

ABSTRACT
“Daqu” is a saccharifying and fermenting agent commonly used in the traditional solid-state fermentation industry (e.g., baijiu and vinegar). The patterns of microbial community succession and flavor formation are highly similar among batches, yet the mechanisms promoting temporal succession in the Daqu microbial ecology remain unclear. Here, we first correlated temporal profiles of microbial community succession with environmental variables (temperature, moisture, and titratable acidity) in medium temperature Daqu (MT-Daqu) throughout fermentation. Temperature dynamics significantly correlated (P < 0.05) with the quick succession of MT-Daqu microbiota in the first 12 d of fermentation, while the community structure was relatively stable after 12 d. Then, we explored the effect of temperature on the MT-Daqu community assembly. In the first 4 d of fermentation, the rapid propagation of most bacterial taxa and several fungal taxa, including Candida, Wickerhamomyces, and unclassified Dipodascaceae and Saccharomycetales species, significantly increased MT-Daqu temperature to 55°C. Subsequently, sustained bio-heat generated by microbial metabolism (53 to 56°C) within MT-Daqu inhibited the growth of most microbes from day 4 to day 12, while thermotolerant taxa, including Bacillus, unclassified Streptophyta, Weissella, Thermoactinomyces, Thermoascus, and Thermomyces survived or kept on growing. Furthermore, temperature as a major driving force on the shaping of MT-Daqu microbiota was validated. Lowering the fermentation temperature by placing the MT-Daqu in a 37°C incubator resulted in decreased relative abundances of thermotolerant taxa, including Bacillus, Thermoactinomyces, and Thermoascus, in the MT-Daqu microbiota. This study revealed that bio-heat functioned as a primary endogenous driver promoting the formation of functional MT-Daqu microbiota.
IMPORTANCE Humans have mastered the Daqu preparation technique of cultivating functional microbiota on starchy grains over thousands of years, and it is well known that the metabolic activity of these microbes is key to the flavor production of Chinese baijiu. The pattern of microbial community succession and flavor formation remains highly similar between batches, yet mechanistic insight into these patterns and into microbial population fidelity to specific environmental conditions remains unclear. Our study revealed that bio-heat was generated within Daqu bricks in the first 4 d of fermentation, concomitant with rapid microbial propagation and metabolism. The sustained bio-heat may then function as a major endogenous driving force promoting the formation of the MT-Daqu microbiota from day 4 to day 12. The bio-heat-driven growth of thermotolerant microorganisms might contribute to the formation of flavor metabolites. This study provides useful information for the temperature-based modulation of microbiota function during the fermentation of Daqu.
KEYWORDS: Daqu, bio-heat, metagenomics, microbial communities
INTRODUCTION
Chinese baijiu is one of the six oldest distilled liquors in the world. Daqu, a type of dried fermentation starter, plays a key role in initiating the solid-state fermentation of baijiu and determines the flavor quality of the final product. Daqu is prepared via an ancient technique of cultivating the microbes endogenous to starchy grains, and is an entirely separate process that precedes the actual fermentation of grains (https://en.wikipedia.org/wiki/Jiuqu#Daqu). Microbes that inhabit Daqu not only participate in the fermentation of baijiu (1, 2), but also provide abundant enzymes (e.g., glucoamylase, cellulose, α-amylase, and esterase) for substrate degradation and flavor compound production (3–5). Meanwhile, microbial metabolites formed during the fermentation process of Daqu may be further converted as intermediates of end flavor components in the baijiu fermentation, or dissolve in the final product, which is referred to in practice as “Daqu aroma” (6).
The industrial Daqu production process usually involves four stages: (i) watering, grinding, and mixing of raw materials (e.g., wheat, barley, peas, and sorghum); (ii) shaping Daqu bricks; (iii) spontaneous solid-state fermentation for about 30 days, which directly determines the shifts in microbial community structure and enriches the functional microbes naturally present in the raw materials and open work environment; and (iv) maturation in a fermentation room without ventilation for 1 to 3 months (see Fig. S1 in the supplemental material). In the third stage of the Daqu production process, once the temperature of Daqu bricks reaches a threshold limit, craftsmen open the windows of the fermentation room to improve its ventilation. Based on the maximum temperature (threshold limit) it reaches during the fermentation process, Daqu is usually classified into three types: low-temperature Daqu (45 to 50°C), medium-temperature Daqu (MT to Daqu) (50 to 60°C), and high-temperature Daqu (60 to 65°C) (7). MT-Daqu is the most widely used starter in the production of traditional Chinese baijiu, especially Luzhou-flavor baijiu (8).
The microbial community underpins the function of Daqu in baijiu fermentation. Microbes that inhabit Daqu bricks reproducibly metabolize nonautoclaved raw materials and synthesize different enzymes and flavor compounds. Thus, the reproducible formation of functional microbiota makes the Daqu microbial ecosystem amenable to be adopted as an experimental model for studying microbial community formation and function in food fermentation (9). Recent studies have focused on the diversity and dynamics of the Daqu microbial community throughout fermentation using culture-dependent and culture-independent methods (1, 2, 5, 9–15). The most common fungi found in Daqu are the filamentous molds, including Aspergillus, Rhizopus, Amylomyces, Monascus, Absidia, Rhizomucor and Mucor, and the yeasts, including Saccharomycopsis, Issatchenkia, Saccharomyces, Pichia, Candida, and Rhodotorula. The dominant bacterial genera in the microbial community of Daqu include Bacillus, Lactobacillus, Leuconostoc, Streptomyces, and Acetobacter. However, the mechanisms underlying community assembly and promotion of temporal succession in microbial ecology of Daqu remain unclear.
In this study, we adopted the fermentation process of MT-Daqu as a research model. Community succession in the microbial ecosystem of the MT-Daqu fermentation process was dissected via high-throughput sequencing. The microbial community structure, succession, and correlations to environmental variables were investigated. This study provides fundamental information about the microbial community succession and can help understand the functionality of MT-Daqu in the solid-state fermentation process.
RESULTS
Temporal changes in environmental variables, cell numbers, and enzyme activity.
In the practical production process, MT-Daqu fermentation is empirically divided into two phases: phase I (days 0 to 12), when the temperature of MT-Daqu is maintained at 50 to 60°C by indoor air ventilation, and phase II (days 12 to 32), when the MT-Daqu fermentation room is no longer ventilated. During phase I, the temperature of MT-Daqu increased rapidly from 29.1 ± 0.7°C to 53.4 ± 1.0°C in the first 4 d, hovered between 53.4 to 56.2°C from day 4 to day 12, and decreased gradually to 32.3 ± 1.4°C in phase II (Fig. 1A). The moisture content of MT-Daqu sharply decreased from 35.4% ± 1.0% to 17.1% ± 1.1% in phase I, and then was maintained at a low content (15.4% to 13.7%) in phase II (Fig. 1A). The titratable acidity of MT-Daqu increased to 0.8 ± 0.1 mmol/g at 4 d, decreased to 0.3 ± 0.0 mmol/g from 4 to 10 d, and then was maintained at about 0.3 mmol/g until the end of fermentation (Fig. 1A).
FIG 1.
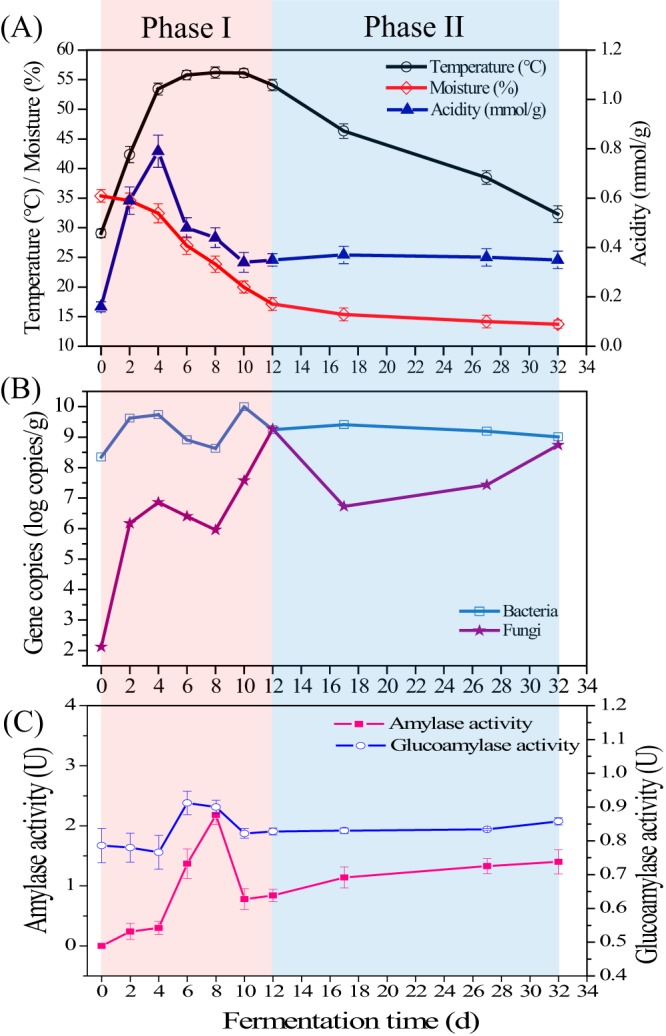
Dynamics of environmental variables, microbial cell number, and amylase and glucoamylase activities in MT-Daqu throughout fermentation. Abundance of bacterial and fungal cell numbers in the MT-Daqu are expressed as the log (10) of 16S rRNA gene and ITS rRNA gene copy numbers, respectively.
Cell numbers of total bacteria and total fungi in MT-Daqu rapidly increased to 9.7 ± 0.1 log 16S rRNA gene copies/g and 6.8 ± 0.0 log ITS rRNA gene copies/g in the first 4 d of fermentation, and decreased to 8.6 ± 0.0 and 5.9 ± 0.3 log copies/g on the 8th day, respectively (Fig. 1B). Then, bacterial cell number increased to 10.0 ± 0.0 log copies/g on day 10, and fungal cell number increased to 9.3 ± 0.1 log copies/g on day 12. Bacterial cell number was maintained at 9.0 to 10.0 log copies/g between 12 and 32 d of fermentation, while fungal cell number gradually increased from 6.7 to 8.7 log copies/g between 17 and 32 d of fermentation (Fig. 1B).
Glucoamylase activity of MT-Daqu slightly decreased in the first 4 d, increased to 0.9 ± 0.0 U within 4 to 6 d, decreased to 0.8 ± 0.0 U within 6 to 10 d, and then was maintained at about 0.8 U until the end of fermentation (Fig. 1C). Amylase activity of MT-Daqu slightly increased in the first 4 d, increased rapidly to 2.2 ± 0.1 U within 4 to 8 d, decreased obviously to 0.8 ± 0.1 U, and then increased gradually to 1.4 ± 0.2 U in phase II (Fig. 1C).
Microbial community pattern of MT-Daqu.
Temporal patterns of microbial community structure during the fermentation process of MT-Daqu are shown in Fig. 2. At the genus level, unclassified bacteria and unclassified Streptophyta were dominant bacteria in MT-Daqu on day 0; however, their abundance dramatically decreased after 2 d of fermentation and relative abundance was maintained at lower levels (<4.4%) until the end of fermentation (Fig. 2A). During the fermentation process, 10 bacterial genera dominated the bacterial community of MT-Daqu, with a total relative abundance of more than 70.0% (Fig. 2A). Lactic acid bacteria related to Weissella, Leuconostoc, and Lactobacillus were dominant bacteria throughout fermentation, while Bacillus became a dominant genus after 10 d of fermentation. Relative abundance of Thermoactinomyces was hardly detected in the first 4 d of fermentation, but was maintained at about 1% after the 8th day. Variation in the relative abundances of bacterial community members at the genus level was similar to that at the order level (see Fig. S3A in the supplemental material).
FIG 2.

Temporal patterns of microbial community structure during the fermentation process of MT-Daqu. (A) Temporal profile for the relative abundance of bacterial taxa represented at the genus level; (B) distance tree based on V1-V3 16S rRNA gene amplicon sequences of bacteria in MT-Daqu constructed by the unweighted pair group method with arithmetic mean (UPGMA); (C) temporal profile for the relative abundance of fungal taxa represented at the genus level; and (D) distance tree based on ITS1 ITS rRNA gene amplicon sequences of fungi in MT-Daqu constructed by UPGMA.
Alternaria and unclassified genera dominated the fungal community in MT-Daqu on day 0; however, their abundance dramatically decreased after 2 d of fermentation (Fig. 2C). Thermoascus, Candida, Wickerhamomyces, and Thermomyces were dominant fungal genera throughout fermentation (Fig. 2C). The relative abundance of Thermoascus dramatically increased from 0.2% on day 6 to 77.1% on day 10 and then gradually decreased to 41.3% from day 12 to day 32. Relative abundances of Candida and Wickerhamomyces reached a maximum in the first 4 d of fermentation and then declined after day 6. The relative abundance of Thermomyces was maintained at a low level (relative abundance, <0.5%) in the first 10 d of fermentation; however, it dramatically increased to 49.5% from day 10 to day 32. A temporal profile for the relative abundance of fungal taxa represented at the order level is shown in Fig. S3B in the supplemental material.
Cluster analysis indicated that bacterial community structures of MT-Daqu from day 12 to day 32 showed high similarity to those from day 0 to day 10 (Fig. 2B). Fungal community structures from day 6 to day 32 showed high similarity to those from day 0 to day 4 (Fig. 2D). Thus, the structure of MT-Daqu microbial community was relatively stable after 12 d of fermentation (Fig. 2).
Correlation between environmental variables and microbiota succession.
The Mantel test was performed to assess significance of the Spearman's correlation between the dynamics of environmental variables and the succession of the microbial community. In phase I of fermentation (day 0 to day 12), temperature correlated significantly with bacterial community succession (r = 0.71, P = 0.02), while no significant correlation was shown between moisture and titratable acidity (P > 0.05) (Table 1). All the tested environmental variables (temperature, moisture, and titratable acidity) were closely associated with the dynamics of fungal cell number (P < 0.05) (Table 1). In phase II (day 12 to day 32), no significant correlation between environmental variables (temperature, moisture, and titratable acidity) and succession of bacterial and fungal communities was observed (P > 0.05) (Table 1). Thus, temperature was determined to be an environmental factor playing a major role in phase I of MT-Daqu fermentation.
TABLE 1.
Correlations between environmental variables and microbial community succession during the fermentation process of MT-Daqu as revealed by the Mantel testa
| Environmental variable | Phase I |
Phase II |
||||||
|---|---|---|---|---|---|---|---|---|
| Bacteria |
Fungi |
Bacteria |
Fungi |
|||||
| r | P | r | P | r | P | r | P | |
| Temperature | 0.71 | 0.02 | 0.76 | 0.01 | −0.50 | 0.83 | −1.00 | 1.00 |
| Moisture | 0.18 | 0.16 | 0.67 | 0.00 | −0.50 | 0.83 | −1.00 | 1.00 |
| Titratable acidity | 0.50 | 0.11 | 0.54 | 0.03 | −0.87 | 1.00 | −0.87 | 1.00 |
r, Spearman correlation coefficient; a P value of <0.05 indicates a significant correlation. Phase I, day 0 to day 12; phase II, day 12 to day 32.
Effect of temperature on the microbiota succession.
We fitted the temperature data of 10 batches of MT-Daqu fermentation by the weighted least square method (Fig. 3). Based on the fitted temperature curves, the first 12 d of MT-Daqu fermentation could be divided into two subphases: phase I-1 (day 0 to day 4) and phase I-2 (day 4 to day 12) (Fig. 3). To further explore the effect of temperature on the shaping of MT-Daqu community assembly, species-level microbiota succession (operational taxonomic units [OTUs] clustered at 97%) was correlated to temperature dynamics in two different fermentation subphases (Fig. 4).
FIG 3.

Variation of temperature in the center of MT-Daqu in 10 batches of fermentation. Temperature data in two different phases (day 0 to 4 and day 4 to 12) were fitted separately by the weighted least square method.
FIG 4.

Heatmaps showing the succession of microbial phylotypes (OTUs clustered at 97%) and its correlation with temperature dynamics in the microbial community of MT-Daqu in the first 12 d of fermentation. (A) Bacterial community succession from day 0 to day 4; (B) bacterial community succession from day 4 to day 12; (C) fungal community succession from day 0 to day 4; and (D) fungal community succession from day 4 to day 12. Only OTUs above 0.05% at total relative abundance are shown.
In subphase I-1, the temperature of MT-Daqu increased rapidly to 55°C. The relative abundance of most bacterial taxa in MT-Daqu (except Cyanobacteria, several genera of Leuconostoc and Proteobacteria, and unclassified Bacteria) showed a positive relation with the increase in temperature (Fig. 4A). Most fungal taxa in MT-Daqu on day 0, including Alternaria, Aspergillus, Davidiella, Debaryomyces, Epicoccum, Kodamaea, Penicillium, Cryptococcus, Cystofilobasidium, and unclassified Fungi, could not grow in subphase I-1, while the relative abundance of Candida, Wickerhamomyces, and unclassified Dipodascaceae and Saccharomycetales showed a positive correlation with temperature (Fig. 4C).
At a relatively high temperature (53 to 56°C) in subphase I-2 of MT-Daqu fermentation, Bacillus, unclassified Streptophyta, Weissella, and Thermoactinomyces were maintained at high relative abundances or kept on growing, while relative abundances of Lactobacillus, Leuconostoc, and all genera in Proteobacteria decreased (Fig. 4B). Most fungal taxa except Thermoascus (OTU 223) and Thermomyces (OTU 118) could not survive during this subphase (Fig. 4D).
Validation of the driving effect of temperature on the shaping of functional microbiota.
To explore the effect of temperature on the shaping of functional microbiota, Daqu bricks after 4 d of fermentation at the core temperature of 52.5 ± 0.8°C were moved to two temperature-controlled fermentation rooms (37°C and 50°C). On the day 12 of fermentation, the temperature in the center of Daqu in the T37 group was 46.7 ± 0.5°C, while that in the T50 group was 56.4 ± 0.4°C.
Multivariate analysis based on Bray-Curtis distance matrices showed that bacterial and fungal community structures between the T37 and T50 groups were significantly different (P < 0.05) (see Fig. S4 in the supplemental material). Lactic acid bacteria related to Lactobacillus, Weissella, and Pediococcus dominated the bacterial community of MT-Daqu in the control (Ctrl) group (Fig. 5A). Lowering the fermentation temperature by placing the MT-Daqu in a 37°C incubator might decrease the relative abundance of several thermotolerant bacteria in the MT-Daqu microbiota on day 12. Relative abundances of Bacillus in the T37 group (13.1%) and T50 group (25.0%) were much higher than that in the Ctrl group (0.1%) (Fig. 5A). Relative abundance of Thermoactinomyces in the T50 group (5.2%) was obviously higher than those in the Ctrl group (undetected) and T37 group (1.7%) (Fig. 5A). The diversity and abundance of fungi were significantly different between the T37 and T50 groups (Fig. 5B). The fungal community in the Ctrl group on day 4 was mainly composed of Candida and Aspergillus. In the T37 group, the dominant fungi were Thermoascus and Aspergillus, and the relative abundance of Candida was significantly lower (0.6%) than in the Ctrl group (94.9%). In the T50 group, the major fungus was Thermoascus, and its relative abundance was above 98.0%; however, the abundances of both Candida and Aspergillus were below 0.7% (Fig. 5B). Microbial OTUs differentially distributed in MT-Daqu microbiota formed at 37°C or 50°C are shown in Tables S3 and S4 in the supplemental material. Statistically, the relative abundances of these microbes showed a significant difference (P < 0.05) between the 37°C and 50°C treatment groups.
FIG 5.

Effect of environmental temperature on the structures of bacterial community (A), fungal community (B), titratable acidity and moisture content (C), and glucoamylase and amylase activities (D) within MT-Daqu after 12 d of fermentation. MT-Daqu bricks in the fermentation room after 4 d of fermentation (Ctrl group, 52.2 ± 0.8°C) were moved to two different fermentation rooms with controlled temperatures at 50°C (T50 group) and 37°C (T37 group), respectively. Values are compared by Duncan's multiple-range test. Columns marked with different letters possess significantly different values (P < 0.05).
After 12 d of incubation, the moisture content of Daqu in the T37 group (20.3% ± 0.3%) was significantly higher than that in the T50 group (17.7% ± 0.5%) (P < 0.05). The titratable acidity in the T37 group (0.28 ± 0.02 mmol/g) was significantly lower than that in the T50 group (0.34 ± 0.01 mmol/g) (P < 0.05) (Fig. 5C). The activities of glucoamylase and amylase in the T37 group were significantly higher than those in the T50 group (P < 0.05) (Fig. 5D).
DISCUSSION
Daqu is a saccharifying and fermenting agent widely used in the traditional solid-state fermentation industry (e.g., baijiu and vinegar) (4). The fermentation of Daqu is a complex process, in which microbes reproducibly metabolize nonautoclaved raw materials and synthesize enzymes and flavor compounds. Thus, it is of interest to elucidate the mechanisms underlying the assembly and succession of the Daqu microbial community.
During the fermentation process of MT-Daqu, a total of 154 bacterial genera and 87 fungal genera were detected. The bacterial community was mainly composed of Lactobacillales (66.2%) and Bacillales (21.9%), while Saccharomycetales (44.1%) and Eurotiales (26.6%) dominated the fungal community. In previous studies based on culture-dependent methods, Lactobacillus, Acetobacter, Bacillus, Candida, Saccharomyces, Aspergillus, Rhizopus, Mucor, and Penicillium were commonly found in Daqu (4). In this study, we found that the relative abundances of Acetobacter, Mucor, and Penicillium maintained at a low level (<0.5%), while those of thermotolerant Thermoascus and Thermomyces were at a high level in the MT-Daqu microbiota (Fig. 2D). These results are similar to those of previous studies (7, 9, 16, 17). It is worth noting that the identification of unclassified bacteria (8.9%) and unclassified fungi (5.4%) relied on available annotations. As the information in reference databases (e.g., Greengenes and RDP) is improved, the taxonomic assignment of high-throughput sequences in this study will be more accurate (15, 18, 19).
Traditional food fermentation (e.g., cheese, wine, Daqu, and pickles) is usually performed in an open work environment. Thus, many environmental factors may affect the microbial diversity and flavor quality of products (4, 20, 21). Previous studies have showed that temperature, acidity, and moisture could affect the microbial composition of Daqu (16, 17, 22). In this study, we found that the increase of temperature was significantly correlated (P < 0.05) with the rapid succession of MT-Daqu microbiota in the first 12 d of fermentation (Table 1), while no significant correlation between temperature dynamics and microbiota succession was observed after 12 d of fermentation (Fig. 2). Besides temperature, it stands to reason that the acidity and humidity of Daqu bricks are also highly correlated with the succession of the fungal community (P < 0.05) (Table 1). An interactive effect between temperature, acidity, and humidity may exist during the fermentation process. The underlying mechanism of these correlations needs further study. Meanwhile, it is of interest to evaluate the effects of other environmental factors, substrate category, and operation conditions on the succession of MT-Daqu microbiota in future studies.
The effect of temperature on the shaping of MT-Daqu community assembly was further studied. Temperature dynamics in phase I of the MT-Daqu fermentation process can be further divided into two subphases: phase I-1 (day 0 to day 4) and phase I-2 (day 4 to day 12) (Fig. 3). In phase I-1, the cell numbers of bacteria and fungi rapidly increased (Fig. 1B), and the Shannon index values of the bacterial and fungal communities increased (see Tables S1 and S2 in the supplemental material). Rapid growth and metabolism of most microbes under appropriate environmental and nutritional conditions produced bio-heat within the Daqu bricks, which resulted in a sharp increase of temperature (Fig. 1). On the other hand, Alternaria and Cyanobacteria, which probably originated from raw materials, gradually disappeared in this phase (Fig. 2). Bio-heat is a by-product of microbial metabolism during the fermentation process. An increase in temperature has been shown to be correlated with rates of CO2 production, which can be taken as concrete evidence of the oxidation process (23). In phase I-2, the temperature of Daqu bricks was maintained at 53 to 56°C (Fig. 1A). Endogenous bio-heat inhibited the growth of most microbes, while thermotolerant taxa, including Bacillus, unclassified Streptophyta, Weissella, Thermoactinomyces, Thermoascus, and Thermomyces could survive or keep on growing (Fig. 4). The descending trends of Shannon index values and microbial cell numbers could also support these microbial dynamics (Fig. 1B; see also Tables S1 and S2). This type of bio-heat enrichment (rapid temperature increase, sustained high temperature, and gradual cooling of Daqu) has also been shown in the microbial community succession in composting and ensiling (24, 25). In phase I-2, many thermotolerant microorganisms survived in the MT-Daqu that could directly participate in the fermentation of grains during the production of baijiu and vinegar; they could also secrete various enzymes such as α-amylase, glucoamylase, proteases, xylanase, CMCase, and β-glucosidase (26–30) to transform starches and proteins to unique flavors (4).
Artificially lowering the temperature in the fermentation room to 37°C resulted in a decrease of the internal temperature of Daqu on day 12 (46.7 ± 0.5°C) that was correlated with the decrease of relative abundances of thermotolerant taxa, including Bacillus, Thermoactinomyces, and Thermoascus (Fig. 5A and B). The results confirmed that the bio-heat produced within Daqu bricks was associated with the composition and function of Daqu microbiota. In addition, artificially lowering the room temperature also resulted in the significant differences between the T37 and T50 groups of Daqu in moisture content, titratable acidity, and activities of glucoamylase and amylase (Fig. 5C and D). Thus, bio-heat is likely to be the primary driver in the progression of Daqu microbiota, but the way the progression takes place might be via other factors such as moisture or acidity. Moreover, ventilation also disturbed the air composition in the fermentation room; an alleviated O2/CO2 ratio could also change the Daqu microbiota. Further studies are needed to explore the effects of other intrinsic and extrinsic factors and their interactive effects on the shaping of functional Daqu microbiota.
To conclude, our study revealed that bio-heat was produced within Daqu bricks in the first 4 d of fermentation, concomitant with rapid microbial propagation. Bio-heat then functioned as a primary endogenous driver promoting the formation of MT-Daqu microbiota from day 4 to day 12. Artificially lowering the bio-heat (37°C) in the Daqu bricks decreased the relative abundances of thermotolerant taxa, including Bacillus, Thermoactinomyces, and Thermoascus in the MT-Daqu microbiota. The bio-heat-driven growth of thermotolerant microorganisms in the MT-Daqu might contribute to the formation of flavor metabolites. This study provides useful information for temperature-based modulation of microbiota function in the production of Daqu.
MATERIALS AND METHODS
Sample collection.
MT-Daqu samples were collected at different time point during fermentation (days 0, 2, 4, 6, 8, 10, 12, 17, 27, and 32) in January 2013 and September 2016 from Luzhou Laojiao Group Co. Ltd. (Sichuan, China). Three MT-Daqu bricks collected at the same time point were mashed to powder, mixed in sterile plastic bags (about 1,000 g), and stored in −80°C before further analysis.
Analyses of environmental variables.
Temperature in the center of Daqu bricks during the fermentation process was monitored using a thermometer. Moisture of Daqu was determined with a gravimetric method by drying MT-Daqu powders at 105°C for 30 min. Titratable acidity, defined as the amount of sodium hydroxide (mmol) consumed per gram of Daqu, was determined using standard sodium hydroxide solution with phenolphthalein as an indicator. All samples were measured in triplicate.
Enzyme assays.
For evaluating the activity and quality of Daqu samples, the activities of amylase (liquefying power) and glucoamylase (saccharifying power) were determined according to the general methods of analysis for Daqu (31). Briefly, a Daqu sample (10 g) was dispersed in a 200 ml sodium acetate buffer (200 mM, pH 4.6) and shaken at 300 rpm and 37°C for 1 h. The filtrate (enzyme source) was obtained using filter papers (Whatman No. 1). Glucoamylase and amylase activities were determined by incubating a mixture of enzyme source and 2% (mass/vol) soluble starch dissolved at 35°C. One unit of glucoamylase activity was defined as the amount of starter required for the liberation of 1 mg glucose per hour under the described conditions. One unit of amylase activity was defined as the amount of starter required for the liquefaction of 1 g starch per hour under the described conditions. Amylase-specific activity was reported as the amount of starch liquefied per hour by 1 g dried Daqu. Glucoamylase-specific activity was reported as the amount of glucose released from soluble starch per hour by 1 g dried Daqu.
Validation of driving effect of temperature on MT-Daqu microbiota formation.
After 4 d of fermentation, the MT-Daqu bricks (Ctrl group) at a temperature of 52.2 ± 0.8°C were moved to two different fermentation rooms with controlled temperatures at 50°C (T50 group) and 37°C (T37 group) separately. During the subsequent fermentation period (day 4 to day 12), temperatures in the fermentation rooms were controlled by mechanical aeration. Microbial community structures of MT-Daqu in the Ctrl, T50, and T37 groups were analyzed by Illumina MiSeq sequencing.
DNA extraction.
An MT-Daqu sample (1.5 g) was ground into a fine powder by using a mortar with liquid nitrogen and mixed with 1.5 ml of cetyltrimethylammonium bromide (CTAB) extraction buffer (pH 8.0; 2% [wt/vol] CTAB, 1.4 mol/liter NaCl, 20 mmol/liter EDTA, 100 mmol/liter Tris-HCl) and 30 μl mercaptoethanol. The mixture was shaken for 30 min at 65°C. Proteinase K was added with a final concentration of 100 μg/ml, and the resulting mixture was incubated at 37°C for 30 min with constant shaking at 220 rpm, and then was centrifuged at 6,000 × g for 10 min. DNA was purified by one round of extraction with phenol-chloroform-isoamyl alcohol (25:24:1). This step was followed by two rounds of extraction with chloroform-isoamyl alcohol (24:1). The resulting samples were incubated with isopropanol at −20°C for 1 h, and the DNA precipitation was collected by centrifugation at 17,400 × g for 10 min. The DNA pellet was washed twice with 70% (vol/vol) ethanol, and after ethanol was completely volatilized the pellet was resuspended in 100 μl Tris-EDTA buffer solution (pH 8.0).
Quantitative real-time PCR (qPCR).
qPCR were performed in a CFX Connect Real-Time system (Bio-Rad), and the SYBR Select master mix for CFX was provided by Life Technologies (USA). Primers Eub338 (5′-ACTCCTACGGGAGGCAGCAG-3′) and Eub518 (5′-ATTACCGCGGCTGCTGG-3′) were used for bacterial cell number analysis, and primers ITS1f (5′-TCCGTAGGTGAACCTGCGG-3′) and 5.8s (5′-CGCTGCGTTCTTCATCG-3′) were used for fungal cell number analysis (32). Each reaction was performed in a reaction volume of 20 μl containing 10 μl SYBR mix, 1 μl of each primer (10 mmol/liter) and 1 μl of DNA template. The qPCR thermocycling steps were set as follows: 95°C for 3 min and 50 cycles of 95°C for 15 s, 54°C for 15 s, and 72°C for 15 s. Melting curve analysis, which was obtained by 0.5°C/s increments from 65 to 95°C with continuous fluorescence collection, was performed to determine the specificity of the PCR products (33).
Bacterial 16S rRNA gene amplification and 454 pyrosequencing.
PCR amplicon libraries of the 16S rRNA gene V1 to V3 hypervariable region were generated for each Daqu sample. PCR was performed using the primers with sequences as follows: forward primer (5′-NNNNNNN-TGGAGAGTTTGATCCTGGCTCAG-3′) and reverse primer (5′-NNNNNNN-TACCGCGGCTGCTGGCAC-3′). Unique multinucleotide sequences (listed as “N” in the sequence) were synthesized at the 5′ end of each pair of primers as barcodes to assign sequences to different samples (34).
The PCR products were purified by the method of Huang et al. (34) and the purified amplicons were pooled in equimolar ratios. Pyrosequencing of PCR amplification was carried out by the Genome Sequencer FLX Titanium.
Amplification and Illumina MiSeq sequencing of bacterial 16S rRNA genes and fungal ITS rRNA genes.
The V3 to V4 region of the bacterial 16S rRNA gene was amplified using the primers 338F (5′-NNNNNNNN-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (38). Unique multinucleotide sequences (listed as “N” in the sequence) were synthesized at the 5′ end of primer 338F as barcodes to assign sequences to different samples. The ITS1 regions of the ITS rRNA genes were amplified using fungal-specific primer ITS1F (5′-NNNNNNNN-CTTGGTCATTTAGAGGAAGTAA-3′) and fungal-universal primer ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) (35–37). Unique multinucleotide sequences (listed as “N” in the sequence) were synthesized at the 5′ end of primer ITS1F as barcodes to assign sequences to different samples. The PCR products were purified according to the methods of Zhang et al. (35) and Hong et al. (38). The purified amplicons were pooled in equimolar ratios, and pyrosequencing of PCR amplification was carried out by the Illumina MiSeq platform.
Data processing and analyses.
Raw sequences from the 454 pyrosequencing platform were analyzed with MOTHUR (version 1.22) for preprocessing, taxonomic assignment, and community structure comparison (39). The raw reads were removed if they met all of the following criteria: shorter than 150 bp, average quality score <20, with an ambiguous base call (N); with any homopolymers of more than eight bases or did not contain the primer sequence; reads were sorted by the tag sequences (34). The preclustering was performed and “contaminant steps” were removed to reduce sequencing noise from pyrosequencing data. Trimmed reads were assigned to clusters using the UCLUST algorithm (http://www.drive5.com/usearch/index.html). A Perl script was used to convert UCLUST output into a format recognized by MOTHUR (34, 39) for further analysis. Reads were assigned to operational taxonomical units (OTUs) (at 3% difference), and moreover, calculation of coverage percentage (Good's coverage), Shannon's diversity index, species richness estimators (Abundance-based Coverage Estimator [ACE] and Chao1), and rarefaction analysis were performed in MOTHUR. The classify.seqs script in MOTHUR was used to classify all trimmed reads. The confidence score threshold was set to 0.8, and sequences with a bootstrap value below 0.8 were assigned to the unclassified category.
Raw sequences from the Illumina MiSeq sequencing platform were demultiplexed and quality-filtered using QIIME (version 1.8.0). The raw reads were processed according to the following criteria: (i) The 300-bp reads were truncated at any site that obtained an average quality score of <20 over a 10-bp sliding window, and truncated reads shorter than 50 bp were discarded; (ii) exact barcode matches, 2-nucleotide mismatches in primer matching, and reads containing ambiguous characters were removed; and (iii) only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence (38). Reads were assigned to OTUs (at 3% difference) using UCLUST. We picked a representative sequence for each OTU and used the RDP classifier to annotate taxonomic information for each representative sequence (35). The confidence score threshold was set to 0.8 and sequences with a bootstrap value below 0.8 were assigned to the unclassified category. Calculation of coverage percentage (Good's coverage), Shannon's diversity index, species richness estimators (ACE and Chao1), and rarefaction analysis were performed in MOTHUR.
Statistical analysis.
The Mantel test was used to assess a Spearman's correlation between entries of the dissimilarity matrix of microbial community succession and the distance matrix of environmental variables in R (version 3.2.4) via the vegan package (version 2.3-4). Distance matrices for relative abundance of bacteria and fungi OTUs were produced by Bray-Curtis dissimilarity transformation. Euclidian distance matrices (P < 0.05) for the environmental variables were calculated.
Spearman's rank correlation was used to analyze the correlation between microbial community succession and temperature dynamics during the MT-Daqu fermentation process in MOTHUR (version 1.35.1).
To compare the structures of bacterial and fungal communities within MT-Daqu treated with different temperatures (37°C and 50°C), OTUs that were differentially distributed (relative abundance) between groups were statistically detected using Metastats via a web interface (http://metastats.cbcb.umd.edu/detection.html) (40). Metastats analysis was performed using 1,000 permutations to compute P values in statistical tests. Analysis of molecular variance (AMOVA) was used to test whether communities from different treatment groups had the same centroid (41, 42). A P value of <0.05 was used as the cutoff for significance.
Statistical differences of the titratable acidity, moisture content, and glucoamylase and amylase activities among the controlled-temperature Daqu samples were obtained by the application of one-way analysis of variance (ANOVA) followed by Duncan's test. A P value of <0.05 was used as the cutoff for significance.
Accession number(s).
The original MT-Daqu sequencing data sets have been deposited in the GenBank database (accession no. PRJNA286831).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Key Research and Development Program of China (no. 2016YFD0400505), a grant from the Key Project of the National Nature Science Foundation of China (no. 31530055), and two grants from the National Engineering Research Center of Solid-State Brewing (no. 2017K-80 and 2017K-79).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01550-17.
REFERENCES
- 1.Chen B, Wu Q, Xu Y. 2014. Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int J Food Microbiol 179:80–84. doi: 10.1016/j.ijfoodmicro.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 2.Wu Q, Xu Y, Chen L. 2012. Diversity of yeast species during fermentative process contributing to Chinese Maotai-flavour liquor making. Lett Appl Microbiol 55:301–307. doi: 10.1111/j.1472-765X.2012.03294.x. [DOI] [PubMed] [Google Scholar]
- 3.Wu XH, Zheng XW, Han BZ, Vervoort J, Robert Nout MJ. 2009. Characterization of Chinese liquor starter, “Daqu”, by flavor type with 1H NMR-based nontargeted analysis. J Agric Food Chem 57:11354–11359. doi: 10.1021/jf902881p. [DOI] [PubMed] [Google Scholar]
- 4.Zheng XW, Tabrizi MR, Robert Nout MJ, Han BZ. 2011. Daqu—a traditional Chinese liquor fermentation starter. J Inst Brewing 117:82–90. doi: 10.1002/j.2050-0416.2011.tb00447.x. [DOI] [Google Scholar]
- 5.Wu Q, Chen LQ, Xu Y. 2013. Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int J Food Microbiol 166:323–330. doi: 10.1016/j.ijfoodmicro.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhang CL, Ao ZH, Chui WQ, Shen CH, Tao WY, Zhang SY. 2012. Characterization of the aroma-active compounds in Daqu: a tradition Chinese liquor starter. Eur Food Res Technol 234:69–76. doi: 10.1007/s00217-011-1616-4. [DOI] [Google Scholar]
- 7.Wang HY, Gao YB, Fan QW, Xu Y. 2011. Characterization and comparison of microbial community of different typical Chinese liquor Daqus by PCR-DGGE. Lett Appl Microbiol 53:134–140. doi: 10.1111/j.1472-765X.2011.03076.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang WX, Qiao ZW, Shigematsu T, Tang YQ, Hu C, Morimura S, Kida K. 2005. Analysis of the bacterial community in Zaopei during production of Chinese Luzhou-flavor liquor. J Inst Brewing 111:215–222. doi: 10.1002/j.2050-0416.2005.tb00669.x. [DOI] [Google Scholar]
- 9.Wolfe BE, Dutton RJ. 2015. Fermented foods as experimentally tractable microbial ecosystems. Cell 161:49–55. doi: 10.1016/j.cell.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 10.Zheng XW, Yan Z, Han BZ, Zwietering MH, Samson RA, Boekhout T, Robert Nout MJ. 2012. Complex microbiota of a Chinese “Fen” liquor fermentation starter (Fen-Daqu), revealed by culture-dependent and culture-independent methods. Food Microbiol 31:293–300. doi: 10.1016/j.fm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Yan Z, Zheng XW, Han BZ, Han JS, Robert Nout MJ, Chen JY. 2013. Monitoring the ecology of Bacillus during Daqu incubation, a fermentation starter, using culture-dependent and culture-independent methods. J Microbiol Biotechnol 23:614–622. doi: 10.4014/jmb.1211.11065. [DOI] [PubMed] [Google Scholar]
- 12.Zhu BF, Xu Y, Fan WL. 2010. High-yield fermentative preparation of tetramethylpyrazine by Bacillus sp. using an endogenous precursor approach. J Ind Microbiol Biotechnol 37:179–186. doi: 10.1007/s10295-009-0661-5. [DOI] [PubMed] [Google Scholar]
- 13.Yao WC, Tang YM, Ren DQ, Liao JM, Shen CH, Xu DF, Ying H, Fan L. 2005. Study on the differences of microbes in the different layers of Guojiao Daqu. Liquor Making 32:35–37. (In Chinese.) [Google Scholar]
- 14.Bengtsson MM, Sjøtun K, Lanzén A, Øvreås L. 2012. Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J 6:2188–2198. doi: 10.1038/ismej.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ercolini D. 2013. High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology. Appl Environ Microbiol 79:3148–3155. doi: 10.1128/AEM.00256-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P, Lin WF, Liu X, Wang XW, Luo LX. 2016. Environmental factors affecting microbiota dynamics during traditional solid-state fermentation of Chinese Daqu starter. Front Microbiol 7:1237. doi: 10.3389/fmicb.2016.01237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang HY, Xu Y. 2015. Effect of temperature on microbial composition of starter culture for Chinese light aroma style liquor fermentation. Lett Appl Microbiol 60:85–91. doi: 10.1111/lam.12344. [DOI] [PubMed] [Google Scholar]
- 18.Wu LH, Lu ZM, Zhang XJ, Wang ZM, Yu YJ, Shi JS, Xu ZH. 2017. Metagenomics reveals flavour metabolic network of cereal vinegar microbiota. Food Microbiol 62:23–31. doi: 10.1016/j.fm.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Lu ZM, Liu N, Wang LJ, Wu LH, Gong JS, Yu YJ, Li GQ, Shi JS, Xu ZH. 2016. Elucidating and regulating the acetoin production role of microbial functional groups in multispecies acetic acid fermentation. Appl Environ Microbiol 82:5860–5868. doi: 10.1128/AEM.01331-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung JY, Lee SH, Kim JM, Park MS, Bae JW, Hahn Y, Madsen EL, Jeon CO. 2011. Metagenomic analysis of kimchi, a traditional Korean fermented food. Appl Environ Microbiol 77:2264–2274. doi: 10.1128/AEM.02157-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfe BE, Button JE, Santarelli M, Dutton RJ. 2014. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158:422–433. doi: 10.1016/j.cell.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng XW, Yan Z, Robert Nout MJ, Smid EJ, Zwietering MH, Boekhout T, Han JS, Han BZ. 2014. Microbiota dynamics related to environmental conditions during the fermentative production of Fen-Daqu, a Chinese industrial fermentation starter. Int J Food Microbiol 182:57–62. doi: 10.1016/j.ijfoodmicro.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Hubbe MA, Nazhad M, Sánchez C. 2010. Composting as a way to convert cellulosic biomass and organic waste into high-value soil amendments: a review. BioRes 5:2808–2854. [Google Scholar]
- 24.Wang X, Cui H, Shi J, Zhao X, Zhao Y, Wei Z. 2015. Relationship between bacterial diversity and environmental parameters during composting of different raw materials. Bioresour Technol 198:395–402. doi: 10.1016/j.biortech.2015.09.041. [DOI] [PubMed] [Google Scholar]
- 25.Jones CM, Heinrichs AJ, Roth GW, Ishler VA. 2004. From harvest to feed: understanding silage management. Pennsylvania State University College of Agricultural Sciences, University Park, Pennsylvania. [Google Scholar]
- 26.Ghribi D, Abdelkefi-Mesrati L, Mnif I, Kammoun R, Ayadi I, Saadaoui I, Maktouf S, Chaabouni-Ellouze S. 2012. Investigation of antimicrobial activity and statistical optimization of Bacillus subtilis SPB1 biosurfactant production in solid-state fermentation. J Biomed Biotechnol 2012:373682. doi: 10.1155/2012/373682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schallmey M, Singh A, Ward OP. 2004. Developments in the use of Bacillus species for industrial production. Can J Microbiol 50:1–17. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- 28.Yoon JH, Kim IG, Shin YK, Park YH. 2005. Proposal of the genus Thermoactinomyces sensu stricto and three new genera, Laceyella, Thermoflavimicrobium and Seinonella, on the basis of phenotypic, phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 55:395–400. doi: 10.1099/ijs.0.63203-0. [DOI] [PubMed] [Google Scholar]
- 29.Collins T, Gerday C, Feller G. 2005. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29:3–23. doi: 10.1016/j.femsre.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Parry NJ, Beever DE, Owen E, Vandenberghe I, Van Beeumen J, Bhat MK. 2001. Biochemical characterization and mechanism of action of a thermostable β-glucosidase purified from Thermoascus aurantiacus. Biochem J 353:117–127. doi: 10.1042/bj3530117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ministry of Light Industry of China. 2011. General methods of analysis for Daqu (QB/T 4257-2011). China Light Industry Press, Beijing, China. [Google Scholar]
- 32.Fierer N, Jackson JA, Vilgalys R, Jackson RB. 2005. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol 71:4117–4120. doi: 10.1128/AEM.71.7.4117-4120.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu W, Huang ZY, Zhang XJ, Li Q, Lu ZM, Shi JS, Xu ZH, Ma YH. 2011. Monitoring the microbial community during solid-state acetic acid fermentation of Zhenjiang aromatic vinegar. Food Microbiol 28:1175–1181. doi: 10.1016/j.fm.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Huang S, Yang F, Zeng XW, Chen J, Li R, Wen T, Li C, Wei W, Liu JQ, Chen L, Davis C, Xu J. 2011. Preliminary characterization of the oral microbiota of Chinese adults with and without gingivitis. BMC Oral Health 11:33. doi: 10.1186/1472-6831-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang LK, Kang MY, Huang YC, Yang LX. 2016. Fungal communities from the calcareous deep-sea sediments in the Southwest India Ridge revealed by Illumina sequencing technology. World J Microbiol Biotechnol 32:78. doi: 10.1007/s11274-016-2030-7. [DOI] [PubMed] [Google Scholar]
- 36.Gardes M, Bruns TD. 1993. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 37.White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p 315–322. In Innis MA, Gelfand DH, Sninsky JJ, White T (ed), PCR protocols and applications: a laboratory manual. Elsevier, London, United Kingdom. [Google Scholar]
- 38.Hong C, Si Y, Xing Y, Li Y. 2015. Illumina MiSeq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas. Environ Sci Pollut Res 22:10788–10799. doi: 10.1007/s11356-015-4186-3. [DOI] [PubMed] [Google Scholar]
- 39.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White JR, Nagarajan N, Pop M. 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Excoffier L, Smouse PE, Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schloss PD. 2008. Evaluating different approaches that test whether microbial communities have the same structure. ISME J 2:265–275. doi: 10.1038/ismej.2008.5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.