899) as a reference sequence using the Geneious mapper method. Settings of medium sensitivity and up to five iterations were used, as recommended by the software. To generate the consensus sequences, SNPs were called within and outside coding regions using the Find variation/SNPs tool with default settings. CSW-1 and V1-99 genomes (GenBank accession numbers JX646899 and JX646898, respectively) were used to annotate the new genome sequences. Mapping to the reference, SNP calling, and annotations were all performed using Geneious V8.1.9 (46).
In order to expand our analysis beyond the six genome sequences of recombinant viruses generated in this study, we also analyzed 35 full-genome sequences available in the NCBI database (see Table S6 in the supplemental material). The sequences were from ILTVs isolated in different geographical regions, including Asia, Australia, Europe, and the United States. Separate recombination analyses were performed for the viruses originated from different geographical regions and for the recombinant viruses generated during this study. To enable analysis for recombination events the sequences were first aligned and curated as previously described (20, 47). Recombination analyses were then performed using reticulate network analysis and the phi test within SplitsTree4 V 4.14.4 (48), as well as seven methods for detection of recombination breakpoints (RDP, GENECONV, 3Seq, SiScan, Chimarea, MaxChi, and Bootscan) within RDP4 V 4.83 (49). As several recombination breakpoints were detected along the full-genome sequences, the same alignments were used to visualize breakpoint distribution plots in order to determine patterns and/or recombination hot spots. The CSW-1, CL9, 1874c5, and WG genome sequences (GenBank accession numbers are provided in Table S6 in the supplemental material) were used as reference strains for the recombination analysis of our in vivo recombinants and Australian, U.S., and Asian strains, respectively. Breakpoint distribution plots were used as a matrix, with 800 permutations and a window size of 200 nucleotides. The recombination breakpoint and the recombination hot spot location analyses were performed as recommended in the RDP4 V 4.83 manual (49).
Accession number(s).
In the present study, the follow genotype pattern codes (and GenBank accession numbers) were determined: 9A (MF156847), 6 (MF156848), 2 (MF156849), 9B (MF156850), 4 (MF156851), and 40 (MF156852).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Mahathi Nadimpalli, Nino Ficorilli, and Mesula Korsa for their help with cell culture and Jose Quinteros, June Daly, Jenece Wheeler, and Cheryl Colson for their help with the in vivo studies.
This work was supported by the Australian Research Council (FT140101287). Carlos A. Loncoman is supported by Becas Chile, CONICYT, Gobierno de Chile.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01532-17.
REFERENCES
- 1.Garcia M, Spatz S, Guy JS. 2013. Laryngotracheitis, p 137–152. In Swayne D, Glisson J, McDougald L, Nolan L, Suarez D, Nair V (ed), Diseases of poultry, 13th ed Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 2.Lee SW, Markham PF, Coppo MJ, Legione AR, Markham JF, Noormohammadi AH, Browning GF, Ficorilli N, Hartley CA, Devlin JM. 2012. Attenuated vaccines can recombine to form virulent field viruses. Science 337:188. doi: 10.1126/science.1217134. [DOI] [PubMed] [Google Scholar]
- 3.Loncoman CA, Vaz PK, Coppo MJ, Hartley CA, Morera FJ, Browning GF, Devlin JM. 2017. Natural recombination in alphaherpesviruses: insights into viral evolution through full genome sequencing and sequence analysis. Infect Genet Evol 49:174–185. doi: 10.1016/j.meegid.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Jetzt AE, Yu H, Klarmann GJ, Ron Y, Preston BD, Dougherty JP. 2000. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J Virol 74:1234–1240. doi: 10.1128/JVI.74.3.1234-1240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes EC. 2003. Error thresholds and the constraints to RNA virus evolution. Trends Microbiol 11:543–546. doi: 10.1016/j.tim.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahy BWJ. 2010. The evolution and emergence of RNA viruses. Emerg Infect Dis 16:899. doi: 10.3201/eid1605.100164. [DOI] [Google Scholar]
- 7.Crute JJ, Lehman IR. 1989. Herpes simplex-1 DNA polymerase. Identification of an intrinsic 5′–3′ exonuclease with ribonuclease H activity. J Biol Chem 264:19266–19270. [PubMed] [Google Scholar]
- 8.McGeoch DJ, Cook S. 1994. Molecular phylogeny of the alphaherpesvirinae subfamily and a proposed evolutionary timescale. J Mol Biol 238:9–22. doi: 10.1006/jmbi.1994.1264. [DOI] [PubMed] [Google Scholar]
- 9.Drake JW, Hwang CBC. 2005. On the mutation rate of herpes simplex virus type 1. Genetics 170:969–970. doi: 10.1534/genetics.104.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Javier RT, Sedarati F, Stevens JG. 1986. Two avirulent herpes simplex viruses generate lethal recombinants in vivo. Science 234:746–748. doi: 10.1126/science.3022376. [DOI] [PubMed] [Google Scholar]
- 11.Thiry E, Meurens F, Muylkens B, McVoy M, Gogev S, Thiry J, Vanderplasschen A, Epstein A, Keil G, Schynts F. 2005. Recombination in alphaherpesviruses. Rev Med Virol 15:89–103. doi: 10.1002/rmv.451. [DOI] [PubMed] [Google Scholar]
- 12.Umene K. 1999. Mechanism and application of genetic recombination in herpesviruses. Rev Med Virol 9:171–182. doi:. [DOI] [PubMed] [Google Scholar]
- 13.Kintner RL, Allan RW, Brandt CR. 1995. Recombinants are isolated at high frequency following in vivo mixed ocular infection with two avirulent herpes simplex virus type 1 strains. Arch Virol 140:231–244. doi: 10.1007/BF01309859. [DOI] [PubMed] [Google Scholar]
- 14.Muylkens B, Farnir F, Meurens F, Schynts F, Vanderplasschen A, Georges M, Thiry E. 2009. Coinfection with two closely related alphaherpesviruses results in a highly diversified recombination mosaic displaying negative genetic interference. J Virol 83:3127–3137. doi: 10.1128/JVI.02474-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dohner DE, Adams SG, Gelb LD. 1988. Recombination in tissue culture between varicella-zoster virus strains. J Med Virol 24:329–341. doi: 10.1002/jmv.1890240310. [DOI] [PubMed] [Google Scholar]
- 16.Fujita K, Maeda K, Yokoyama N, Miyazawa T, Kai C, Mikami T. 1998. In vitro recombination of feline herpesvirus type 1. Arch Virol 143:25–34. doi: 10.1007/s007050050265. [DOI] [PubMed] [Google Scholar]
- 17.Henderson LM, Katz JB, Erickson GA, Mayfield JE. 1990. In vivo and in vitro genetic recombination between conventional and gene-deleted vaccine strains of pseudorabies virus. Am J Vet Res 51:1656–1662. [PubMed] [Google Scholar]
- 18.Loncoman CA, Hartley CA, Coppo MJC, Vaz PK, Diaz-Méndez A, Browning GF, Lee S-w, Devlin JM. 2017. Development and application of a TaqMan single nucleotide polymorphism genotyping assay to study infectious laryngotracheitis virus recombination in the natural host. PLoS One 12:e0174590. doi: 10.1371/journal.pone.0174590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davison AJ. 2010. Herpesvirus systematics. Vet Microbiol 143:52–69. doi: 10.1016/j.vetmic.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaz PK, Horsington J, Hartley CA, Browning GF, Ficorilli NP, Studdert MJ, Gilkerson JR, Devlin JM. 2016. Evidence of widespread natural recombination among field isolates of equine herpesvirus 4 but not among field isolates of equine herpesvirus 1. J Gen Virol 97:747–755. doi: 10.1099/jgv.0.000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coppo MJ, Hartley CA, Devlin JM. 2013. Immune responses to infectious laryngotracheitis virus. Dev Comp Immunol 41:454–462. doi: 10.1016/j.dci.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 22.Coppo MJ, Noormohammadi AH, Browning GF, Devlin JM. 2013. Challenges and recent advancements in infectious laryngotracheitis virus vaccines. Avian Pathol 42:195–205. doi: 10.1080/03079457.2013.800634. [DOI] [PubMed] [Google Scholar]
- 23.Collet S. 2013. Principles of disease prevention, diagnosis and control introduction, p 4–40. In Swayne D, Glisson J, McDougald L, Nolan L, Suarez D, Nair V (ed), Diseases of poultry, 13th ed Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 24.Luo Y, Li N, Cong X, Wang C-H, Du M, Li L, Zhao B, Yuan J, Liu D-D, Li S, Li Y, Sun Y, Qiu H-J. 2014. Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet Microbiol 174:107–115. doi: 10.1016/j.vetmic.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Jas D, Aeberle C, Lacombe V, Guiot AL, Poulet H. 2009. Onset of immunity in kittens after vaccination with a non-adjuvanted vaccine against feline panleucopenia, feline calicivirus and feline herpesvirus. Vet J 182:86–93. doi: 10.1016/j.tvjl.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi M, Otsuka T, Okuno Y, Asano Y, Yazaki T, Isomura S. 1974. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet 304:1288–1290. doi: 10.1016/S0140-6736(74)90144-5. [DOI] [PubMed] [Google Scholar]
- 27.Kindt R, Coe R. 2005. Tree diversity analysis: a manual and software for common statistical methods for ecological and biodiversity studies. World Agroforestry Centre, Nairobi, Kenya: http://www.worldagroforestry.org/resources/databases/tree-diversity-analysis. [Google Scholar]
- 28.Tóthmérész B. 1995. Comparison of different methods for diversity ordering. J Veg Sci 6:283–290. doi: 10.2307/3236223. [DOI] [Google Scholar]
- 29.Zhao Y, Kong C, Wang Y. 2015. Multiple comparison analysis of two new genomic sequences of ILTV strains from China with other strains from different geographic regions. PLoS One 10:e0132747. doi: 10.1371/journal.pone.0132747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agnew-Crumpton R, Vaz PK, Devlin JM, O'Rourke D, Blacker-Smith HP, Konsak-Ilievski B, Hartley CA, Noormohammadi AH. 2016. Spread of the newly emerging infectious laryngotracheitis viruses in Australia. Infect Genet Evol 43:67–73. doi: 10.1016/j.meegid.2016.05.023. [DOI] [PubMed] [Google Scholar]
- 31.Dutch RE, Bruckner RC, Mocarski ES, Lehman IR. 1992. Herpes simplex virus type 1 recombination: role of DNA replication and viral a sequences. J Virol 66:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutch RE, Bianchi V, Lehman IR. 1995. Herpes simplex virus type 1 DNA replication is specifically required for high-frequency homologous recombination between repeated sequences. J Virol 69:3084–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K, Kolb AW, Sverchkov Y, Cuellar JA, Craven M, Brandt CR. 2015. Recombination analysis of herpes simplex virus 1 reveals a bias toward GC content and the inverted repeat regions. J Virol 89:7214–7223. doi: 10.1128/JVI.00880-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S-W, Devlin JM, Markham JF, Noormohammadi AH, Browning GF, Ficorilli NP, Hartley CA, Markham PF. 2013. Phylogenetic and molecular epidemiological studies reveal evidence of multiple past recombination events between infectious laryngotracheitis viruses. PLoS One 8:e55121. doi: 10.1371/journal.pone.0055121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirkpatrick NC, Mahmoudian A, O'Rourke D, Noormohammadi AH. 2006. Differentiation of infectious laryngotracheitis virus isolates by restriction fragment length polymorphic analysis of polymerase chain reaction products amplified from multiple genes. Avian Dis 50:28–34. doi: 10.1637/7414-072205R.1. [DOI] [PubMed] [Google Scholar]
- 36.Fahey KJ, Bagust TJ, York JJ. 1983. Laryngotracheitis herpesvirus infection in the chicken: the role of humoral antibody in immunity to a graded challenge infection. Avian Pathol 12:505–514. doi: 10.1080/03079458308436195. [DOI] [PubMed] [Google Scholar]
- 37.Devlin JM, Browning GF, Hartley CA, Kirkpatrick NC, Mahmoudian A, Noormohammadi AH, Gilkerson JR. 2006. Glycoprotein G is a virulence factor in infectious laryngotracheitis virus. J Gen Virol 87:2839–2847. doi: 10.1099/vir.0.82194-0. [DOI] [PubMed] [Google Scholar]
- 38.Bagust TJ, Calnek BW, Fahey KJ. 1986. Gallid-1 herpesvirus infection in the chicken. 3. Reinvestigation of the pathogenesis of infectious laryngotracheitis in acute and early post-acute respiratory disease. Avian Dis 30:179–190. [PubMed] [Google Scholar]
- 39.Kirkpatrick NC, Mahmoudian A, Colson CA, Devlin JM, Noormohammadi AH. 2006. Relationship between mortality, clinical signs and tracheal pathology in infectious laryngotracheitis. Avian Pathol 35:449–453. doi: 10.1080/03079450601028803. [DOI] [PubMed] [Google Scholar]
- 40.Kawaguchi T, Nomura K, Hirayama Y, Kitagawa T. 1987. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res 47:4460–4464. [PubMed] [Google Scholar]
- 41.Devlin JM, Browning GF, Gilkerson JR. 2006. A glycoprotein I- and glycoprotein E-deficient mutant of infectious laryngotracheitis virus exhibits impaired cell-to-cell spread in cultured cells. Arch Virol 151:1281–1289. doi: 10.1007/s00705-005-0721-8. [DOI] [PubMed] [Google Scholar]
- 42.National Health and Medical Research Council. 2013. Australian code for the care and use of animals for scientific purposes, 8th ed National Health and Medical Research Council, Canberra, Australia. [Google Scholar]
- 43.Mahmoudian A, Kirkpatrick NC, Coppo M, Lee SW, Devlin JM, Markham PF, Browning GF, Noormohammadi AH. 2011. Development of a SYBR Green quantitative polymerase chain reaction assay for rapid detection and quantification of infectious laryngotracheitis virus. Avian Pathol 40:237–242. doi: 10.1080/03079457.2011.553582. [DOI] [PubMed] [Google Scholar]
- 44.Oksanen J. 2015. Multivariate analysis of ecological communities in R: vegan tutorial. http://vegan.r-forge.r-project.org/.
- 45.Hill MO. 1973. Diversity and evenness: a unifying notation and its consequences. Ecology 54:427–432. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 46.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaz PK, Job N, Horsington J, Ficorilli N, Studdert MJ, Hartley CA, Gilkerson JR, Browning GF, Devlin JM. 2016. Low genetic diversity among historical and contemporary clinical isolates of felid herpesvirus 1. BMC Genomics 17:704. doi: 10.1186/s12864-016-3050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 49.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution 1(1):vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.