

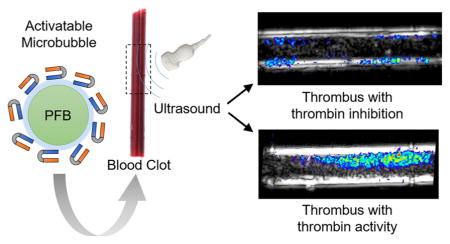
Abstract
Acute deep vein thrombosis (DVT) is the formation of a blood clot in the deep veins of the body that can lead to fatal pulmonary embolism. Acute DVT is difficult to distinguish from chronic DVT by ultrasound (US), the imaging modality of choice, and is therefore treated aggressively with anticoagulants, which can lead to internal bleeding. Here we demonstrate that conjugating perfluorobutane-filled (PFB-filled) microbubbles (MBs) with thrombin-sensitive activatable cell-penetrating peptides (ACPPs) could lead to the development of contrast agents that detect acute thrombosis with US imaging. Successful conjugation of ACPP to PFB-filled MBs was confirmed by fluorescence microscopy and flow cytometry. Fluorescein-labeled ACPP was used to evaluate the efficiency of thrombin-triggered cleavage by measuring the mean fluorescence intensity of ACPP-labeled MBs (ACPP-MBs) before and after incubation at 37 °C with thrombin. Lastly, control MBs and ACPP-MBs were infused through a tube containing a clot, and US contrast enhancement was measured with or without the presence of a thrombin inhibitor after washing the clot with saline. With thrombin activity, 91.7 ± 14.2% of the signal was retained after ACPP-MB infusion and washing, whereas only 16.7 ± 4% of the signal was retained when infusing ACPP-MBs in the presence of hirudin, a potent thrombin inhibitor.
Keywords: ultrasound, microbubble, cell-penetrating peptide, DVT, activatable, thrombin, thrombosis
Graphical Abstract
INTRODUCTION
Patients with acute deep vein thrombosis (DVT), which require anticoagulation therapy, are difficult to recognize by ultrasound (US) imaging alone, the screening modality of choice, from those with chronic DVT for which anticoagulation is ineffective.1,2 Chronic thrombosis, when there is no active clotting, is a transition phase in the healing process when acute clots are either dissolved or organized into a fibrous scar.3 Since acute clots have no internal signal, they are invisible on US. Diagnosis is made by venous compression where the presence of clot prevents venous collapse.4,5 In the setting of chronic DVT when echoes may be visible inside the vein, venous collapse is no longer diagnostic. At present, ultrasound cannot exclude the presence of acute clot when chronic DVT is present, requiring empirical treatment with anticoagulant. The ability to make the distinction between acute and chronic DVT is important, since anticoagulation carries the risk of internal bleeding including cerebral hemorrhage. We therefore aim to distinguish acute from chronic DVT by detecting the presence of thrombin, which is indicative of active thrombosis, by developing a thrombin-specific US molecular imaging agent6 to help recognize patients that would benefit from anticoagulation. In recent years, many efforts have been dedicated to the development of activatable contrast agents that are responsive to environmental stimuli such as low pH,7,8 elevated levels of reactive oxygen species (ROS),9–11 hypoxia,12,13 enzymatic activity,14–17 or a combination of multiple stimuli.18 Active clotting, which is linked to acute DVT is known to have a thrombin-rich environment.19 In addition to the importance of thrombin in DVT, this enzyme has been associated with renal injury,20 cardiovascular diseases, stroke,21 and cancer.22–24 Researchers have used this biomarker with success to activate imaging probes to study a variety of conditions including atherosclerotic disease.25 We are using the enzymatic activity of thrombin as the trigger to activate our contrast agent for molecular-based US recognition of active thrombosis. Our approach holds the advantage of binding specifically to active thrombus compared to other US contrast agents that target individual components of thrombi26 such as platelets27–29 or fibrin30–32 and are not specific to active thrombosis.
Several reports of thrombin-sensitive imaging probes for biomedical imaging using fluorescence25,33–36 and magnetic resonance imaging37 have been published recently. One example of a thrombin-sensitive US contrast agent has been reported.15,38 Specifically, this activatable contrast agent used thrombin-specific aptamers to cross-link the microbubble (MB) shell increasing its stiffness and decreasing its volumetric oscillation, the dominant mechanism for harmonic signal generation during contrast-enhanced US imaging.39 Upon aptamer removal by thrombin, MB elasticity and its harmonic signal were restored increasing the signal by several dB.15
Here we employed activatable cell-penetrating peptides (ACPPs) described by Prof. R. Tsien and co-workers.40 ACPP has a polycationic cell-penetrating arm linked to a polyanionic inhibitory domain through a thrombin-cleavable peptide substrate (Figure 1A).14 We used the sequence (Nle)-TPRSFL as the thrombin-sensitive substrate as it showed the highest sensitivity toward thrombin.34 The thrombin-triggered cleavage occurs between the arginine and serine, which leads to the release of the polyanionic inhibitory domain and the unmasking of the polycationic cell-penetrating peptide (Figure 1B).
Figure 1.

(A) Chemical structure of the thrombin-sensitive activatable cell-penetrating peptide (ACPP) constituted of a polycationic cell-penetrating peptide (blue) linked to a polyanionic inhibitory domain (orange) via a thrombin-sensitive substrate. The arrow indicates the site of cleavage of the peptide substrate by thrombin. (B) Schematic representation of the peptide cleavage yielding the polycationic cell-penetrating peptide.
The C-terminus cysteine allows an easy conjugation of ACPP to maleimide-functionalized microbubbles (mal-MBs). After conjugation and upon exposure of MBs to thrombin, ACPP is cleaved and the polyanionic inhibitor released, leaving positively charged MBs that subsequently adhere to adjacent negatively charged surfaces. Since acute clots are rich with red blood cells, MBs accumulate in the clot allowing its detection on US imaging (Figure 2).
Figure 2.
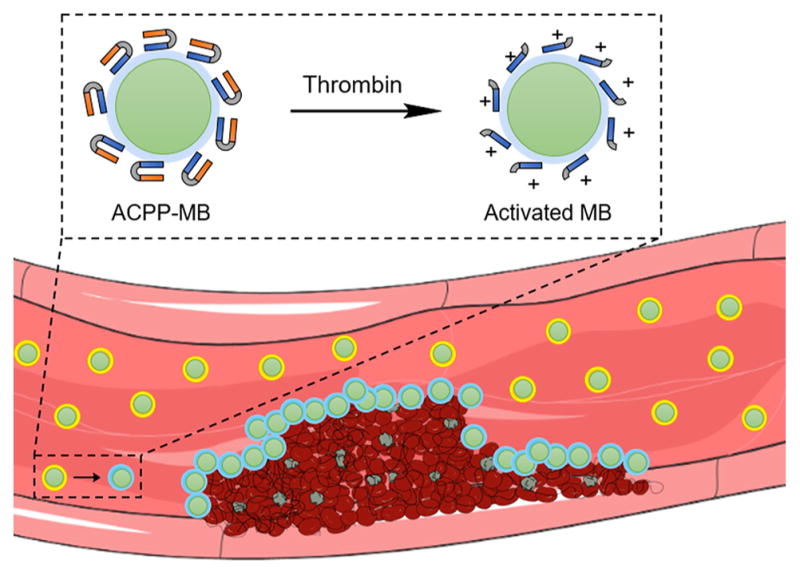
Schematic representation of thrombin-triggered activation of perfluorobutane-filled (PFB-filled) ACPP-labeled microbubbles (ACPP-MBs). After ACPP cleavage, the ACPP-MB surface becomes positively charged and adheres to the negatively charged surfaces, such as those of red blood cells, fibrin, and platelets. Adapted from Servier Medical Art.
Herein, we developed a new thrombin-sensitive US contrast agent and evaluated its potential for the detection of acute thrombosis in vitro. We used hirudin, a specific and potent thrombin inhibitor,41–45 as a method to mimic chronic DVT (no thrombin activity) while preserving the physical properties of the clot to allow direct comparison of ACPP-labeled MB (ACPP-MB) performance. We used unmodified commercial MBs used clinically (Definity, Lantheus Medical Imaging, North Billerica, MA) as a control.
RESULTS AND DISCUSSION
Synthesis of ACPP and Fl-ACPP
We synthesized ACPP and its fluorescein-labeled analog (Fl-ACPP) using a peptide synthesizer (see the Supporting Information). We purified both peptides by HPLC and characterized ACPP and Fl-ACPP by mass spectrometry (Figures S1 and S2). We confirmed their cleavage in the presence of thrombin by LC-MS (Figure S3).
Conjugation of ACPP and Fl-ACPP to MBs
ACPP or Fl-ACPP was conjugated to microbubbles (ACPP-MBs or Fl-ACPP-MBs) by first formulating perfluorobutane-filled (PFB-filled) MBs containing 5 mol % of maleimide-bearing phospholipids (see the Experimental Section for details). Briefly, lipid films were solvated in a mixture of PBS 1×/propylene glycol/glycerol (80:10:10) and sonicated at 70 °C. PFB vapor was then introduced in the solution, and the resulting mixture was mixed with an amalgamator for 45 s before being cooled down. The resulting MB formulation was washed through successive low-speed centrifugations to yield PEGylated MBs with terminal maleimide functions (mal-MBs; Figure 3A). Finally, ACPP or Fl-ACPP was added and gently mixed with mal-MBs for 3 h at room temperature. Free unconjugated ACPP or Fl-ACPP was removed through multiple low-speed centrifugations, and the final size distribution and particle count were determined using a Coulter counter (see the Experimental Section for details). In addition, the size and count of ACPP-MBs were monitored over 60 min of incubation at 37 °C with or without addition of proteins and demonstrated that there were no significant changes in size or loss of MBs (Figure S5).
Figure 3.

Conjugation of ACPP to microbubbles (MBs). (A) Schematic representation of the chemical constituents of Fl-ACPP-MBs. The Fl-labeled ACPP (green star) attaches to MBs through a thiol–maleimide coupling reaction. (B) Flow cytometry data of MBs before (left panel) and after (right panel) conjugation with Fl-ACPP showing the characteristic scatter plot of MBs (left) and the increase in fluorescence intensity (right) after conjugation. (C) Bright field (left) and fluorescence (right) microscopy of Fl-ACPP-MBs at 20×; scale bar is 50 μm.
Characterization of ACPP-MBs
To evaluate the efficiency of ACPP conjugation to MBs, we used the fluorescent derivative Fl-ACPP for conjugation. This fluorescent marker was only used for the validation steps of this study (characterization of the conjugation and cleavage of ACPP attached to MBs) and is not part of the final design for use in blood clots. The resulting Fl-ACPP-MBs and nonconjugated MBs were characterized using flow cytometry (Figure 3B). The microbubbles were easily identified on the scatter plot by their characteristic signal that appears because of the nonlinearity of forward and side scatter (a phenomenon that occurs in particles of similar size magnitude to the laser wavelength and dimensions of the focal region),46 and their mean fluorescence intensity (MFI) was measured before and after conjugation. As expected, the conjugation of Fl-ACPP was quantitative with 98.4% of microbubbles expressing a fluorescent signal. The conjugation was further confirmed by fluorescence microscopy (Figure 3C). The fluorescein channel showed green fluorescence that colocalized with the shell of the microbubbles visible in bright field imaging. The amount of conjugated Fl-ACPP molecules was quantified by fluorescence spectroscopy and was measured at more than 300 000 peptides per MB (see the Experimental Section).
Thrombin-Triggered Cleavage
We assessed thrombin-triggered cleavage of ACPP-MBs, using a new formulation of Fl-ACPP-MBs that incorporate DiD, a lipophilic dye that intercalates in the lipid layer, to label the MB shell. (Figure 4A). We observed colocalization between the DiD red fluorescence and the fluorescein green fluorescence in the Fl-ACPP-MB sample (Figure 4B, top). After addition of thrombin to the suspension of Fl-ACPP-MBs, a dramatic decrease in MB green fluorescence was observed accompanied by a dramatic increase of the background green fluorescence (Figure 4B, bottom) due to the release of the Fl-labeled polyanionic arm.
Figure 4.

(A) Schematic representation of Fl-ACPP conjugated to MBs labeled with a lipidic dye (DiD, red star) and its cleavage in the presence of thrombin. (B) Representative bright field (left), DiD (center), and fluorescein (right) fluorescence microscopy images before (top) and after (bottom) cleavage of Fl-ACPP in the presence of thrombin.
To better understand cleavage kinetics of the ACPP peptide conjugated to MBs, we added known amounts of thrombin to reach final concentrations ranging from 28 to 280 nM and incubated Fl-ACPP-MBs in PBS 1× at 37 °C. We selected this concentration range as free thrombin concentration in coagulation reactions can range from 1 to greater than 100–500 nM.47,48 In the case of healthy individuals, the concentration of thrombin is around 100 pM,49 which is 2 orders of magnitude lower than the lowest thrombin concentration evaluated. We do not anticipate any nonspecific activation of ACPP-MBs at this very low concentration. The thrombin-triggered cleavage of the ACPPs was monitored by flow cytometry by measuring MFI over time (Figure S6). To verify that no cleavage occurs in the absence of thrombin activity, we incubated Fl-ACPP-MBs with 280 nM of both thrombin and hirudin. Immediately after the addition of thrombin, even at the lowest concentration, a substantial amount of ACPPs were cleaved resulting in a decrease in fluorescence intensity while no decrease in fluorescence intensity was observed in the presence of hirudin (Figure 5). The percent of MBs with fluorescence intensity above the initial MFI immediately decreased to 39.8 ± 1.2% when exposed to 28 nM of thrombin, and continued to decrease to 26.5 ± 1.9% after 15 min of incubation at 37 °C. At the highest thrombin concentration (280 nM), this percentage was 1.6 ± 0.1% at 15 min, representing a 65.3 ± 4.4% loss of its mean fluorescence intensity (Figure S6). To demonstrate that the thrombin-directed cleavage of ACPP at the surface of MBs changes the surface charge of ACPP-MBs, we measured the ζ potential of ACPP-MBs before and after incubation with thrombin. We found out that ACPP-MBs have a ζ potential of −8.83 mV before cleavage and yield a positive signal at +5.60 mV after incubation with thrombin.
Figure 5.

Fl-ACPP-MB cleavage at 37 °C when incubated with increasing thrombin concentrations (28, 56, 112, 196, and 280 nM) and with 280 nM thrombin plus hirudin. Percent of Fl-ACPP-MBs with fluorescence intensity above the initial MFI (Pre), immediately after adding thrombin (t = 0), and after 2, 5, 10, and 15 min of incubation (n = 3). Insert: dose–response effect of thrombin concentration on ACPP cleavage after 10 min of incubation.
To assess the adherence of thrombin-sensitive activatable US contrast agent to acute clot, we designed an in vitro experiment in which we prepared thrombin-rich blood clots and infused them with a suspension of MBs at 37 °C while imaging with real-time US (for a schematic representation of the experiment design, see Figure S9). After washing the clots formed in the neck of a transfer pipet with saline, we infused three different MB suspensions: (1) ACPP-MBs in the presence of thrombin; (2) a hirudin solution first, followed by ACPP-MBs mixed with hirudin; and (3) control MBs (Definity). Following MB infusions, clots were washed by infusing a large amount of saline to remove all nonadherent MBs. To assess MB adherence, we compared the US image intensity of the clots before and after the final saline wash. We expect that, during ACPP-MB infusion, MBs circulate through and mostly around the clot, are activated by thrombin, bind to the clot, and progressively accumulate. Conversely, control MBs or ACPP-MBs, in the presence of thrombin-inhibiting hirudin, should have a lower binding to the clot, limited to the nonspecific adhesion of MBs.50 Since the clot and transducer are mechanically fixed, images are perfectly coregistered throughout the experiment. We subtracted the baseline US images acquired before MB infusion from the images acquired after the final saline wash, and the signal on a pixel-by-pixel basis was converted to a color-coded heat map and overlaid on the baseline grayscale B-mode images (Figure 6A). Similarly, we compared the US images acquired before and after the final saline wash to determine the signal loss following washing of unbound MBs. The mean intensities are summarized in Figure 6B,C. While there were no significant differences in the signal collected at the end of MB infusion among the three suspensions (Figure 6C, Infusion), there were statistically significant differences in the molecular signal (trapped MBs) between ACPP-MBs, regular MBs (p = 0.0058), and ACPP-MBs + hirudin (p = 0.0012), indicative of thrombin specificity (Figure 6C, Binding). Remarkably, the microbubble signal of ACPP-MBs retained after the saline wash indicated that 91.7 ± 14.2% of ACPP-MBs were adherent (Figure 6B). This binding percentage was significantly higher than those of regular MBs (33 ± 22.9%, p = 0.0196) and ACPP-MBs with thrombin inhibition (16.7 ± 4%, p = 0.0009). The difference in ACPP-MBs bound to clot in the presence of thrombin was 4.7-fold greater than when thrombin was inhibited (Figure 6C, Binding). We also validated the binding of ACPP-MBs in the presence of thrombin using microscopy (see the Experimental Section).
Figure 6.

(A) Spatial maps of the molecular ultrasound (US) signal color-coded and overlaid on B-mode images acquired at baseline after infusion of MBs and washing with saline using ACPP-MBs (top), regular MBs (middle), and ACPP-MBs coinjected with hirudin (bottom). The tube has an inner diameter of 4 mm. (B) Percentage US video-intensity signal remaining (bound MBs) after washing unbound MBs. (C) Clot signal intensity (arbitrary unit, a.u.) measured after MB infusion (Infusion, left) and after final wash to remove unbound MBs (Binding, right). Experiments done in triplicate. Error bars represent standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001.
CONCLUSIONS
Recognizing active thrombosis in DVT is critical for patient management. Acute DVT requires aggressive systemic anticoagulation treatment to stop its progression and the potential pulmonary embolism that could be fatal. In this study, we demonstrated that a thrombin-sensitive activatable US contrast agent specifically accumulates in blood clots rich with thrombin activity. Specifically, when thrombin was present in the clot, 91.7 ± 14.2% of the molecular US signal was retained after the clot was washed, while only 16.7 ± 4% of the molecular US signal was retained when thrombin was inhibited by hirudin. These differences in binding translated to a 4.7-fold increase in US signal intensity, suggesting that ACPP-MBs may allow the recognition of acute thrombosis. We are now translating this agent to a DVT animal model. We also plan to convert this smart imaging agent to a theranostic51 to promote sonothrombolysis,50,52 targeted anticoagulation53 or the combination of both.54
EXPERIMENTAL SECTION
Materials and Methods
Lipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and Corden Pharma. Perfluorobutane was purchased from F2 Chemicals Ltd. DiD was purchased from Thermo Fisher Scientific. For regular MBs, Definity (Perflutren Lipid Microsphere) was purchased from Lantheus Medical Imaging, Inc. (North Billerica, MA). Recombinant Hirudin (16 500 U/mg) was purchased from Creative BioMart Inc. (Shirley, NY). Other chemicals were purchased from Fisher Scientific and Sigma-Aldrich and used without further purification. Microbubble sizing and counting were performed with a Multisizer 4 Coulter counter system (Beckman Coulter Inc., Brea, CA). Microbubbles were emulsified with a VIALMIX amalgamator (Bristol-Myers-Squibb, New York, NY). Flow cytometry experiments were performed with a multiwavelength BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Microscopy experiments were performed with an Axio A1 upright fluorescence microscope (Carl Zeiss AG) equipped with a mercury lamp and a variety of filter sets (DAPI, FITC, TRITC, Cy5). Photographs of the clot and its cross sections were taken with a DSLR camera (Canon EOS Rebel T5) equipped with a macro lens (EFS 60 mm f2.8 Macro USM). US imaging experiments were performed at 7 MHz using a clinical Siemens Acuson Sequoia 512 US system (Siemens Healthcare, Mountain View, CA) equipped with a 15L8 imaging transducer. Cine were recorded through the S-video output of the US system via an Imaging Source video-to-USB converter (The Imaging Source, Charlotte, NC). They were digitally recorded to a computer at 30 frames per second and 640 × 480 pixels with the manufacturer’s software (IC Capture). All data is presented as mean ± standard deviation.
Synthesis and Characterization of ACPP and Fl-ACPP
Both ACPPs and Fl-ACPPs were synthesized by solid-phase peptide synthesis using a peptide synthesizer (Applied Biosystems 433 synthesizer) at a 0.1 mmol scale with the following sequence for ACPP: NH2-e9-o-Nle-T-P-R-S-F-L-r9-c-CONH2 (26 amino acids + linker).34 This peptide is constituted of a polycationic cell-penetrating peptide (sequence of nine positively charged D-arginine; r9) linked to a polyanionic inhibitory domain (sequence of nine negatively charged D-glutamic acid; e9) via a thrombin-sensitive substrate (L-norleucine, L-threonine, L-proline, L-arginine, L-serine, L-phenylalanine, and L-leucine; Nle-T-P-R-S-F-L) that is cleaved between the L-arginine and L-serine amino acids.
Peptides were prepared by conventional solid-phase peptide synthesis using fmoc-protected amino acids (Novabiochem, P3Bio), where upper-case letters denote L-amino acids and lower-case letters denote D-amino acids, o denotes the hydrophilic spacer fmoc-5-amino-3-oxapentanoic acid (CAS 260367-12-2). Fl-ACPP was prepared by modifying the N-terminal amino acid with a 2-fold excess of NHS–fluorescein in N-methylpyrrolidine in the presence of N,N-diisopropylethylamine overnight at room temperature.
After the synthesis, the peptides were purified with high-performance liquid chromatography (HPLC, Waters Model 600) using a reverse-phase C18 column (250 × 10 mm, Vydac) with an acetonitrile/water eluent mixture (0–100% gradient, 120 min) with 0.1% TFA as additive. The characterization of ACPP (Figure S1) and Fl-ACPP (Figure S2) was confirmed by electrospray ionization mass spectrometry (ESI-MS, Agilent 6540) after deconvolution or MALDI-TOF (Waters LR MALDI-TOF), and the purified product was lyophilized.
MS ACPP: found, 3602.92 Da; calcd, 3602.9 Da. m/z: 1201.8057 (z = 3), 901.6133 (z = 4), 601.4877 (z = 6). Fl-ACPP: found, 3960.81 Da; calcd, 3960.1 Da.
Thrombin-Directed Cleavage of ACPP
For confirmation of the thrombin-directed cleavage of ACPP between the arginine and serine amino acids of the substrate, thrombin (1 μM) was added to a solution of 100 μM ACPP in PBS 1×, and the resulting solution was mixed for 15 min at 37 °C. After this time, a 100 μL aliquot was injected in the LC-MS (Waters Alliance 2695 HPLC with UV diode array and ELSD) with an acetonitrile/water eluent mixture (2:98 to 99:1 gradient, 30 min). After 15 min of reaction with thrombin, no peak was visible in UV at 8.67 min, and a new peak appeared at 9.72 min (Figure S3A). Both peaks were analyzed by ES-MS, and fragments were identified for the starting material ACPP and for the cleaved polyanionic inhibitory domain (PID) confirming the cleavage of ACPP with thrombin (Figure S3B).
MS ACPP: found, 3602.92 Da; calcd, 3602.9 Da. m/z: 1201.2661 (z = 3), 901.2019 (z = 4), 721.1588 (z = 5), 601.1233 (z = 6), 515.2764 (z = 7), 451.2456 (z = 8). PID: found, 1748.6909 Da; [M + 9H]+ calcd, 1748.73 Da. m/z: 874.3293 (z = 2), 583.2474 (z = 3).
Formulation of mal-MBs
Lipid films containing a mixture of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N[maleimide(polyethylene glycol)-5000] (DSPE-PEG(5000)-mal) with a 95:5 molar ratio were prepared. These films were prepared by dissolving DSPC and DSPE-PEG(5000)-mal in 100 μL of chloroform and slowly evaporating the mixture with a rotary evaporator (Büchi Rotavapor R-100) until mostly dry. The resulting films were then dried overnight under vacuum and stored at −20 °C for later use. The lipid films were solvated in a mixture of PBS 1×/propylene glycol/glycerol (80:10:10 v/v/v, 2 mL total) and sonicated at 70 °C until clear or for 15 min. Perfluorobutane (PFB) vapor was then introduced in the solution, and the resulting mixture was mixed with an amalgamator for 45 s before being cooled down in an ice bath. The resulting MB formulation was washed through three successive low-speed centrifugations (300g, 3 min) to yield PEGylated MBs with terminal maleimide functions (mal-MBs). Four washes were performed with PBS 1× pH 6.5 + 1 mM EDTA to allow optimal conditions for mal-MB conjugation to ACPP through a thiol–maleimide coupling reaction performed in a later step. Microbubbles were counted (4.90 × 109 MBs/mL in 0.1 mL, N = 3) and sized (mean diameter 1.6 μm, N = 3), and the number of moles of maleimide (0.9 nmol) was directly calculated from these values (Figure S4C).
Conjugation of ACPP or Fl-ACPP to mal-MBs
ACPP or Fl-ACPP was added to the mal-MB mixture and gently mixed for 3 h at room temperature. The resulting MB formulation was washed with PBS 1× through four successive low-speed centrifugations (300g, 3 min) to yield PEGylated MBs with terminal ACPP (ACPP-MBs; Figure S4A). After each centrifugation, MBs were rising to the top of the syringe because of their buoyancy, and the infranatant was removed. The MBs were then dispersed in 3 mL and washed again. The washing process was repeated three times for a total of 4 washes to remove all liposomes and free peptide (Figure S4B). Microbubbles were counted (4.48 × 109 MBs/mL in 0.1 mL) and sized (mean diameter 2.1 μm), which corresponds to a loss of only 9% of the microbubble population after conjugation of ACPP and purification (Figure S4C).
Flow Cytometry Study of ACPP Conjugation to MBs
A suspension with 6 × 106 Fl-ACPP-MBs/mL was analyzed by flow cytometry. The region on the scatter plot corresponding to the MBs was identified and used to gate the fluorescence signal (P1, Figure 3B) before and after conjugation with Fl-ACPP. To evaluate labeling efficiency, we set a threshold below which 100% of the nonfluorescent mal-MBs remained (V1-L: 100%, Figure 3B). The percent of Fl-ACPP-conjugated MBs was then obtained by looking at the percentage of MBs that have a fluorescence intensity over this threshold (V1-R: 98.4%, Figure 3B).
Quantification of ACPP Conjugation to MBs by Fluorescence Spectroscopy
For quantification of the amount of Fl-ACPP conjugated to MBs, a standard curve was performed by diluting a solution of Fl-ACPP first in DMSO (1:20 dilution) and then in 5 mM NaOH (1:20 dilution). Serial dilutions were performed to obtain between 4 and 32 nM Fl-ACPP in the wells (N = 4). The linear regression of the calibration curve provided an equation of Y = 111.1X − 80.61 with an R2 value of 0.9988 (Figure S4D). Fl-ACPP-MB samples were prepared by diluting 5 μL of MB suspension in DMSO and 5 mM NaOH similarly to the standards. The Fl-ACPP-MB sample was then sonicated for 20 s and vortexed to make sure that MBs were destroyed and that Fl-ACPP was homogeneously distributed in solution. Samples were excited at 488 nm, and fluorescence intensity was read at 520 nm. The concentration of the sample in the well (1:400 dilution) was measured at 6.3 nM, which corresponds to 2.5 μM in the undiluted MB sample. As the sample had a concentration of 4.48 × 109 MBs/mL, that corresponds to 5.63 × 10−10 nmol/MB, which is 338 815 Fl-ACPP molecules conjugated per MB (N = 4).
Fluorescence Microscopy of Fl-ACPP-MBs
Suspensions of MBs or Fl-ACPP-MBs were prepared by diluting 2 μL of the MB suspensions in 198 μL of PBS 1×. Samples were placed on a microscopy slide inside a thin silicone barrier to keep fluid in place and increase the volume of the well. The resulting suspension was then imaged with bright field, Cy 5 (for DiD dye) and FITC filter sets. Photographs were taken at 20× or 63×, and images were processed with ImageJ (public domain, NIH).55 To observe the effects of Fl-ACPP cleavage with thrombin, we prepared a sample with 10 μL of Fl-ACPP-MBs in 500 μL of PBS 1× and observed the sample by fluorescence microscopy before and after immediate addition of 6 μM thrombin.
Stability of ACPP-MBs at 37 °C
A suspension of ACPP-MBs (2.45 ± 0.15 × 109 mL−1, 2.4 μm) in 1× PBS was incubated at 37 °C, and 2 μL aliquots were analyzed at different time points (0, 5, 30, and 60 min) with the multisizer for size and concentration (N = 3). No significant changes in concentration and size were observed after 60 min of incubation at 37 °C (Figure S5A,B). The stability of ACPP-MBs was also assessed in the presence of thrombin and hirudin. For a demonstration that thrombin cleavage did not affect the stability of ACPP-MBs, a suspension of ACPP-MBs (2.40 × 108 mL−1, 2.1 μm) in 1× PBS was incubated at 37 °C with 280 nM thrombin (16 500 U/mg), and 2 μL aliquots were analyzed at different time points (0, 5, 30, and 60 min) with the multisizer for size and concentration (N = 3). No significant change in concentration and size were observed after 60 min of incubation at 37 °C (Figure S5C). For a demonstration that the presence of hirudin did not affect the stability of ACPP-MBs, a suspension of ACPP-MBs (1.30 × 108 mL−1, 2.0 μm) in 1× PBS was incubated in the presence of 1 μg of recombinant hirudin (16 500 U/mg) at 37 °C, and 2 μL aliquots were analyzed at different time points (0, 5, 30, and 60 min) with the multisizer for size and concentration (N = 3). No significant changes in concentration and size were observed after 60 min of incubation at 37 °C (Figure S5D).
Study of ACPP-MBs Cleavage Kinetics with Flow Cytometry
A suspension of 6 × 106 Fl-ACPP-MBs in 1 mL of PBS 1× was prepared, and a 100 μL aliquot was analyzed by flow cytometry (“Pre” time point). Thrombin was then added in the suspension, and another 100 μL aliquot was immediately analyzed by flow cytometry (t = 0 min). The rest of the suspension was slowly rotated in an incubator at 37 °C, and 100 μL aliquots were taken and analyzed by flow cytometry after 2, 5, 10, and 15 min of incubation. This experiment was performed with thrombin concentrations of 28, 56, 112, 196, and 280 nM. A sample containing 280 nM thrombin and hirudin was used as a negative control to confirm that no cleavage is observed in the absence of thrombin activity. Briefly, 10 μg of recombinant hirudin in 10 μL of PBS 1× was mixed with 10 μg of thrombin in 10 μL of PBS 1× at 37 °C prior to the addition to Fl-ACPP-MBs. We gated the signal using the scatter plot and measured the MFI. To evaluate the efficacy of the cleavage, we set a threshold at the MFI of the sample before thrombin cleavage (V1-L, 50%; V1-R, 50%; Figure S6). The percentage of MBs that have a fluorescence intensity over the initial MFI was reported (Figure 5A), and the MFI (Figure S7A) as well as its normalized value were reported (Figure S7B) using various concentrations of thrombin, as a function of the incubation time.
ζ Potential of ACPP-MBs before and after Incubation with Thrombin
A suspension of ACPP-MBs (3.6 × 106) in 10 mM NaCl buffer (940 μL) was incubated at 37 °C for 30 min after addition of 60 μg of thrombin in 60 μL of 10 mM NaCl buffer. The suspension was added in disposable folded capillary cells (Malvern Instruments Inc., CAT DTS1070), and the electrophoretic mobility of the particles was measured by dynamic light scattering (Zetasizer ZS, Malvern Instruments Inc.). The ζ potential was calculated from the electrophoretic mobility by DLS software (average of 100 measurements) and yielded a positive signal of +5.60 mV. A negative control was performed without addition of thrombin and yielded a ζ potential value of −8.83 mV.
In Vitro US Imaging of ACPP-MBs in the Presence of Thrombin or Hirudin
A suspension of ACPP-MBs (1.04 ± 0.32 × 105 MBs, 2.1 μm) in 1× PBS (3 mL) was placed at 37 °C in a plastic pipet bulb inside a tissue-mimicking sample holder composed of 1% agar and 0.5% corn starch. We then added to the sample either thrombin to reach a concentration of 280 nM or 30 μg of recombinant hirudin (16 500 U/mg). The sample was imaged with a clinical ultrasound scanner (Siemens Acuson Sequoia 512) equipped with a 15L8 imaging transducer (Mountain View, CA). We used an MB-sensitive contrast pulsing sequence (CPS) imaging mode that was displayed side-by-side with a brightness-modulated (B-mode) image for bulb visualization and placement. Regarding the former nonlinear US imaging mode, the system transmitted US pulses at 7 MHz and received at 14 MHz with a mechanical index (MI) of 0.06, a pulse duration of 1 cycle, and a framerate of 16 Hz. Cine loops (10 s) were recorded before and after addition of protein after 0, 2, 5, 10, 15, 20, 25, and 30 min of incubation at 37 °C. Ultrasound pulses were stopped between acquisitions to limit MB destruction due to ultrasound exposure. We used ImageJ to quantify the ultrasound signal of ACPP-MBs over time. Ellipsoidal regions-of-interest (ROIs) were drawn on the grayscale US image (Figure S8A), and the log–compressed mean video intensity (a.u.) was measured from an average of 10 frames for each time point. Each experiment was done in triplicate. No significant change in ultrasound signal was observed after 30 min of incubation at 37 °C (Figure S8B,C). The small decrease in signal intensity was attributed to the destruction of ACPP-MBs in this diluted suspension by ultrasound exposure.
In Vitro US Imaging of Blood Clots
Blood was drawn from rabbits and collected in tubes containing 3.2% sodium citrate buffer to avoid spontaneous coagulation. Blood samples were stored at 4 °C and used in under 48 h. Blood clots were prepared by adding 0.5 mL of blood containing 280 nM of thrombin into the neck (i.d. = 4 mm) of a plastic transfer pipet (see photo, Figure S9). The plastic bulb was pierced, and tubing was fitted through and glued to the pipet bulb to avoid any leakage. This tubing was connected to a T-connector that was attached to a syringe containing the MB suspension (5 mL at 106 MBs/mL). The plastic pipet was then immersed in a water bath at 37 °C and was imaged with a clinical Siemens Acuson Sequoia 512 US system equipped with a 15L8 imaging transducer. We used an MB-sensitive contrast pulsing sequence (CPS) imaging mode that was displayed side-by-side with a brightness-modulated (B-mode) image for pipet visualization and placement. Regarding the former nonlinear US imaging mode, the system transmitted US pulses at 7 MHz and received at 14 MHz with a mechanical index (MI) of 0.06, a pulse duration of 1 cycle, and a framerate of 16 Hz.
Each of the following experiments used a different clot, and each experiment was performed in triplicate. Following saline infusion at 2 mL/min to wash the clot, three different MB suspensions were infused: ACPP-MBs with thrombin, ACPP-MBs with hirudin, a specific thrombin inhibitor, and standard MBs used clinically (Definity). A suspension of ACPP-MBs was made that had a final thrombin concentration of 280 nM. Another suspension of ACPP-MBs was made that had 0.1 mg of recombinant hirudin (16 500 U/mg) added to ensure that no thrombin activity is present during infusion. All MB suspensions were made to contain 106 MBs/mL that were infused at 0.4 mL/min (residence time of 10 s in a 2.5 cm clot) for a total volume of 3.5 mL. Prior to infusion of the ACPP-MBs + hirudin, clots were infused with 10 mL of PBS 1× containing 1.4 μM hirudin to ensure that all circulating and clot-bound thrombin was inhibited.41 US cine loops were recorded every minute during MB infusion. At the completion of MB infusions, a syringe containing saline was connected to the T-connector and flow system, and saline was infused at a rate of 2 mL/min for 20 min, until no more circulating MBs were seen on US, and only bound MBs were present. MBs that are bound to the clot are very stable as no signal decrease was observed during the washing step of the clot, which lasted 20 min. MBs were then cleared from the image plane using an MB destruction pulse sequence (MI = 1.9, t = 5 s). Cine loops were recorded during saline washing and MB destruction for offline analysis.
Image processing of all US data sets was performed using custom Matlab programs (Mathworks Inc., Natick, MA). After all frame data is loaded for a particular experiment, both B-mode and CPS images are displayed for visual review. The clot space is then isolated using manual segmentation (polygonal region-of-interest, ROI) from either image type. The resultant binary mask is applied to the time sequence of contrast-enhanced US images. A temporal average of 10 frames before MB injection was then used as a background image and subtracted from all subsequent contrast-enhanced US images. An average of 10 frames before MB destruction were digitally subtracted from those obtained after MB clearance and represents the molecular US signal.56 This spatial map was then overlaid with the grayscale image of the clot without MBs to generate the color-coded spatial maps of the molecular US signal presented in Figure 6.57
We used ImageJ to quantify the signal enhancement from the MBs. Polygonal ROIs were drawn on the contrast-enhanced US image, and the log–compressed mean video intensity (a.u.) was measured from an average of 10 frames during MB infusion (Figure 6C, Infusion) and after washing with saline (Figure 6C, Binding). The mean intensity of the same ROIs was measured from an average of 10 frames without MBs and was subtracted to remove the background signal from the blood clot and walls of the tube. The binding percentage is defined as the percentage of the signal intensity remaining after the clot was washed with saline (Figure 6B) compared to the signal intensity measured at the end of the MB infusion. Representative CPS images of the sample during diffusion, after saline wash with or without ACPP-MBs, as well as the B-mode image of the clot are shown in Figure S10.
Statistical Analysis
Signal intensity data was grouped for each MB suspension (n = 3) and also before and after saline wash. Data is represented as mean ± standard deviation. Statistical analysis was performed as a paired Student’s parametric two-tailed t-test when the US video-intensity signal for the same clot was compared before and after washing. Statistical analysis was performed as an unpaired Student’s parametric two-tailed t-test when different clots were compared: *p < 0.05, **p < 0.01, ***p < 0.001.
Optical Imaging of ACPP-MBs Binding to Blood Smear
Rabbit blood (10 μL) was added to a microscopy slide, and 1 μg of thrombin in 1 μL of 1× PBS was added to the blood and smeared uniformly on the microscopy slide using a coverslip; the blood was allowed to clot over several minutes. Two suspensions of DiD-labeled ACPP-MBs (6.33 × 105 mL−1, 2.3 μm) were prepared with no thrombin in the first suspension and a final thrombin concentration of 280 nM in the second suspension. Blood smears were then immersed in the ACPP-MB suspensions for 2 min with gentle shaking and were then removed and immersed successively in two beakers filled with 1× PBS (100 mL) to wash away unbound MBs. Prior to exposing the blood smear to the ACPP-MB not exposed to thrombin, 10 μg of recombinant hirudin (16 500 U/mg) in 10 μL of 1× PBS was spread across the blood smear to inhibit any remaining thrombin to ensure that microbubbles would not be activated. A coverslip was then added to the blood smear to seal the system and keep the MBs in a wet environment (Figure S11A). Seven fields of view were randomly selected on each microscopy slide before imaging. Microscopy slides were then observed at 20-fold magnification, and microbubbles present in the fields of view were counted; the average number of MBs per field of view was compared when the smears where immersed in activated or nonactivated ACPP-MBs (Figure S11B), and 80 ± 36 activated ACPP-MBs adhered to the smear per field of view after washing, whereas only 20 ± 10% of nonactivated ACPP-MBs were counted (Figure S11C). It is interesting to note that microbubbles with activated ACPP were adherent to the smear and not moving while microbubbles with nonactivated ACPP were drifting over the blood smear, confirming binding to the clot surface. Representative images of ACPP-MBs were taken at 63× magnification demonstrating DiD fluorescence on the MBs (Figure S11D).
Statistical Analysis
Data is represented as mean ± standard deviation. Statistical analysis was performed as an unpaired Student’s two-tailed t-test between the group with activated and nonactivated microbubbles (N = 7 for each): **p < 0.01.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by the Cancer Prevention and Research Institute of Texas (CPRIT) Grant RR150010 and NIH Grants K25EB017222 and R21CA212851. R.F.M. is a CPRIT Established Investigator. A.M.V. was supported in part by T32-EB005970 and the Radiological Society of North America Presidents Circle Research Resident Grant RR 1472. Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under the Center for Translational Medicine’s award UL1TR001105. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors thank Prof. Haydn Ball and the protein chemistry technology core at UT Southwestern for the syntheses and characterizations of ACPP and Fl-ACPP. The authors would also like to thank Dr. Khaled Nasr for his help with LC-MS training.
ABBREVATIONS
- ACPP
activatable cell-penetrating peptide
- DSPC
1,2-distearoyl-sn-glycero-3-phosphocholine
- DSPE-PEG(5000)-mal
1,2-distearoyl-sn-glycero-3-phos-phoethanolamine-N[maleimide(polyethylene glycol)5000]
- DVT
deep vein thrombosis
- EDTA
ethylenediaminetetraacetic acid
- Fl
fluorescein
- MB
microbubble
- PFB
perfluorobutane
- ROS
reactive oxygen species
- US
ultrasound
Footnotes
Author Contributions
J.L. and R.F.M. designed the experiments. J.L., A.M.V., A.M.A., and S.R.A. performed the experiments. J.L., A.M.V., and A.M.A. collected the data. J.L. and K.H. processed the imaging data. J.L., K.H., and R.F.M. analyzed and interpreted the data. S.R.S. contributed to the MB formulation. J.L., S.R.S., and R.F.M. performed the literature review. J.L. and R.F.M. wrote and edited the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
DEDICATION
Dedicated to the memory of Prof. Roger Y. Tsien, who contributed to the design of ACPP-MBs.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.7b10592.
HPLC chromatograms and MS spectra, LC-MS traces (UV and MS), fluorescence spectra from flow cytometry experiments, MFI graphs, and a schematic representation of the experiment design (PDF)
References
- 1.Coley BD, Trambert MA, Mattrey RF. Perfluorocarbon-Enhanced Sonography - Value in Detecting Acute Venous Thrombosis in Rabbits. AJR, Am J Roentgenol. 1994;163(4):961–964. doi: 10.2214/ajr.163.4.8092043. [DOI] [PubMed] [Google Scholar]
- 2.Tan CH, Bedi D, Vikram R. Sonography of Thrombosis of the Deep Veins of the Extremities: Clinical Perspectives and Imaging Review. J Clin Ultrasound. 2012;40(1):31–43. doi: 10.1002/jcu.20904. [DOI] [PubMed] [Google Scholar]
- 3.Puskas A, Balogh Z, Hadadi L, Imre M, Orban E, Kosa K, Brassai Z, Mousa SA. Spontaneous Recanalization in Deep Venous Thrombosis: a Prospective Duplex Ultrasound Study. Int J Angiol. 2007;26(1):53–63. [PubMed] [Google Scholar]
- 4.Keogan MT, Paulson EK, Paine SS, Hertzberg BS, Carroll BA. Bilateral Lower Extremity Evaluation of Deep Venous Thrombosis with Color Flow and Compression Sonography. J Ultrasound Med. 1994;13(2):115–118. doi: 10.7863/jum.1994.13.2.115. [DOI] [PubMed] [Google Scholar]
- 5.Zierler BK. Ultrasonography and Diagnosis of Venous Thromboembolism. Circulation. 2004;109(12-1):I9–14. doi: 10.1161/01.CIR.0000122870.22669.4a. [DOI] [PubMed] [Google Scholar]
- 6.Saini R, Hoyt K. Recent Developments in Dynamic Contrast-Enhanced Ultrasound Imaging of Tumor Angiogenesis. Imaging Med. 2014;6(1):41–52. doi: 10.2217/iim.13.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou KJ, Wang YG, Huang XN, Luby-Phelps K, Sumer BD, Gao JM. Tunable, Ultrasensitive pH-Responsive Nanoparticles Targeting Specific Endocytic Organelles in Living Cells. Angew Chem, Int Ed. 2011;50(27):6109–6114. doi: 10.1002/anie.201100884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou KJ, Liu HM, Zhang SR, Huang XN, Wang YG, Huang G, Sumer BD, Gao JM. Multicolored pH-Tunable and Activatable Fluorescence Nanoplatform Responsive to Physiologic pH Stimuli. J Am Chem Soc. 2012;134(18):7803–7811. doi: 10.1021/ja300176w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee D, Bae S, Ke Q, Lee J, Song B, Karumanchi SA, Khang G, Choi HS, Kang PM. Hydrogen Peroxide-Responsive Copolyoxalate Nanoparticles for Detection and Therapy of Ischemia-Reperfusion Injury. J Controlled Release. 2013;172(3):1102–1110. doi: 10.1016/j.jconrel.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinstain R, Savariar EN, Felsen CN, Tsien RY. In Vivo Targeting of Hydrogen Peroxide by Activatable Cell-Penetrating Peptides. J Am Chem Soc. 2014;136(3):874–877. doi: 10.1021/ja411547j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin C, Zhen X, Fan Q, Huang W, Pu K. Degradable Semiconducting Oligomer Amphiphile for Ratiometric Photoacoustic Imaging of Hypochlorite. ACS Nano. 2017;11(4):4174–4182. doi: 10.1021/acsnano.7b01092. [DOI] [PubMed] [Google Scholar]
- 12.Okuda K, Okabe Y, Kadonosono T, Ueno T, Youssif BG, Kizaka-Kondoh S, Nagasawa H. 2-Nitroimidazole-Tricarbocyanine Conjugate as a Near-Infrared Fluorescent Probe for in Vivo Imaging of Tumor Hypoxia. Bioconjugate Chem. 2012;23(3):324–329. doi: 10.1021/bc2004704. [DOI] [PubMed] [Google Scholar]
- 13.Krohn KA, Link JM, Mason RP. Molecular Imaging of Hypoxia. J Nucl Med. 2008;49(2):129S–148S. doi: 10.2967/jnumed.107.045914. [DOI] [PubMed] [Google Scholar]
- 14.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable Cell Penetrating Peptides Linked to Nanoparticles as Dual Probes for In Vivo Fluorescence and MR Imaging of Proteases. Proc Natl Acad Sci U S A. 2010;107(9):4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakatsuka MA, Mattrey RF, Esener SC, Cha JN, Goodwin AP. Aptamer-Crosslinked Microbubbles: Smart Contrast Agents for Thrombin-Activated Ultrasound Imaging. Adv Mater. 2012;24(45):6010–6016. doi: 10.1002/adma.201201484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malone CD, Olson ES, Mattrey RF, Jiang T, Tsien RY, Nguyen QT. Tumor Detection at 3 T with an Activatable Cell Penetrating Peptide Dendrimer (ACPPD-Gd), alpha T1Magnetic Resonance (MR) Molecular Imaging Agent. PLoS One. 2015;10(9):1–15. doi: 10.1371/journal.pone.0137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nejadnik H, Ye DJ, Lenkov OD, Donig JS, Martin JE, Castillo R, Derugin N, Sennino B, Rao JH, Daldrup-Link H. Magnetic Resonance Imaging of Stem Cell Apoptosis in Arthritic Joints with a Caspase Activatable Contrast Agent. ACS Nano. 2015;9(2):1150–1160. doi: 10.1021/nn504494c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viger ML, Collet G, Lux J, Nguyen Huu VA, Guma M, Foucault-Collet A, Olejniczak J, Joshi-Barr S, Firestein GS, Almutairi A. Distinct ON/OFF Fluorescence Signals from Dual-Responsive Activatable Nanoprobes Allows Detection of Inflammation with Improved Contrast. Biomaterials. 2017;133:119–131. doi: 10.1016/j.biomaterials.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coughlin SR. Thrombin Signalling and Protease-Activated Receptors. Nature. 2000;407(6801):258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 20.Gupta A, Williams MD, Macias WL, Molitoris BA, Grinnell BW. Activated Protein C and Acute Kidney Injury: Selective Targeting of PAR-1. Curr Drug Targets. 2009;10(12):1212–1226. doi: 10.2174/138945009789753291. [DOI] [PubMed] [Google Scholar]
- 21.Chen B, Cheng Q, Yang K, Lyden PD. Thrombin Mediates Severe Neurovascular Injury During Ischemia. Stroke. 2010;41(10):2348–2352. doi: 10.1161/STROKEAHA.110.584920. [DOI] [PubMed] [Google Scholar]
- 22.Nierodzik ML, Plotkin A, Kajumo F, Karpatkin S. Thrombin Stimulates Tumor-Platelet Adhesion Invitro and Metastasis Invivo. J Clin Invest. 1991;87(1):229–236. doi: 10.1172/JCI114976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nierodzik ML, Karpatkin S. Thrombin Induces Tumor Growth, Metastasis, and Angiogenesis: Evidence for a Thrombin-Regulated Dormant Tumor Phenotype. Cancer Cell. 2006;10(5):355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Lopez MT, Gutierrez-Rodriguez M, Herranz R. Thrombin-Activated Receptors: Promising Targets for Cancer Therapy? Curr Med Chem. 2010;17(2):109–128. doi: 10.2174/092986710790112639. [DOI] [PubMed] [Google Scholar]
- 25.Olson ES, Whitney MA, Friedman B, Aguilera TA, Crisp JL, Baik FM, Jiang T, Baird SM, Tsimikas S, Tsien RY, Nguyen QT. In Vivo Fluorescence Imaging of Atherosclerotic Plaques with Activatable Cell-Penetrating Peptides Targeting Thrombin Activity. Integr Biol. 2012;4(6):595–605. doi: 10.1039/c2ib00161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klibanov AL. Microbubble Contrast Agents: Targeted Ultrasound Imaging and Ultrasound-Assisted Drug-Delivery Applications. Invest Radiol. 2006;41(3):354–362. doi: 10.1097/01.rli.0000199292.88189.0f. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Hagemeyer CE, Hohmann JD, Leitner E, Armstrong PC, Jia F, Olschewski M, Needles A, Peter K, Ahrens I. Novel Single-Chain Antibody-Targeted Microbubbles for Molecular Ultrasound Imaging of Thrombosis: Validation of a Unique Non-Invasive Method for Rapid and Sensitive Detection of Thrombi and Monitoring of Success or Failure of Thrombolysis in Mice. Circulation. 2012;125(25):3117–3126. doi: 10.1161/CIRCULATIONAHA.111.030312. [DOI] [PubMed] [Google Scholar]
- 28.Hagisawa K, Nishioka T, Suzuki R, Takizawa T, Maruyama K, Takase B, Ishihara M, Kurita A, Yoshimoto N, Ohsuzu F, Kikuchi M. Enhancement of Ultrasonic Thrombus Imaging Using Novel Liposomal Bubbles Targeting Activated Platelet Glycoprotein IIb/IIIa Complex–In Vitro and In Vivo Study. Int J Cardiol. 2011;152(2):202–206. doi: 10.1016/j.ijcard.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Schumann PA, Christiansen JP, Quigley RM, McCreery TP, Sweitzer RH, Unger EC, Lindner JR, Matsunaga TO. Targeted-Microbubble Binding Selectively to GPIIb IIIa Receptors of Platelet Thrombi. Invest Radiol. 2002;37(11):587–593. doi: 10.1097/00004424-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Hall CS, Marsh JN, Scott MJ, Gaffney PJ, Wickline SA, Lanza GM. Time Evolution of Enhanced Ultrasonic Reflection Using a Fibrin-Targeted Nanoparticulate Contrast Agent. J Acoust Soc Am. 2000;108(6):3049–3057. doi: 10.1121/1.1322566. [DOI] [PubMed] [Google Scholar]
- 31.Marsh JN, Senpan A, Hu G, Scott MJ, Gaffney PJ, Wickline SA, Lanza GM. Fibrin-Targeted Perfluorocarbon Nanoparticles for Targeted Thrombolysis. Nanomedicine (London, U K) 2007;2(4):533–543. doi: 10.2217/17435889.2.4.533. [DOI] [PubMed] [Google Scholar]
- 32.Tiukinhoy-Laing SD, Buchanan K, Parikh D, Huang SL, MacDonald RC, McPherson DD, Klegerman ME. Fibrin Targeting of Tissue Plasminogen Activator-Loaded Echogenic Liposomes. J Drug Targeting. 2007;15(2):109–114. doi: 10.1080/10611860601140673. [DOI] [PubMed] [Google Scholar]
- 33.Jaffer FA, Tung CH, Gerszten RE, Weissleder R. In Vivo Imaging of Thrombin Activity in Experimental Thrombi With Thrombin-Sensitive Near-Infrared Molecular Probe. Arterioscler, Thromb, Vasc Biol. 2002;22(11):1929–1935. doi: 10.1161/01.atv.0000033089.56970.2d. [DOI] [PubMed] [Google Scholar]
- 34.Whitney M, Savariar EN, Friedman B, Levin RA, Crisp JL, Glasgow HL, Lefkowitz R, Adams SR, Steinbach P, Nashi N, Nguyen QT, Tsien RY. Ratiometric Activatable Cell-Penetrating Peptides Provide Rapid In Vivo Readout of Thrombin Activation. Angew Chem, Int Ed. 2013;52(1):325–330. doi: 10.1002/anie.201205721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Shi L, Hua X, Huang Y, Su S, Fan Q, Wang L, Huang W. Target-Induced Conjunction of Split Aptamer Fragments and Assembly with a Water-Soluble Conjugated Polymer for Improved Protein Detection. ACS Appl Mater Interfaces. 2014;6(5):3406–3412. doi: 10.1021/am405550j. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Hua X, Fan Q, Chao J, Su S, Huang YQ, Wang L, Huang W. Thioflavin T as an Efficient G-Quadruplex Inducer for the Highly Sensitive Detection of Thrombin Using a New Foster Resonance Energy Transfer System. ACS Appl Mater Interfaces. 2015;7(30):16458–16465. doi: 10.1021/acsami.5b03662. [DOI] [PubMed] [Google Scholar]
- 37.Hua N, Baik F, Pham T, Phinikaridou A, Giordano N, Friedman B, Whitney M, Nguyen QT, Tsien RY, Hamilton JA. Identification of High-Risk Plaques by MRI and Fluorescence Imaging in a Rabbit Model of Atherothrombosis. PLoS One. 2015;10(10):1–15. doi: 10.1371/journal.pone.0139833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakatsuka MA, Barback CV, Fitch KR, Farwell AR, Esener SC, Mattrey RF, Cha JN, Goodwin AP. In Vivo Ultrasound Visualization of Non-Occlusive Blood Clots with Thrombin-Sensitive Contrast Agents. Biomaterials. 2013;34(37):9559–9565. doi: 10.1016/j.biomaterials.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Church CC. The Effects of an Elastic Solid-Surface Layer on the Radial Pulsations of Gas-Bubbles. J Acoust Soc Am. 1995;97(3):1510–1521. [Google Scholar]
- 40.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor Imaging by Means of Proteolytic Activation of Cell-Penetrating Peptides. Proc Natl Acad Sci U S A. 2004;101(51):17867–17872. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weitz JI, Hudoba M, Massel D, Maraganore J, Hirsh J. Clot-Bound Thrombin is Protected from Inhibition by Heparin-Antithrombin-III but is Susceptible to Inactivation by Antithrombin-III-Independent Inhibitors. J Clin Invest. 1990;86(2):385–391. doi: 10.1172/JCI114723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weitz JI, Hirsh J, Samama MM. New Anticoagulant Drugs. Chest. 2004;126(3):265s–286s. doi: 10.1378/chest.126.3_suppl.265S. [DOI] [PubMed] [Google Scholar]
- 43.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis Associates With Inflammation in Early-Stage Atherosclerosis Evaluated by Molecular Imaging In Vivo. Circulation. 2007;116(24):2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 44.Nutescu EA, Shapiro NL, Chevalier A. New Anticoagulant Agents: Direct Thrombin Inhibitors. Cardiol Clin. 2008;26(2):169–187. doi: 10.1016/j.ccl.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Spyropoulos AC. Brave New World: The Current and Future Use of Novel Anticoagulants. Thromb Res. 2008;123:S29–S35. doi: 10.1016/j.thromres.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Satinover SJ, Dove JD, Borden MA. Single-Particle Optical Sizing of Microbubbles. Ultrasound Med Biol. 2014;40(1):138–147. doi: 10.1016/j.ultrasmedbio.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 47.Wolberg AS. Thrombin Generation and Fibrin Clot Structure. Blood Rev. 2007;21(3):131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 48.Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci. 2008;38(1):15–23. doi: 10.1016/j.transci.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruhl H, Muller J, Harbrecht U, Fimmers R, Oldenburg J, Mayer G, Potzsch B. Thrombin Inhibition Profiles in Healthy Individuals and Thrombophilic Patients. Thromb Haemostasis. 2012;107(5):848–853. doi: 10.1160/TH11-10-0719. [DOI] [PubMed] [Google Scholar]
- 50.Bader KB, Gruber MJ, Holland CK. Shaken and Stirred: Mechanisms of Ultrasound-Enhanced Thrombolysis. Ultrasound Med Biol. 2015;41(1):187–196. doi: 10.1016/j.ultrasmedbio.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Gkanatsas Y, Palasubramaniam J, Hohmann JD, Chen YC, Lim B, Hagemeyer CE, Peter K. Thrombus-Targeted Theranostic Microbubbles: A New Technology towards Concurrent Rapid Ultrasound Diagnosis and Bleeding-free Fibrinolytic Treatment of Thrombosis. Theranostics. 2016;6(5):726–738. doi: 10.7150/thno.14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bader KB, Bouchoux G, Holland CK. Sonothrombolysis. Adv Exp Med Biol. 2016;880:339–362. doi: 10.1007/978-3-319-22536-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bader KB, Bouchoux G, Peng T, Klegerman ME, McPherson DD, Holland CK. Thrombolytic Efficacy and Enzymatic Activity of rt-PA-Loaded Echogenic Liposomes. J Thromb Thrombolysis. 2015;40(2):144–155. doi: 10.1007/s11239-015-1204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laing ST, Moody MR, Kim H, Smulevitz B, Huang SL, Holland CK, McPherson DD, Klegerman ME. Thrombolytic Efficacy of Tissue Plasminogen Activator-Loaded Echogenic Liposomes in a Rabbit Thrombus Model. Thromb Res. 2012;130(4):629–635. doi: 10.1016/j.thromres.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ Ecosystem: An Open Platform for Biomedical Image Analysis. Mol Reprod Dev. 2015;82(7–8):518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sorace AG, Saini R, Mahoney M, Hoyt K. Molecular Ultrasound Imaging Using a Targeted Contrast Agent for Assessing Early Tumor Response to Antiangiogenic Therapy. J Ultrasound Med. 2012;31(10):1543–1550. doi: 10.7863/jum.2012.31.10.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoyt K, Warram JM, Wang D, Ratnayaka S, Traylor A, Agarwal A. Molecular Ultrasound Imaging of Tissue Inflammation Using an Animal Model of Acute Kidney Injury. Mol Imaging Biol. 2015;17(6):786–792. doi: 10.1007/s11307-015-0860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.