

Abstract
G protein–coupled receptor kinases (GRKs) phosphorylate activated receptors to promote arrestin binding, decoupling from heterotrimeric G proteins, and internalization. GRK2 and GRK5 are overexpressed in the failing heart and thus have become therapeutic targets. Previously, we discovered two classes of GRK2-selective inhibitors, one stemming from GSK180736A, a Rho-associated coiled-coil containing kinase 1 (ROCK1) inhibitor, the other from paroxetine, a selective serotonin-reuptake inhibitor. These two classes of compounds bind to the GRK2 active site in a similar configuration but contain different hinge-binding “warheads”: indazole and benzodioxole, respectively. We surmised from our prior studies that an indazole would be the stronger hinge binder and would impart increased potency when substituted for benzodioxole in paroxetine derivatives. To test this hypothesis, we synthesized a series of hybrid compounds that allowed us to compare the effects of inhibitors that differ only in the identity of the warhead. The indazole-paroxetine analogs were indeed more potent than their respective benzodioxole derivatives but lost selectivity. To investigate how these two warheads dictate selectivity, we determined the crystal structures of three of the indazole hybrid compounds (CCG224061, CCG257284, and CCG258748) in complex with GRK2–Gβγ. Comparison of these structures with those of analogous benzodioxole-containing complexes confirmed that the indazole-paroxetine hybrids form stronger interactions with the hinge of the kinase but also stabilize a distinct conformation of the kinase domain of GRK2 compared with previous complexes with paroxetine analogs. This conformation is analogous to one that can be assumed by GRK5, at least partially explaining the loss in selectivity.
Introduction
Heart failure is broadly described as an inability of the heart to effectively pump and supply blood to the body. In response to insufficient blood flow, the sympathetic nervous system produces increased amounts of catecholamines to stimulate β-adrenergic receptors, which are part of the G protein–coupled receptor (GPCR) superfamily (Salazar et al., 2007; Belmonte and Blaxall, 2011); however, prolonged stimulation of GPCRs initiates the upregulation and phosphorylation by GPCR kinases (GRKs), which targets these receptors for arrestin binding and internalization (Ferguson et al., 1996; Ribas et al., 2007). Receptor internalization reduces the number of receptors present on the cell surface, thereby diminishing the responsiveness to agonists (Ferguson et al., 1996).
The seven different GRKs found in humans are divided into the GRK1 (GRK1 and 7), GRK2 (GRK2 and 3), and GRK4 (GRK4, 5, and 6) subfamilies, all of which belong to the protein kinase A (PKA), G, and C family of Ser/Thr protein kinases. GRKs 2 and 5 are the dominant GRKs in the myocardium and are upregulated during heart failure (Dzimiri et al., 2004; Montó et al., 2012). A series of studies have shown that selective inhibition of GRK2 activity is a promising approach to improving cardiac function during heart failure (Rockman et al., 1998; Raake et al., 2013; Schumacher et al., 2015).
We previously identified paroxetine, a Food and Drug Administration–approved serotonin-reuptake inhibitor, and GSK180736A (Fig. 1) as GRK2 inhibitors (Thal et al., 2012; Homan et al., 2015). Cocrystal complexes of these compounds bound to GRK2 revealed that they both bind similarly in the active site (Fig. 2A) (Thal et al., 2012; Homan et al., 2015). The A rings of both compounds occupy the adenine subsite and bind the hinge (Fig. 2A). The core of the scaffold (ring B), either a dihydropyrimidinone (GSK180736A) or piperidine (paroxetine), occupies the ribose subsite, and the C ring occupies the polyphosphate subsite; however, neither occupies the hydrophobic pocket of the active site, as observed with the Takeda compound (Fig. 2B), known as CMPD101 or compound 115h (Ikeda et al., 2007; Thal et al., 2011; Okawa et al., 2017). We subsequently used a rational drug design approach to generate derivatives of both GSK180736A and paroxetine with substituents appended onto the C ring via an amide linker to occupy the hydrophobic pocket (Waldschmidt et al., 2016, 2017).
Fig. 1.

Chemical structures of GRK2 inhibitor scaffolds and activity against GRK2, GRK5, and ROCK1, expressed as either IC50 or percent inhibition.
Fig. 2.

Comparison of GRK2 inhibitor scaffolds bound in the GRK2 active site. (A) Overlay of paroxetine (gray carbons) and GSK180736A (purple carbons) bound to the GRK2–Gβγ complex (PDB IDs: 3V5W and 4PNK, respectively). (B) Overlay of paroxetine (gray carbons) and Takeda CMPD101 (light blue carbons) bound to the GRK2–Gβγ complex (PDB IDs: 3V5W and 3PVW, respectively). Ring systems are labeled according to the A, B, C, and D convention shown in Fig. 1. Nitrogens are colored blue, oxygens red, and fluorines cyan.
We hypothesized that, owing to the similar binding modes of GSK180736A and paroxetine (Fig. 2), their structure-activity relationships would be translatable. Although this turned out generally not to be the case, overall, the addition of amide-linked D-ring substituents to these scaffolds did lead to increases in potency in each class. In the case of GSK180736A, several substituents enabled us to build in selectivity for GRK2 over other kinases, especially Rho-associated coiled-coil containing kinase 1 (ROCK1), for which this compound was originally designed to inhibit (Sehon et al., 2008). We were also interested in understanding whether the hinge-binding moiety, an indazole in the case of GSK180736A and a benzodioxole in the case of paroxetine, was interchangeable; however, in the case of the GSK180736A scaffold, exchanging indazole for a benzodioxole resulted in >600-fold loss of activity (Waldschmidt et al., 2016).
We next hypothesized that exchanging the benzodioxole moiety in the paroxetine scaffold with an indazole would instead result in increased potency owing to its stronger interactions with the hinge (two hydrogen bonds for indazole vs. a hydrogen bond and a carbon-oxygen hydrogen bond for benzodioxole). Thus, we synthesized a series of paroxetine hybrids in which the benzodioxole ring was exchanged for indazole. These indazole-paroxetine hybrids in general showed an increase in GRK2 potency relative to their benzodioxole counterparts but lost selectivity. To understand the molecular basis for enhanced potency and loss of selectivity, we crystallized several of these hybrid compounds in complex with GRK2–Gβγ. These and prior crystallographic studies have shown that the indazole warhead locks the kinase domain into a specific conformation that leads to one particular crystal form, whereas the benzodioxole-based compounds lead to a variety of crystal forms with subtly different conformations of the kinase domain. Our results indicate that the identity and strength of the hinge-binding moiety are important for dictating the overall conformation of the GRK2 kinase domain and that weaker hinge interactions allow access to a broader distribution of subtly different conformational states. The selectivity generally exhibited by benzodioxole-based compounds for GRK2 may therefore result from the ability of its kinase domain to access conformational states not readily available to closely related enzymes.
Materials and Methods
Synthesis.
Synthetic procedures are provided in the Supplemental Materials. 1H nuclear magnetic resonance spectra were taken in dimethylsulfoxide -d6, MeOD, or CDCl3 at room temperature on Varian Inova 400 MHz or Varian Inova 500-MHz instruments. Reported chemical shifts for the 1H nuclear magnetic resonance spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Mass spectrometry data were measured using a Waters Corporation (Milford, MA) Micromass LCT or Agilent 6230 Q-TOF (Agilent Technologies, Santa Clara, CA). High-performance liquid chromatography was used to determine the purity of biologically tested compounds on an Agilent 1100 series with an Agilent Zorbax Eclipse Plus C18 column. A gradient of 10%–90% acetonitrile/water over 6 min, followed by 90% acetonitrile/water for 7 min, was used with detection at 254 nm. All tested compounds had purity >95%.
Kinase Assays.
PKA and ROCK1 inhibition was assayed using the ADP-Glo Kinase Assay system (Promega, Madison, WI), as previously described (Homan et al., 2015). Compounds were tested against protein kinase A (PKA) in duplicate using an eight-point concentration range, and the experiment was repeated in triplicate on separate days. Compounds were screened at 10 μM for ROCK1 inhibition in triplicate, the experiment was repeated in triplicate on separate days, and the percent inhibition was calculated. A BMG Labtech PHERAstar imaging system (BMG LABTECH, Ortenberg, Germany) was used to measure luminescence. Inhibition of GRK1, 2, and 5 was determined in 20 mM HEPES, pH 7.0, 2 mM MgCl2, and 0.025% n-Dodecyl-β-D-maltoside with 50 nM of the respective GRK and 500 nM tubulin. Kinetic reactions were initiated by the addition of [γ-32P]ATP (500 μCi, 5 μM), allowed to proceed for 5 min, and quenched by the addition of SDS loading buffer. Each compound was tested in duplicate using an eight-point concentration range, and the experiment was repeated in triplicate on separate days. Samples were then separated by SDS-PAGE, and gels were dried and exposed with a phosphor-imaging screen. The images were then scanned with a Typhoon imager and quantified using ImageQuant (GE Healthcare Life Sciences, Pittsburgh, PA) as previously described (Thal et al., 2012). Data were fit to a three-parameter dose-inhibitor response curve with a fixed Hill slope of 1 and the bottom constrained to 0 using GraphPad Prism (GraphPad Software, San Diego, CA). A maximum of two outliers were removed per individual data set, and in some cases, an entire data set was excluded from the analysis.
Mouse Liver Microsome Assays.
Metabolic stability was determined using CD-1 mouse liver microsomes. Reactions consisted of 1 μM compound, 0.5 mg/ml microsomes, 1.7 mM NADPH in 0.1 M phosphate buffer, pH 7.4, supplemented with 3.3 mM MgCl2, and incubated at 37°C. Aliquots of 40 μl were taken at 0, 5, 10, 15, 30, 45, and 60 min and quenched in 3 volumes of cold acetonitrile containing 100 ng/ml internal standard. Samples were centrifuged at 15,000 rpm for 10 min, and the supernatant was analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Protein Expression and Purification.
Human GRK2 S670A with a C-terminal hexahistidine tag was expressed in High-Five Cells (Boyce Thompson Institute for Plant Research, Ithaca, NY) using the Bac-to-Bac insect cell expression system (Life Technologies, Carlsbad, CA). Cells were harvested 48 h postinfection and lysed. GRK2 was purified from the clarified lysate, as described previously for GRK1 using nickel-nitrilotriacetic acid affinity and cation-exchange chromatography (Singh et al., 2008). Fractions containing GRK2 were pooled and further purified on a Sephadex 200 column into 20 mM HEPES, pH 7.5, 100 mM NaCl, and 1 mM DTT. Soluble human Gβ1γ2 (C68S mutant) containing an N terminal hexahistidine tag on the Gβ1 subunit was expressed using a dual-promoter insect cell-expression vector in High-Five cells. The cells were harvested 48 h postinfection and lysed. Gβ1γ2 was purified from the clarified lysate, as described previously, using nickel-nitrilotriacetic acid affinity and anion exchange chromatography (Kozasa, 2004). Fractions containing Gβ1γ2 were pooled and further purified on a Sephadex 200 column into 20 mM HEPES, pH 8.0, 100 mM NaCl, and 1 mM DTT.
Crystal Structure Determination.
Purified GRK2 (S670A) and soluble Gβ1γ2 (C68S) were mixed in a 1.2:1 molar ratio with a final protein concentration of approximately 10 mg/ml. Inhibitor (500 μM) and MgCl2 (2 mM) were added to the protein mixture and allowed to incubate on ice for 30 min before filtration through a 0.2 μm Nanosep centrifugal device (Pall Laboratory, Port Washington, NY). Inhibitor complexes were crystallized as previously described by hanging drop vapor diffusion at 4°C with drops consisting of 0.8 μl of protein and 0.8 μl of reservoir solution, which consisted of 50 mM MES, pH 6.0, 0.8–1.2 M NaCl, and 8%–16% PEG3350 (Thal et al., 2011, 2012). Crystals generally appeared after 2–3 d and grew for 1 to 2 weeks. Crystals were harvested in a cryoprotectant solution consisting of the reservoir solution supplemented with 25% ethylene glycol and 500 μM inhibitor before being flash-frozen in liquid nitrogen. Diffraction data were collected on the LS-CAT beamlines 21-ID-G and 21-ID-D at wavelengths of 0.97857 and 1.0332 Å, respectively. Data integration and scaling were performed with XDS (Sauter et al., 2013) or DIALS (Gildea et al., 2014). The structures were solved using Phaser (McCoy et al., 2007) with PDB ID 4PNK as the search model. Reciprocal-space refinement was performed with PHENIX (Afonine et al., 2012) and alternated with local real-space refinement and model-building using Coot (Emsley and Cowtan, 2004; http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/). Crystal refinement statistics are listed in Supplemental Table 1. The final models were validated using MolProbity (Chen et al., 2010; http://molprobity.biochem.duke.edu/) before deposition in the Protein Data Bank under accession codes: 5WG3, 5WG4, and 5WG5.
Principal Component Analysis.
Principal component (PC) analysis has been previously performed for the kinase domains of PKA, GRK1, GRK4 family members, and GRK2, which were previously deposited on the Protein Data Bank (Yao et al., 2017). The kinase domains were aligned using core residues identified to be structurally invariant. Previous structural analysis of 49 GRK and 201 PKA structures revealed two distinct structural motions, which were grouped into PC1 (72% of the structural variance) and PC2 (approximately 10% of the structural variance). These structural motions corresponded to an opening and closing of the kinase domain mediated by the hinge connecting the small and large lobes (PC1) and a twisting motion between these domains (PC2). The GRK2 structures from this study were incorporated into the same analysis to benchmark their conformational states with respect to these previously published structures.
Myocyte-Shortening Assays.
Mouse cardiomyocytes were isolated from left-ventricular free wall and septum of C57/Bl6 mice as previously described (Song et al., 2008). Cells were used within 2–8 h of isolation and plated on laminin-coated coverslips and bathed in HEPES-buffered (20 mM, pH 7.4) medium 199 containing 1.8 mM extracellular Ca2+. Coverslips were then mounted in the Dvorak-Stotler chamber for recording and bathed in 0.7 ml of fresh medium. Imaging was performed with a variable field-rate camera (Zeiss IM35, Ionoptix, Westwood, MA) using edge detection and sarcomere length. Peak contraction was measured as the percentage of cell shortening. Cells were paced at 1 Hz and treated with isoproterenol (0.5 μM) for 2 min, with pretreatment of either PBS as vehicle or CCG224061 (0.5 and 1 μM) for 10 min and the contractions recorded (Thal et al., 2012).
Mouse Pharmacokinetic (PK) Studies.
All animal experiments were approved and conducted in accordance with standards set by the University of Michigan Committee on the Use and Care of Animals and Unit for Laboratory Animal Medicine. Preliminary PK of CCG224061 was determined in female CD-1 mice after intraperitoneal injection at 10 mg/kg. The compound was dissolved in 15% (v/v) dimethylsulfoxide, 15% (v/v) PEG-400, and 70% (v/v) phosphate-buffered saline. Blood samples of 50 μl were collected at 0.5, 2, 4, and 7 h; centrifuged at 3500 rpm for 10 min; and the plasma frozen at −80°C for later analysis. Plasma compound concentrations were quantified using LC-MS/MS. The LC-MS/MS method consisted of a Shimadzu high-performance liquid chromatography system with a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm) for chromatographic separation of the compound. An AB Sciex QTrap 4500 mass spectrometer was equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring mode for detection. All PK parameters were calculated by noncompartmental methods using WinNonLin software, version 3.2 (Pharsight Corporation, Mountain View, CA).
Results
Synthesis.
Paroxetine hybrid compounds in which there is an indazole in place of the benzodioxole were prepared by a convergent approach (Scheme 1). Intermediate 7b was synthesized from commercially available 2-fluoro-5-methylphenol 1. Nitration of compound 1 proceeded first through the nitroso via sulfuric acid and sodium nitrite, and then nitric acid was used to afford nitro 2 (Flaugh et al., 1979). Amine 3 was readily accessed via palladium reduction of nitro 2. Acetylation, followed by cyclization of the amine with isoamyl nitrite and subsequent hydrolysis, yielded 6-fluoro-1H-indazol-5-ol 4b (Iwakubo et al., 2007). Silyl protection of the alcohol gave 5b. In the presence of catalytic dimethylaminopyridine, the indazole was then Boc-protected, giving regioisomers of 6b. Final silyl deprotection gave the free alcohol 7b. Intermediate 7a was synthesized from the commercially available 1H-indazol-5-ol 4a as described for 7b.
Scheme 1.

Synthesis of intermediate 7. Reagents and conditions: a) AcOH, H2SO4, 0°C, then NaNO2; b) 20% HNO3, 45°C; c) 10% Pd/C, H2, EtOH, THF; d) KOAc, Ac2O, CHCl3, 0°C, and then isoamyl nitrite and 80°C; e) 6N HCl, MeOH; f) TBSCl, DIEA, imidazole, DCM; g) Boc2O, DIEA, DMAP, THF; h) TBAF, THF.
Analogs 11a and 11b were synthesized from intermediate 8, which was obtained as previously described (Scheme 2) (Waldschmidt et al., 2017). Mesylation of alcohol 8, followed by displacement by 7a or 7b, gave intermediates 9a/b. Base-mediated hydrolysis of 9a/b, followed by amide coupling and Boc deprotection, yielded analogs 11a/b.
Scheme 2.

Synthesis of indazole-substituted paroxetine hybrids. Reagents and conditions: a) Ms2O, DIEA, DCM; b) NaH, DMF, 7a or 7b; c) 1N NaOH, MeOH; d) DIEA, EDC, HOBt, R2NH2; e) HCl/dioxanes.
Synthesis of the indazole substituted nonhybrid analogs are shown in Scheme 3. Alcohol 12, which has previously been reported (Waldschmidt et al., 2017), was then N-alkylated via two different methods. The N-ethyl 13a was achieved through substitution using ethyl iodide under basic conditions. The N-isopropyl 13b was prepared through reductive amination with acetone. Respective N-substituted analogs (compound 14 is commercially available, and 15 has been previously reported) were then mesylated at the benzylic alcohol, which was subsequently displaced with alcohols 7a or 7b to give intermediates 16a-e. Final Boc deprotection yielded analogs 17a–e.
Scheme 3.

Synthesis of analogs 17a–e. Reagents and conditions: a) K2CO3, EtI, DMF; b) NaCNBH3, acetone, THF, AcOH; c) Ms2O, DIEA, DCM; d) NaH, DMF, 7a or 7b; e) 4M HCl/dioxanes; f) 20% TFA/DCM.
Structure-Activity Relationships.
To investigate whether binding of paroxetine hybrids could be improved, we exchanged the benzodioxole moiety for an indazole, analogous to what is present in GSK180736A (Table 1). CCG224061 showed a 20-fold increase in potency for GRK2 (IC50 = 66 nM) over paroxetine (IC50 = 1.4 μM) (Thal et al., 2012); however, this change also resulted in increased activity against GRK1 and 5, PKA, and ROCK1. Although CCG224061 was still selective for GRK2, the selectivity factor over ROCK1 was only 5-fold versus more than 50-fold selectivity observed with paroxetine (Waldschmidt et al., 2017). In an effort to improve selectivity, 2,6-dimethoxybenzylamide was appended to the fluorophenyl C-ring (CCG232406). Previously, this adduct gave the largest improvement in the GSK180736A scaffold (Waldschmidt et al., 2016). Compound CCG232406 indeed showed improvement in GRK2 selectivity over the other GRKs and PKA and approximately 20-fold selectivity over ROCK1.
TABLE 1.
Kinase activity and half-life in mouse liver microsomes of indazole-paroxetine hybrids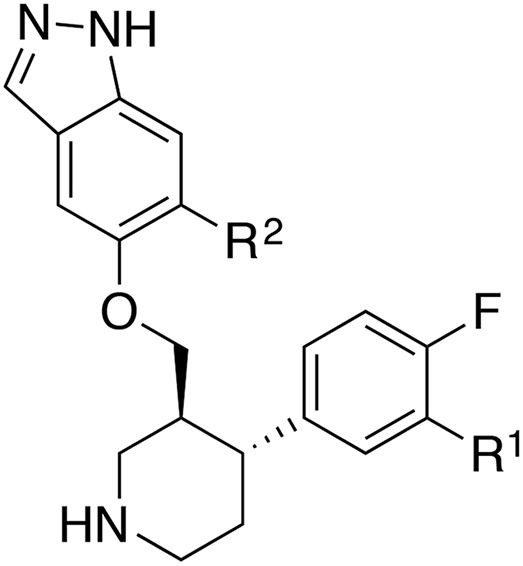
| CCG# | R1 | R2 | GRK2 IC50 (μM)a | GRK1 IC50 (μM)a | GRK5 IC50 (μM)a | PKA IC50 (μM)a | ROCK1b (%) | MLM t1/2 (min) |
|---|---|---|---|---|---|---|---|---|
| 224061 | H | H | 0.066 (1.1) | 6.4 (1.3) | 1.3 (1.2) | 3.1 (1.4) | 100 ± 1.5 | 30 |
| 258002 | H | F | 0.13 (1.6) | 3.6 (1.3) | 0.81 (1.2) | 3.4 (1.6) | 48 ± 5.8 | 22 |
| 232406 | 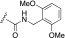 |
H | 0.36 (1.8) | >100 | 28.8 (1.7) | 9.8 (1.2) | 63 ± 6.2 | 8.3 |
| 258003 | 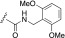 |
F | 0.36 (1.7) | 40 (1.1) | 8.0 (1.2) | 24 (1.9) | 80 ± 8.4 | 13 |
| 257284 | 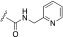 |
H | 0.10 (1.2) | 3.9 (1.5) | 0.49 (1.1) | 15 (1.3) | 100 ± 5.7 | 2.7 |
| 258748 | 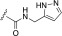 |
H | 0.008 (1.3) | 4.4 (1.3) | 0.24 (1.2) | 62 (1.6) | 83 ± 2.4 | 8.1 |
All IC50 measurements are reported as the geometric mean of three separate experiments run in duplicate. Error is expressed in parenthesis as the geometric S.D. factor (multiplied or divided by the mean).
Percent inhibition at an inhibitor concentration of 10 μM, error is expressed as the S.D.
In a parallel attempt to improve selectivity, we appended two favorable adducts, 2-pyridylmethylamide and 3-pyrazolylmethylamide, discovered from our previous efforts using the paroxetine scaffold (Waldschmidt et al., 2017) to generate CCG257284 and CCG258748, respectively. CCG257284 showed no improvement in GRK2 potency relative to CCG224061 but resulted in pan-GRK inhibition with potencies similar to that of the analogous GSK180736A-based analog (CCG215022) (Waldschmidt et al., 2016). CCG258748 exhibited our most potent GRK2 inhibition to date with an IC50 of 8 nM. Although this compound displayed greater than 30-fold selectivity over the other GRKs and PKA, the IC50 for GRK5 was still quite potent at 240 nM.
Lastly, we investigated alkylating the piperidine nitrogen of CCG224061 to improve selectivity (Table 2). N-methylation (CCG258001) resulted in a 4-fold decrease in GRK2 potency but much more dramatic decreases in potency for GRK1, GRK5, and PKA, resulting in >100 fold-selectivity for GRK2 over all three kinases. Increasing the size of the methyl to ethyl and isopropyl (CCG258211 and CCG258746, respectively) was not as well tolerated in that the GRK2 potency dropped with increasing substituent size. Therefore, larger alkyl groups were not explored.
TABLE 2.
Kinase activity of alkylated indazole-paroxetine hybrids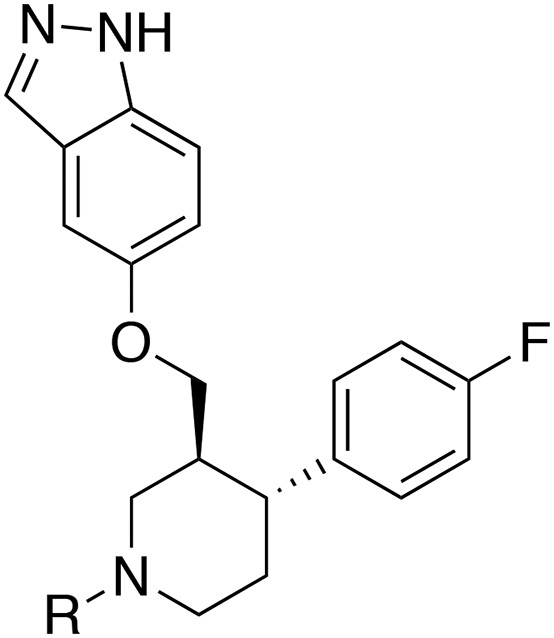
| CCG# | R | GRK2 IC50 (μM)a | GRK1 IC50 (μM)a | GRK5 IC50 (μM)a | PKA IC50 (μM)a | ROCK1b (%) |
|---|---|---|---|---|---|---|
| 224061 | H | 0.066 (1.1) | 6.4 (1.3) | 1.3 (1.2) | 3.1 (1.4) | 100 ± 1.5 |
| 258001 | Me | 0.28 (1.5) | 52 (1.1) | 33 (1.2) | 85 (1.2) | 61 ± 5.3 |
| 258211 | Et | 0.96 (1.3) | >100 | >100 | >100 | 45 ± 1.5 |
| 258746 | iPr | 1.8 (1.2) | >100 | >100 | NT | 33 ± 10.3 |
All IC50 measurements are reported as the geometric mean of three separate experiments run in duplicate. Error is expressed in parenthesis as the geometric S.D. factor (multiplied or divided by the mean).
Percent inhibition at an inhibitor concentration of 10 μM, error is expressed as the S.D.
Metabolic Stability.
In addition to improving the in vitro potency and selectivity for GRK2, we also aimed to generate analogs with more favorable PK properties, and thus hopefully efficacy, because the paroxetine scaffold is much better tolerated than GSK180736A (Waldschmidt et al., 2017). The compounds were incubated with mouse liver microsomes (MLMs) to test for metabolic stability (Table 1). The initial hit compound, paroxetine, had a half-life of 24.1 min in this assay (Waldschmidt et al., 2017). Replacing the benzodioxole ring with an indazole (CCG224061) resulted in a longer half-life of 30.3 min; however, CCG257584 and CCG258748 did not show any improvement in metabolic stability compared with their respective benzodioxole analogs (CCG211998 and CCG258208). We also explored fluorination of the C6 position of the indazole ring to improve potency and improve metabolic stability, as this addition was previously shown to be successful with similar compounds (Goodman et al., 2007; Sehon et al., 2008). The addition of a fluoro substituent to the A ring (CCG258002) did not however improve GRK2 potency or have a significant effect on the metabolic stability (Table 1).
Crystallography.
Previously, we determined crystal structures of GRK2–Gβγ complexes bound to three paroxetine analogs containing a benzodioxole warhead and paroxetine (Thal et al., 2012; Waldschmidt et al., 2017). These complexes crystallized in three different crystal forms (two C2 and one P2), whereas inhibitors based off the GSK180736A scaffold (which contain an indazole warhead) all crystallized in a C2221 crystal form (Waldschmidt et al., 2016) (Supplemental Table 1). A previously reported benzolactam derivative of paroxetine (PDB ID: 4MK0) also crystallized with the same packing as the GSK180736A-based compounds (Homan et al., 2014). Therefore, we hypothesized that the identity of the hinge-binding moiety is critical for not only determining potency toward GRK2 in these compounds but also for dictating the conformation of the kinase domain and consequently the crystal form. The indazole and benzolactam moieties seem to be able to form stronger hinge interactions via multiple conventional hydrogen bonds, thereby stabilizing a consistent kinase conformation that allows GRK2 to crystallize in a reproducible way (Thal et al., 2012; Homan et al., 2014, 2015).
Consistent with this reasoning, the three indazole-paroxetine hybrids (CCG224061, CCG257284, and CCG258748) in complex with GRK2–Gβγ also crystallized in the previously observed C2221 crystal form. In all three structures, the indazole forms two hydrogen bonds to the carbonyl oxygen of Asp272 and the backbone nitrogen of Met274 in the hinge (Fig. 3). As previously observed in other paroxetine complexes, there is an additional highly conserved hydrogen bond between the piperidine nitrogen of the paroxetine scaffold and the backbone carbonyl of Ala321. As expected, the D-ring amide linker of CCG258748 and CCG257284 made additional interactions. In the case of CCG258748, the carbonyl oxygen and amide nitrogen formed hydrogen bonds with the backbone nitrogen of Gly201 and the sidechain of Asp335, respectively (Fig. 3C). In the case of CCG257284, a hydrogen bond with the sidechain of Asp335 was not observed because this residue is shifted slightly away from the ligand. Finally, in the case of CCG258748, the pyrazole is able to make additional hydrogen bonds with the sidechains of Glu239 and Lys220 that could explain its higher potency (Fig. 3C). The pyridine nitrogen of CCG257284 does not appear to be able to make any interactions with the protein and is modeled with the nitrogen pointing out of the active site so that it can favorably interact with solvent (Fig. 3B).
Fig. 3.

Electron density |Fo|–|Fc| omits maps contoured at 3.0 σ and binding modes for ligands cocrystallized with GRK2–Gβγ. (A) GRK2–Gβγ·CCG2224061. Carbon atoms for ligand are shown in cyan. (B) GRK2–Gβγ·CCG257284. Carbon atoms for ligand are shown in light blue. (C) GRK2–Gβγ·CCG258748. Carbon atoms for ligand are shown in green. Hydrogen bonds are shown as black dashed lines, and backbone nitrogens are shown as blue spheres.
Structural Comparisons with Analogous GRK2-Inhibitor Complexes.
We have previously reported complexes with paroxetine, CCG211998, and CCG258208 (PDB IDs: 3V5W, 5UKK, and 5UKM, respectively), which are the benzodioxole analogs of the complexes of CCG224061, CCG257284, and CCG258784, respectively (Thal et al., 2012; Waldschmidt et al., 2017). Comparison of these structures thus gives insights into how substituting indazole for benzodioxole in the paroxetine scaffold influences the overall conformation of the small and large lobes of the kinase domain and clues into the molecular basis for the high selectivity underlying the benzodioxole-based compounds.
Alignment of the small lobes (residues 185–271) of the CCG224061 (indazole) and paroxetine (benzodioxole) complexes yielded a root mean-squared deviation (r.m.s.d.) value of 0.32 Å for the Cα atoms. The two compounds superimpose almost exactly. Their A rings are slightly offset but make analogous hydrogen bonds with the hinge (to Asp272 and Met274) (Fig. 4A); however, in the case of paroxetine, the interaction with Asp272 is weaker because it is a CH—O hydrogen bond. In the CCG224061 complex, the backbone of the hinge is pushed out a maximum of 0.6 Å at Asn275 compared with the paroxetine complex, and the P-loop (β1-β2 turn) exhibits a more closed conformation, exhibiting a 2.1 Å difference at the Cα of Gly201. Differences were also observed in the β2-β3 and β4-β5 loops of up to 0.6 and 0.8 Å, respectively.
Fig. 4.

Structural comparison of analogous GRK2–Gβγ complexes with indazole- or benzodioxole-substituted paroxetine derivatives. Key structural changes in the P-loop and/or αB-αC loop are highlighted with red circles and black dashed lines linking Cα carbons with the greatest displacement in each region discussed in the text. (A) GRK2–Gβγ·CCG224061 is shown in cyan, and GRK2–Gβγ·paroxetine (PDB ID: 3V5W) in gray. (B) GRK2–Gβγ·CCG257284 is shown in blue, and GRK2–Gβγ·CCG211998 (PDB ID: 5UKK) is shown in magenta. (C) GRK2–Gβγ·CCG258748 is shown in green, and GRK2–Gβγ·CCG258208 (PDB ID: 5UKM) is shown in orange.
Comparison of the CCG257284 (indazole) and CCG211998 (benzodioxole) complexes yielded the largest r.m.s.d. value of the three pairs of complexes (0.70 Å), which likely reflects the unusual space group of the CCG211998 complex (P2); however, the two compounds bind similarly, and their protein-ligand interactions are conserved (Fig. 4B). The largest differences are observed in the A and D rings. The indazole forms stronger interactions with the hinge via shorter hydrogen bonds with the backbone of Met274 and Asp272 (3.3 and 2.8 Å vs. 3.6 and 3.4 Å, respectively). The tighter interaction between the indazole and hinge is paired with a shift of the Cα of Asp272 by 0.7 Å, which propagates into a shift of the β2-β3 and β4-β5 loops of 1.0 and 0.9 Å, respectively. The P-loop is shifted upwards in the CCG257284 complex by up to 1.0 Å (for the Cα of Gly201) and the αB-αC loop is pushed outwards by a maximum of 1.3 Å. Additionally, a large portion of the active-site tether (AST, residues 493–500) is ordered in the CCG257284 complex relative to the CCG211998 complex.
The CCG258748 (indazole) and CCG258208 (benzodioxole) complexes displayed a r.m.s.d. deviation of 0.41 Å for the Cα atoms of the small lobe. The compounds bind similarly (Fig. 4C), although the hydrogen bonds with the hinge are again stronger (shorter) in the case of CCG258748 (distances of 3.7 and 2.9 Å vs. 3.8 and 3.1 Å, respectively). The hinge is displaced a maximum of 0.5 Å away from the active site in CCG258748 compared with the CCG2528208 complex. Additionally, the 3-pyrazolylmethylamide shows slightly different conformation and interactions in the hydrophobic subsite. Although in both CCG258748 and CCG258208 the two pyrazole nitrogens form hydrogen bonds with Lys220 and Glu239, CCG258208 forms an additional contact with Asp335 via its amide linker. The P-loop is shifted up to 0.6 Å at Gly201 away from the active site in the CCG258748 complex. Hinge interactions formed by CCG258748 also induce up to a 0.8 Å change of the β4–β5 loop and a large shift in the β2-β3 loop (1.2 Å for the Cα of Asp212). There is a smaller change in the αB-αC loop compared with analogous complexes, with a 0.7 Å shift for the Cα of Glu233. Additionally, the highly mobile AST is more ordered in the CCG258748 complex, allowing an additional five residues (491–496) to be modeled.
The buried surface area (BSA) of each of these six ligands and their hinge-binding moieties was then compared (Table 3). In general, the BSA of the indazole warhead was greater than that of benzodioxole; however, the entire BSA of CCG258748 and CCG257284 was less than observed with their respective benzodioxole analogs (CCG258208 and CCG211998, respectively) as a result of the differences in packing of the D rings and outward movements of the αB–αC loop.
TABLE 3.
Buried surface area calculation for paroxetine analogs crystallized with GRK2
| Compound | Hinge-Binding Moiety | Total BSA of Ligand (Å2) | BSA of Hinge-Binding Ring (Å2) |
|---|---|---|---|
| Paroxetine | 759 | 354 | |
| CCG211998 | Benzodioxole | 1056 | 362 |
| CCG258208 | 1030 | 347 | |
| CCG224061 | 798 | 369 | |
| CCG257284 | Indazole | 1051 | 368 |
| CCG258748 | 1025 | 371 | |
PC analysis was then performed to plot the conformation of the new ligand complexes in the 2D space spanned by PC1 and PC2, in which PC1 represents a concerted opening/closing of the kinase domain describing the largest variance across published GRK structures and PC2 is a twisting motion representing smaller structural variance (Yao et al., 2017). This approach allows us to analyze in an unbiased way the global conformational changes of the kinase domain in response to binding different classes of inhibitors (in this case, indazole vs. benzodioxole warheads). This analysis showed that the GRK2 complexes with the indazole-paroxetine hybrids (CCG224061, CCG257284, and CCG258748) all cluster closely together with several previously determined indazole-containing complexes (Fig. 5). More specifically, all GRK2 complexes with indazole-containing compounds display similar PC1 compositions but exhibit greater variance along the PC2 coordinate (especially for entries 5HE0 and 5HE3). The GRK2 complexes with CCG224061 and CCG257284 showed almost identical PC1/PC2 compositions and ended up quite close to the CCG224406 (a GSK analog, entry 5HE2) and GSK180736A (entry 4PNK) complexes. The GRK2 complex with CCG258748 was closest to 4MK0 (complex with a benzolactam-paroxetine analog) in PC space. On the other hand, the various benzodioxole complexes inhabit a much broader spectrum of conformational space. The CCG211998 (5UKK) complex showed the smallest displacement from the mean along PC1, perhaps consistent with the fact that this was the only complex thus far to crystallize in the P2 crystal form.
Fig. 5.

PC analysis of GRK and PKA structures. PC1 represents the opening and closing of the small and large lobes at the hinge, whereas PC2 corresponds to a smaller twisting motion of the small lobe relative to the large lobe. Circles indicate the PCA coordinates for the kinase domains in deposited crystal structures of GRKs and PKA: KD-closed and -open GRK4 subfamily members (black and red, respectively), GRK1 (blue), and closed and open PKA (gray and pink, respectively). GRK2–Gβγ complexes with compounds containing a benzodioxole or an indazole warhead are shown in bright yellow and green, respectively. The remaining GRK2 structures are indicated in dark green. Key structures are labeled with their PDB codes (for previously published structures) or compound codes (for new structures reported in this article). The axes indicate the displacement from the mean conformation along either PC1 or PC2, with numbers in axis labels showing the percentage of total structural variance captured by PC1 or PC2.
Myocyte-Shortening Assays.
To investigate how indazole-paroxetine hybrids perform in a more in vivo context, we tested CCG224061 with mouse cardiomyocytes and measured their ability to produce a contraction. The cardiomyocytes were harvested from adult mice and incubated with varying does of the inhibitors followed by a dose of the β-AR agonist isoproterenol. Paroxetine needed to be given at a dose of 10 μM to produce a significant response, whereas GSK180736A showed a similar response at 1 μM (Thal et al., 2012; Waldschmidt et al., 2016) consistent with its 10-fold improvement in potency over paroxetine measured in vitro. Similarly, CCG224061 produced a significant increase in the maximum contraction response when dosed at 1 μM, consistent with its 20-fold higher potency for GRK2 relative to paroxetine (Table 4).
TABLE 4.
Mouse cardiomyocyte contractility
| Concentration | CCG224061 | ||
|---|---|---|---|
| DMSO | 0.5 μM | 1 μM | |
| Baseline before isoproterenol | |||
| Max contraction (% cell length) | 4.6 ± 0.7 | 4.5 ± 0.8 | 6.4 ± 0.6 |
| After isoproterenol | |||
| Max contraction (% cell length) | 11 ± 1.7 | 16 ± 1.1 | 17 ± 1.2* |
| % Increase in contraction | 150 ± 45 | 280 ± 92 | 180 ± 26 |
Values represent the mean contraction amplitude (as a percentage of cell length) ± S.E.M. for six to eight cardiomyocytes. Statistical significance (*P < 0.05) was determined in comparison with the dimethylsulfoxide (DMSO) control by a one-way ANOVA followed by Dunnett’s test.
Mouse PK Studies.
CCG224061 was then evaluated in a abbreviated mouse PK study (Table 5). In this study, the compound was injected intraperitoneally at 10 mg/kg and plasma samples collected after 30 min, 2, 4, and 7 h. The plasma levels of compound were quantified using MS/MS and a calibration curve. At 7 h the levels of CCG224061 were still quantifiable at 13 ng/ml (0.04 μM), however this is below the IC50 of 0.066 μM. This compound showed a much lower area under the curve than CCG258208, which was previously evaluated (Waldschmidt et al., 2017). However, in the mouse liver microsome assays CCG224061 showed a 3-fold longer half-life than CCG258208. This suggests that CCG224061 may be cleared renally in addition to undergoing P450-mediated metabolism.
TABLE 5.
In vivo exposure after 10 mg/kg intraperitoneal administration in mice
| Time | [224061] |
|---|---|
| h 0.5 | ng/ml 440 ± 62 |
| 2 | 93 ± 8.6 |
| 4 | 29 ± 7.7 |
| 7 | 13 ± 6.6 |
| AUC0–7 h (ng·h/ml) | AUC0–∞ (ng·h/ml) |
| 695 | 731 |
Plasma concentrations are the mean from three mice for each time point, error is expressed as the S.D.
AUC, area under the curve.
Discussion
Understanding the molecular bases for potency and selectivity among closely related protein kinases is key to the rational design of improved GRK chemical probes. In this study, we hypothesized that substituting the benzodioxole moiety of paroxetine or its derivatives for an indazole would result in higher potency for GRK2. The hope was that they would retain the same or better selectivity as mediated by other substituents in each compound (namely the B, C and D rings). Indeed, these compounds showed corresponding increases in GRK2 potency. Additionally, this class of indazole-paroxetine hybrids showed similar metabolic stability and efficacy in a mouse cardiomyocyte assay compared with previous paroxetine analogs; however, when CCG224061 was tested in mice, it showed worse PK parameters than a previous benzodioxole analog (CCG258208), suggesting a renal clearance mechanism. The expected increase in potency was also met with corresponding increases in GRK5, PKA, and ROCK1 potency and consequently the indazole series of compounds suffered from a 200- to 20-fold loss in selectivity compared with their benzodioxole-containing analogs.
To investigate the reasons for these changes in potency and selectivity, we determined the crystal structures of GRK2–Gβγ bound to three pairs of analogs in each series. The stronger hydrogen bond interactions and small increase in BSA for the indazole moiety likely lead to stronger interactions in the adenine-binding pocket next to the hinge, leading to higher stability and a well defined kinase domain conformation that consistently leads to the same crystal form (Supplemental Table 2). The indazole-paroxetine analogs that contained amide-linked D rings also appeared to further stabilize the AST region, which passes over the active site. On the other hand, selectivity conferred by the benzodioxole hinge-binding moiety seems to be due to looser interactions in the adenine pocket, as typified by weaker hydrogen bonds to the hinge (including one CH-O bond) and less BSA. GRK2 can alter its configuration to conform to each particular analog, yielding three distinct crystal forms when in complex with benzodioxole, warhead ligands (one paroxetine analog not reported in this study was observed to crystallize in the same crystal form as the indazole-hybrids). Given their highly conserved active sites, the molecular basis underlying selectivity for GRK2 over other GRKs and more distantly related AGC kinases is therefore most easily explained by the innate ability of GRK2 to accommodate these conformational changes.
Changing the identity of the hinge-binding moiety also affects how the D-ring packs, thereby influencing the conformation of residues in the αC helix and αB-αC loop that contacts the D ring, if present. Indeed, there was a consistent shift of the αB-αC loop away from the active site in the indazole-hybrid complexes relative to those of the benzodioxole analogs; however, in the case of the CCG258748 complex, this change in the αB-αC loop is subtler, which is most likely the result of the interactions formed with the pyrazole D-ring of these compounds. In particular, the hydrogen bond formed between a pyrazole nitrogen and Glu239 in αC would restrict the movement of this loop. The outward movement of the αB-αC loop creates a slightly broader active site that can accommodate larger substituents, which could explain why bulky D-ring substituents like a 2,6-dimethoxybenzylamide showed high potency in the GSK180736A series but not in the benzodioxole-paroxetine compounds (Waldschmidt et al., 2016, 2017). Additionally, this shift of the αB–αC loop may contribute to a kinase domain conformation that is more similar to that observed with other kinases such as GRK5 or PKA.
PC analysis revealed that the GRK2–Gβγ complexes with indazole-paroxetine hybrid analogs adopt similar kinase domain conformations as complexes with GSK180736A analogs and the benzolactam-paroxetine analog (which all crystallized in the C2221 crystal form). The kinase domain conformation in these complexes is also closely related to that of GRK5 bound to sangivamycin (PDB entry 4TNB) (Komolov et al., 2015). This result could help explain why indazole-paroxetine hybrids are able to inhibit GRK5 more potently than the corresponding benzodioxole analogs, as they are able to trap the kinase domains of GRK2 and GRK5 in a similar conformational state. It should, however, be noted that the structure of GRK5 in complex with another indazole-based GSK180736A analog, CCG215022 (PDB entry 4WNK), a pan-GRK inhibitor, adopted a distinct kinase conformation (Fig. 5). Thus, the structure of a complex between GRK5 and an indazole-paroxetine hybrid would help to further elaborate on how GRK5 potency is gained. On the other hand, the benzodioxole analogs allow more conformational flexibility in the GRK2 kinase domain, as evidenced by the wider spread in PC space of 3V5W, 5UKK, 5UKM, and 5UKL, especially along the PC1 axis, which indicates once again that, with respect to benzodioxole moieties, selectivity is likely driven by the apparently unique ability of GRK2 to mold itself to these ligands. Notably, another highly selective GRK2 inhibitor, CMPD101 (115h), also forms relatively weak interactions with the hinge via a pyridine moiety and was crystallized in complex with GRK2 in one of the C2 crystal forms (Thal et al., 2011; Okawa et al., 2017). Given that the unique conformational space sampled by the kinase domains of GRK2 and GRK5 seems to be a major determinant of selectivity, it will be important to take their differing landscapes into account as one looks toward the rational design of GRK5-selective inhibitors.
Acknowledgments
The MLM stability and short PK studies were conducted by the University of Michigan Pharmacokinetics Core.
Abbreviations
- BSA
buried surface area
- GPCR
G protein–coupled receptor
- GRK
G protein–coupled receptor kinase
- LC-MS/MS
liquid chromatography tandem mass chromatography
- MLM
mouse liver microsome
- PC
principal component
- PK
pharmacokinetics
- PKA
protein kinase A
- r.m.s.d.
root mean squared deviation
- ROCK1
Rho-associated coiled-coil containing kinase 1
Authorship Contributions
Participated in research design: Koch, Larsen, Tesmer.
Conducted experiments: Bouley, Waldschmidt, Cato, Cannavo, Yao.
Contributed new reagents or analytic tools: Song, Cheung.
Performed data analysis: Bouley, Waldschmidt, Cato, Cannavo, Yao.
Wrote or contributed to the writing of the manuscript: Bouley, Waldschmidt, Tesmer.
Footnotes
The work at University of Michigan was supported by the National Institutes of Health ((NIH) National Heart, Lung, and Blood Institute [Grants HL071818 and HL122416] to J.J.G.T.; the American Heart Association [Grant 15PRE22730028] to H.V.W.; and the University of Michigan Chemistry-Biology Interface (CBI) training program NIH [Grant 5T32GM008597] to M.C.C. The work at Temple University was supported by the NIH [Grants R37 HL061690, P01 HL075443, P01 HL108806, and P01 HL091799] to W.J.K. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357, and the use of LS-CAT sector 12 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant [Grant 085P1000817].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte SL, Blaxall BC. (2011) G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res 109:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzimiri N, Muiya P, Andres E, Al-Halees Z. (2004) Differential functional expression of human myocardial G protein receptor kinases in left ventricular cardiac diseases. Eur J Pharmacol 489:167–177. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE, III, Colapietro AM, Barak LS, Ménard L, Caron MG. (1996) Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271:363–366. [DOI] [PubMed] [Google Scholar]
- Flaugh ME, Crowell TA, Clemens JA, Sawyer BD. (1979) Synthesis and evaluation of the antiovulatory activity of a variety of melatonin analogues. J Med Chem 22:63–69. [DOI] [PubMed] [Google Scholar]
- Gildea RJ, Waterman DG, Parkhurst JM, Axford D, Sutton G, Stuart DI, Sauter NK, Evans G, Winter G. (2014) New methods for indexing multi-lattice diffraction data. Acta Crystallogr D Biol Crystallogr 70:2652–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KB, Cui H, Dowdell SE, Gaitanopoulos DE, Ivy RL, Sehon CA, Stavenger RA, Wang GZ, Viet AQ, Xu W, et al. (2007) Development of dihydropyridone indazole amides as selective Rho-kinase inhibitors. J Med Chem 50:6–9. [DOI] [PubMed] [Google Scholar]
- Homan KT, Larimore KM, Elkins JM, Szklarz M, Knapp S, Tesmer JJG. (2015) Identification and structure-function analysis of subfamily selective G protein-coupled receptor kinase inhibitors. ACS Chem Biol 10:310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan KT, Wu E, Wilson MW, Singh P, Larsen SD, Tesmer JJG. (2014) Structural and functional analysis of g protein-coupled receptor kinase inhibition by paroxetine and a rationally designed analog. Mol Pharmacol 85:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Keneko M, and Fujiwara S (2007) inventors, Takeda Pharmaceutical Company Ltd, assignee. Cardiotonic agent comprising GRK inhibitor. World patent WO2007034846. 2007 Mar 29.
- Iwakubo M, Takami A, Okada Y, Kawata T, Tagami Y, Ohashi H, Sato M, Sugiyama T, Fukushima K, Iijima H. (2007) Design and synthesis of Rho kinase inhibitors (II). Bioorg Med Chem 15:350–364. [DOI] [PubMed] [Google Scholar]
- Komolov KE, Bhardwaj A, Benovic JL. (2015) Atomic structure of GRK5 reveals distinct structural features novel for G protein-coupled receptor kinases. J Biol Chem 290:20629–20647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozasa T. (2004) Purification of G protein subunits from Sf9 insect cells using hexahistidine-tagged alpha and beta gamma subunits. Methods Mol Biol 237:21–38. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montó F, Oliver E, Vicente D, Rueda J, Agüero J, Almenar L, Ivorra MD, Barettino D, D’Ocon P. (2012) Different expression of adrenoceptors and GRKs in the human myocardium depends on heart failure etiology and correlates to clinical variables. Am J Physiol Heart Circ Physiol 303:H368–H376. [DOI] [PubMed] [Google Scholar]
- Okawa T, Aramaki Y, Yamamoto M, Kobayashi T, Fukumoto S, Toyoda Y, Henta T, Hata A, Ikeda S, Kaneko M, et al. (2017) Design, synthesis, and evaluation of the highly selective and potent G-protein-coupled receptor kinase 2 (GRK2) inhibitor for the potential treatment of heart failure. J Med Chem 60:6942–6990. [DOI] [PubMed] [Google Scholar]
- Raake PWJ, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, Pleger S, Mier W, Haberkorn U, Koch WJ, et al. (2013) AAV6.βARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J 34:1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, García-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr (2007) The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta 1768:913–922. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ. (1998) Control of myocardial contractile function by the level of beta-adrenergic receptor kinase 1 in gene-targeted mice. J Biol Chem 273:18180–18184. [DOI] [PubMed] [Google Scholar]
- Salazar NC, Chen J, Rockman HA. (2007) Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim Biophys Acta 1768:1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter NK, Hattne J, Grosse-Kunstleve RW, Echols N. (2013) New python-based methods for data processing. Acta Crystallogr D Biol Crystallogr 69:1274–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, Tesmer JJ, Koch WJ. (2015) Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med 7:277ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehon CA, Wang GZ, Viet AQ, Goodman KB, Dowdell SE, Elkins PA, Semus SF, Evans C, Jolivette LJ, Kirkpatrick RB, et al. (2008) Potent, selective and orally bioavailable dihydropyrimidine inhibitors of Rho kinase (ROCK1) as potential therapeutic agents for cardiovascular diseases. J Med Chem 51:6631–6634. [DOI] [PubMed] [Google Scholar]
- Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJG. (2008) Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J Biol Chem 283:14053–14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhang X-Q, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. (2008) Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange versus Na+ -K+ -ATPase. Am J Physiol Heart Circ Physiol 295:H1615–H1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, Chuprun JK, Song J, Gao E, Cheung JY, et al. (2012) Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol 7:1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Yeow RY, Schoenau C, Huber J, Tesmer JJG. (2011) Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol Pharmacol 80:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldschmidt HV, Homan KT, Cato MC, Cruz-Rodríguez O, Cannavo A, Wilson MW, Song J, Cheung JY, Koch WJ, Tesmer JJG, et al. (2017) Structure-based design of highly selective and potent G protein-coupled receptor kinase 2 inhibitors based on paroxetine. J Med Chem 60:3052–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldschmidt HV, Homan KT, Cruz-Rodríguez O, Cato MC, Waninger-Saroni J, Larimore KM, Cannavo A, Song J, Cheung JY, Kirchhoff PD, et al. (2016) Structure-based design, synthesis, and biological evaluation of highly selective and potent G protein-coupled receptor kinase 2 inhibitors. J Med Chem 59:3793–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X-Q, Cato MC, Labudde E, Beyett TS, Tesmer JJG, Grant BJ. (2017) Navigating the conformational landscape of G protein-coupled receptor kinases during allosteric activation. J Biol Chem 292:16032–16043 . [DOI] [PMC free article] [PubMed] [Google Scholar]