

Abstract
Nonalcoholic steatohepatitis (NASH) is the advanced form of nonalcoholic fatty liver disease (NAFLD) that sets the stage for further liver damage. The mechanism for the progression of NASH involves multiple parallel hits, including oxidative stress, mitochondrial dysfunction, inflammation, and others. Manipulation of any of these pathways may be an approach to prevent NASH development and progression. Arachidyl‐amido cholanoic acid (Aramchol) is presently in a phase IIb NASH study. The aim of the present study was to investigate Aramchol's mechanism of action and its effect on fibrosis using the methionine‐ and choline‐deficient (MCD) diet model of NASH. We collected liver and serum from mice fed an MCD diet containing 0.1% methionine (0.1MCD) for 4 weeks; these mice developed steatohepatitis and fibrosis. We also collected liver and serum from mice receiving a control diet, and metabolomes and proteomes were determined for both groups. The 0.1MCD‐fed mice were given Aramchol (5 mg/kg/day for the last 2 weeks), and liver samples were analyzed histologically. Aramchol administration reduced features of steatohepatitis and fibrosis in 0.1MCD‐fed mice. Aramchol down‐regulated stearoyl‐coenyzme A desaturase 1, a key enzyme involved in triglyceride biosynthesis and the loss of which enhances fatty acid β‐oxidation. Aramchol increased the flux through the transsulfuration pathway, leading to a rise in glutathione (GSH) and the GSH/oxidized GSH ratio, the main cellular antioxidant that maintains intracellular redox status. Comparison of the serum metabolomic pattern between 0.1MCD‐fed mice and patients with NAFLD showed a substantial overlap. Conclusion: Aramchol treatment improved steatohepatitis and fibrosis by 1) decreasing stearoyl‐coenyzme A desaturase 1 and 2) increasing the flux through the transsulfuration pathway maintaining cellular redox homeostasis. We also demonstrated that the 0.1MCD model resembles the metabolic phenotype observed in about 50% of patients with NAFLD, which supports the potential use of Aramchol in NASH treatment. (Hepatology Communications 2017;1:911–927)
Abbreviations
- 0.1MCD
methionine‐ and choline‐deficient diet containing 0.1% methionine
- ACC
acetylcoenzyme A carboxylase
- ALT
alanine aminotransferase
- Aramchol
arachidyl‐amido cholanoic acid
- AST
aspartate aminotransferase
- CBS
cystathionine β‐synthase
- CD
choline‐deficient
- CD64
cluster of differentiation 64
- CoA
coenzyme A
- COL1A1
collagen, type I, alpha 1
- CPT1A
carnitine palmitoyltransferase 1A
- DG
diglyceride
- DMSO
dimethyl sulfoxide
- FA
fatty acid
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GPX1
glutathione peroxidase 1
- GS
glutathione synthetase
- GSH
glutathione
- GSR
glutathione reductase
- GSSG
oxidized glutathione
- GSTM1‐3
glutathione S‐transferases 1‐3
- Lyso
lysophospholipid
- Mat1a
methionine adenosyltransferase‐1a
- MCD
methionine and choline deficient
- MEM
minimum essential medium
- mRNA
messenger RNA
- MUFA
monounsaturated fatty acid
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- oxLA
oxidized derivatives of linoleic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PPARγ
peroxisome proliferator‐activated receptor gamma
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- RT‐PCR
quantitative real‐time polymerase chain reaction
- SAH
S‐adenosylhomocysteine
- SAMe
S‐adenosylmethionine
- SCD1
stearoyl‐coenzyme A desaturase 1
- TG
triglyceride
- TXNRD1
thioredoxin reductase
- VLDL
very low density lipoprotein
Introduction
Nonalcoholic steatohepatitis (NASH), the advanced form of nonalcoholic fatty liver disease (NAFLD), is defined histologically by the presence of hepatic fat (steatosis) with inflammation and hepatocellular ballooning.1, 2 NASH is a progressive disease that can lead to further liver injury, advanced fibrosis, cirrhosis, and hepatocellular carcinoma.1, 2 During the last decades, the incidence of NAFLD has multiplied, and it is now the most common disease of the liver, with a prevalence between 10% and 40% in Western countries and approximately 10%‐30% of them progressing to NASH. At present, there is no approved therapy for NASH; current treatments focus on weight loss and control of medical conditions associated with the disease, such as obesity, diabetes, and hyperlipidemia.1, 2
Several therapies with diverse mechanisms of action are currently in clinical development.3 One of these therapies is arachidyl‐amido cholanoic acid (Aramchol). In a phase IIa, Aramchol administered to patients with NAFLD for 3 months was shown to be safe, well tolerated, and significantly reduced liver fat content.4 Currently, a large phase IIb study in patients with NASH is ongoing (ARamchol for the REsolution of STeatohepatitis [ARREST] study, NCT 02279524). Aramchol targets stearoyl‐coenzyme A (CoA) desaturase 1 (SCD1),5 the rate‐limiting step in the synthesis of monounsaturated fatty acids (MUFAs), the major fatty acid (FA) of triglycerides (TGs), cholesteryl esters, and membrane phospholipids.6 SCD1 deficiency in mice has been demonstrated to reduce lipid synthesis and increase mitochondrial FA β‐oxidation and insulin sensitivity in various tissues, including the liver.7, 8 Accordingly, SCD1 deficiency has been demonstrated to prevent liver steatosis in several mouse models of NAFLD, including mice fed high‐carbohydrate and high‐fat diets.9 The aim of this study was to investigate Aramchol's effect in liver lipid metabolism in NASH. To answer this question, we used mice fed a methionine‐ and choline‐deficient (MCD) diet, a widely used mouse model of NASH,10 in a treatment mode. Aramchol down‐regulated SCD1, which led to a decrease in hepatic FAs and TGs, ameliorated inflammation, and reversed fibrosis. Aramchol improved β‐oxidation by increasing the flux through the transsulfuration pathway, leading to a rise in glutathione (GSH) and the GSH/oxidized GSH (GSSG) ratio, the main cellular antioxidant that maintains intracellular redox status. The data support the potential use of Aramchol in NASH treatment.
Materials and Methods
ANIMAL EXPERIMENTS
We purchased C57BL/6 male mice (Charles River, St Germain sur l'Arbresle, France) at 8 weeks of age and allowed them to acclimate for a period of 1 week. The mice were placed in groups of 10 on a diet devoid of choline and with 0.1% methionine (0.1MCD diet) for a period of 4 weeks. A control group was maintained on a regular diet with 1,030 mg/kg choline and 0.3% methionine (Teklad Global 14% Protein Rodent Maintenance diet; Envigo RMS Spain, Sant Feliu de Codines, Spain). A pilot experiment (Supporting Fig. S1) demonstrated that the mice subjected to the 0.1MCD diet developed signs of steatosis, inflammation, and fibrosis comparable to those induced by a diet completely devoid of methionine and choline without the associated weight loss. After 2 weeks of feeding on the 0.1MCD diet, mice were divided into groups of 10 and treated by intragastric gavage with a formulation of 5 mg/kg/day of Aramchol in vehicle (1.7% carboxymethyl cellulose; BUFA, IJsselstein, the Netherlands) and 0.2% sodium lauryl sulfate (Sigma Aldrich, Steinheim, Germany) or vehicle alone. Animals kept on the normal diet were also provided the vehicle preparation. The mice were analyzed for transaminases in blood at 0, 2, and 4 weeks of exposure to the 0.1MCD diet. All mice were sacrificed at 4 weeks. Livers were then removed and snap frozen in liquid nitrogen, optical coherence tomography cryo‐compound embedded, or formalin fixed. All experiments with animals were approved by CIC bioGUNE's Biosafety and Bioethics Committee and the Country Council of Bizkaia.
CELLULAR EXPERIMENTS
Primary hepatocytes were isolated from C57BL/6 mice livers by collagenase perfusion,11 seeded over collagen‐coated tissue culture dishes or cover slips, and cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) in a 5% CO2–95% air incubator at 37°C. After 2 hours of attachment, the culture medium was removed and replaced by serum‐free MEM. After 3 hours in culture, the medium was replaced by serum‐free MEM or MCD medium, with or without Aramchol (10 μM), and cultured for an additional 48 hours. The human hepatic stellate cell line LX‐2 (Millipore Corporation, Darmstadt, Germany)12 was cultured in Dulbecco's modified Eagle's medium supplemented with 2% FBS for 30 hours, after which the medium was replaced by serum‐free Dulbecco's modified Eagle's medium. After 12 hours in culture, LX‐2 cells were treated with Aramchol (10 μM) for 24 hours. For cell cultures, 100 mM Aramchol solution was prepared by dissolving 70 mg in 1 mL dimethyl sulfoxide (DMSO). This stock solution was then further diluted 10 times in DMSO before treatment of cells at a final concentration of 10 μM. The final concentration of DMSO in culture media was 0.1%.
Hepatocytes in culture were incubated with Molecular Probes BODIPY 493/503 (4,4‐difluoro‐1,3,5,7,8‐pentamethyl‐4‐bora‐3a,4a‐diaza‐s‐indacene) (Invitrogen, Carlsbad, CA) at a concentration of 10 μg/mL for 45 minutes prior to fixation (4% paraformaldehyde). Exposure settings were unchanged throughout acquisition. Quantification of lipid bodies was performed using FRIDA software (http://bui3.win.ad.jhu.edu/frida/; Johns Hopkins University) as detailed elsewhere.13 Cellular reactive oxygen species (ROS) production in primary hepatocytes was assessed using CellROX Deep Green Reagent (ThermoFisher, Waltham, MA). The hepatocytes were loaded with 1.5 μM CellROX in 10% FBS‐MEM for 30 minutes at 37°C in a CO2 incubator. The hepatocytes were then carefully washed 3 times with phosphate‐buffered saline, collected, and analyzed by flow cytometry.
METABOLOMICS ANALYSIS
Metabolome Analysis
Serum and liver metabolic profiles were semiquantified as described.14, 15 Metabolomics data were preprocessed using the TargetLynx application manager for MassLynx 4.1 (Waters Corp., Milford, MA). Intrabatch and interbatch normalization followed the procedure described.16
Quantification of Methionine Metabolites
Extraction of the main metabolites belonging to the methionine pathway were performed as described.17, 18
Quantification of Total Lipids
Livers (40 mg) were homogenized and lipids extracted as described.19 TGs were quantified using a diagnostic kit (A. Menarini Diagnostics, Italy). Phosphatidylethanolamine (PE), phosphatidylcholine (PC), and diglycerides (DGs) were separated by thin layer chromatography and quantified as described.20
PROTEOMICS
Proteins were digested by the filter‐assisted sample preparation method.21 Peptides were speed vacuumed in a RVC2 25 speedvac concentrator (Christ GmbH, Osterode, Germany), resuspended in 3% acetonitrile and 0.1% formic acid, and sonicated for 5 minutes prior to analysis. Acquisition of data and differential protein analysis were carried out as described.22 Briefly, peptide mixtures were separated on a nanoACQUITY ultra performance liquid chromatography system (Waters Corp., Manchester, United Kingdom) connected to an LTQ Orbitrap XL ETD mass spectrometer (Thermo Electron, Bremen, Germany) using a 120‐minute gradient from 3% to 50% of mobile phase B (acetonitrile, 0.1% formic acid) at 300 nL/minute. Full mass spectrometry survey spectra (m/z 400‐2,000) were acquired in the Orbitrap, and the six most intense ions (charge states of 2 and 3) were subjected to collision‐induced dissociation fragmentation in the linear ion trap. Progenesis liquid chromatography–mass spectrometry software (Nonlinear Dynamics Ltd., Newcastle upon Tyne, United Kingdom) was used for differential protein expression analysis following the sequential software steps. Protein identification was carried out using Mascot Search engine (www.matrixscience.com; Matrix Science, London, United Kingdom). Carbamidomethylation of cysteine was selected as the fixed modification, and oxidation of methionine was selected as the variable. Peptide mass tolerance of 10 ppm and 0.5 Da fragment mass tolerance were adopted as search parameters. Spectra were searched against the UniProt/Swiss‐Prot database, limiting the search to Mus musculus (version 2016_09, 17,239 sequences; 9,744,969 residues), and a decoy search was carried out to estimate the false discovery rate. Only peptides with a false discovery rate <1% were further considered. Relative abundance comparison was carried out using the Student t test (P < 0.05).
WESTERN BLOT
Frozen liver tissue samples or cultured cells were homogenized in lysis buffer (10 mM Tris/HCl pH 7.6, 5 mM ethylene diamine tetraacetic acid, 50 mM NaCl, 1% Triton X‐100, complete protease inhibitor cocktail, and 50 mM NaF) and centrifuged (10,000g, 20 minutes, 4°C). Protein concentration was determined by using the BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). After quantification, 10‐25 μg of protein was electrophoresed on sodium dodecyl sulfate‐polyacrylamide gels and transferred onto membranes. Membranes were incubated with the following antibodies: SCD1 (C12H5) rabbit monoclonal antibody (2794S; Cell Signaling Technology, Danvers, MA), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) antibody [6C5] (ab8245; Abcam, Cambridge, United Kingdom), β‐ACTIN antibody clone AC‐15 (A5441; Sigma‐Aldrich, St. Louis, MO), and COLLAGEN I type I antibody [C‐18] (sc‐8784; Santa Cruz Biotechnology, Dallas, TX). As secondary antibodies, we used anti‐rabbit immunoglobulin G‐horseradish peroxidase‐linked and anti‐mouse immunoglobulin G‐horseradish peroxidase‐linked (Cell Signaling Technology) antibodies. Densities were analyzed by Image J (National Institutes of Health, Bethesda, MD) software. For densitometric quantification, levels of each protein for each sample were normalized to loading controls (GAPDH or β‐ACTIN).
RNA ISOLATION AND REAL‐TIME POLYMERASE CHAIN REACTION
Total RNA was isolated with Trizol (Invitrogen, Carlsbad, CA). One μg of total RNA was treated with DNAse (Invitrogen) and reverse transcribed into complementary DNA using M‐MLV Reverse Transcriptase (Invitrogen). Quantitative real‐time polymerase chain reaction (RT‐PCR) was performed using SYBR Select Master Mix (Applied Biosystems, Foster City, CA) and the Viia 7 Real‐Time PCR System (Applied Biosystems). PCR was executed with the following primers: Collagen, type I, alpha 1F (COL1A1F), 5′‐TTGACCAACCGAACATGACC‐3′; COL1A1R, 5′‐GCAGAAAGGGACTTACCCCC‐3′; peroxisome proliferator‐activated receptor gamma F (PPARγF), 5′‐GCTGTGCAGGAGATCACAGA‐3′; PPARγR, 5′‐GGGCTCCATAAAGTCACCAA‐3′; GAPDHF, 5′‐AATGAAGGGGTCATTGATGG‐3′; GAPDHR, 5′‐AAGGTGAAGGTCGGAGTCA‐3′. Expression levels were normalized to the level of GAPDH messenger RNA (mRNA) in each sample.
HISTOLOGICAL STAINING
Liver samples were stained with Sudan Red III (251731.1606; Panreac, Castellar del Vallés, Barcelona, Spain) for lipid accumulation, Sirius Red (Direct Red 385548, Fast Green FCF:F7258, picric acid 197378; Sigma‐Aldrich) for fibrosis, and hematoxylin (HHS128‐4L; Sigma‐Aldrich) and eosin (HT110232‐1L; Sigma‐Aldrich), as described.18 Additionally, liver sections were immunostained for detecting F4/80 (MCA497BB; Bio‐Rad, Hercules, CA) and cluster of differentiation 64 (CD64; ab119843; Abcam, Cambridge, United Kingdom) for inflammation, as described.18 Quantification of staining areas was performed using FRIDA software and expressed as percentage of stained area.
STATISTICAL ANALYSIS
Data were represented as mean ± SEM. Differences between groups were tested using the Student t test. Significance was defined as P < 0.05. All calculations were performed using statistical software package R v.3.1.1 (R Development Core Team, 2011; https://cran.r‐project.org/). Hierarchical clustering analysis based on metabolite ion intensities was performed with the cluster R package, including Ward's minimum variance method as the agglomeration method, as described.18
Results
HEPATIC S‐ADENOSYLMETHIONINE METABOLISM IN MICE FED A 0.1MCD DIET
We used a modification of the canonical MCD diet model,10 adding 0.1% methionine to the diet. This choline‐deficient (CD) diet containing 0.1% methionine (0.1MCD diet) induced steatosis, inflammation, and fibrosis, just like the MCD diet model (Supporting Fig. S1). The increase in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was also similar compared to the MCD diet model. Body weight loss, however, was less and stabilized after the first 2 or 3 days compared to the control group. Feeding the 0.1MCD diet to mice resulted in a reduction in hepatic S‐adenosylmethionine (SAMe) and an increase in S‐adenosylhomocysteine (SAH) (Fig. 1A,B). A reduction in the SAMe/SAH ratio is known to cause inhibition of PE N‐methyltransferase, the enzyme that converts PE rich in polyunsaturated fatty acids (PUFAs), such as PE(22:6), to PC rich in PUFAs by N‐methylation of PE.23 Accordingly, 0.1MCD‐fed mice showed an increase in hepatic PE(22:6) and a reduction in PC(22:6) (Fig. 1A,B), the main phospholipid comprising the outer coat of very low density lipoproteins (VLDLs), which consequently impairs VLDL formation and export, producing TG accumulation. The reduction in serum TG levels induced by feeding mice the 0.1MCD diet is presumably due to the deficient hepatic secretion of VLDLs.24 Consistently, the serum content of PC(22:6) was diminished in 0.1MCD‐fed mice compared to mice fed a control diet (Fig. 1B). Feeding the 0.1MCD diet to mice also led to a reduction in GSH hepatic concentration, the main output of the transsulfuration pathway, and of its precursor cystathionine (Fig. 1A,B). Conversely, GSSG increased, which translated into a lower GSH/GSSG ratio (Fig. 1A,B), a biomarker of oxidative stress.25
Figure 1.
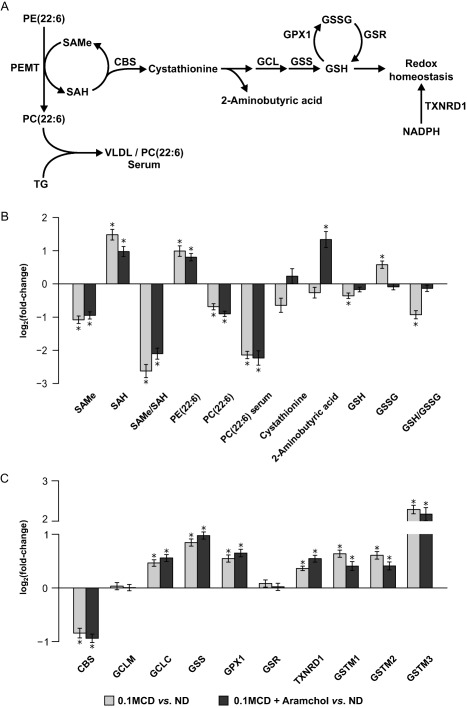
Hepatic S‐adenosylmethionine metabolism in mice fed a 0.1MCD diet and treated with vehicle or with Aramchol. (A) Schematic representation of hepatic SAMe metabolism. SAMe, the main biological methyl donor, is generated from methionine in an ATP‐dependent process catalyzed by methionine adenosyltransferase. One of the enzymes that contributes most to the hepatic transmethylation flux is PEMT, which converts PE rich in PUFAs into PC rich in PUFAs, using SAMe and generating SAH. PCs rich in PUFAs are the main phospholipids comprising the outer coat of VLDLs, whereas TGs can be incorporated and eliminated in the form of VLDLs. SAH is routed toward the transsulfuration pathway, generating 2‐aminobutyric acid and GSH, which plays a key role in maintaining redox homeostasis together with NADPH. (B) Relative fold change (log2) in the hepatic content of the main metabolites involved in hepatic SAMe metabolism in mice fed a 0.1MCD diet compared to mice fed a normal diet. Mice that were fed the 0.1MCD diet and treated with vehicle or with Aramchol showed reduced hepatic SAMe levels, increased SAH levels, and a reduction in the SAMe/SAH ratio. These mice also showed an increase in hepatic PE(22:6) and a reduction in hepatic and serum content of PC(22:6). Feeding the mice a 0.1MCD diet also led to a reduction in the hepatic concentration of cystathionine and GSH and to an increase in GSSG, which in turn led to a reduction in the GSH/GSSG ratio. The 2‐aminobutyric acid content remained constant. (C) Relative fold change (log2) in the protein content of enzymes involved in hepatic SAMe metabolism in 0.1MCD‐fed mice treated with vehicle or with Aramchol compared to mice fed a normal diet. A 0.1MCD diet led to changes in the main enzymes involved in GSH synthesis and catabolism. Mice fed a 0.1MCD diet showed a reduced content of CBS, normal levels of GCLM, and increased content of GCLC and GS. These mice also showed increased levels of GPX1, GSTM1‐3, and TXNRD1 and unchanged levels of GSR. *P < 0.05. Abbreviations: ATP, adenosine triphosphate; GCLC, GCL catalytic subunit; GCLM, modifier subunit; NADPH, nicotinamide dinucleotide reduced phosphate; PEMT, phosphatidylethanolamine‐N‐methyltransferase; ND, normal diet. Data were represented as mean ± SEM.
At the protein expression level, compared to animals fed a control diet, mice fed the 0.1MCD diet showed reduced content of cystathionine β‐synthase (CBS), the first enzyme linking SAMe metabolism with the transsulfuration pathway; increased levels of glutamate–cysteine ligase (GCL) catalytic subunit, the rate‐limiting step in GSH synthesis; normal content of GCL modifier subunit, the regulatory subunit of GCL; and increased content of GSH synthetase (GS), the last step in GSH synthesis (Fig. 1A,C). The protein content of various GSH‐consuming enzymes, such as GSH peroxidase 1 (GPX1), GSH S‐transferases 1‐3 (GSTM1‐3), and thioredoxin reductase 1 (TXNRD1), was increased, while the content of GSH reductase (GSR), the enzyme that regenerates GSH from GSSG, was unchanged, compared to mice fed a control diet (Fig. 1A,C). These results suggest that the fall in GSH and the reduction in the GSH/GSSG ratio are largely a result of oxidative stress and increased use because all GSH‐utilizing enzymes are increased.
To investigate if patients with NAFLD presented alterations in hepatic metabolism similar to those observed in 0.1MCD‐fed mice, we compared the liver metabolome of 0.1MCD‐fed mice and animals fed a control diet and generated a list with the fold change and P values for each metabolite (Supporting Table S1). Next, we did the same for the serum metabolome and generated a second list (Supporting Table S1). Finally, we analyzed if the serum metabolic profile reflected liver metabolism in the 0.1MCD‐fed mice by comparing both sets of data, observing that the system may be defined by two linear regression plots, one made by the pool of TGs (rho = 0.865, P = 2.5E‐12) and the second integrated by all lipids except TGs (rho = 0.576, P = 2.2E‐16) (Supporting Fig. S2). We then selected the top 50 serum metabolites that more significantly differentiated between the 0.1MCD‐fed mice and animals fed a control diet (Fig. 2A) and used this metabolic signature to classify a cohort of 535 patients with biopsy‐proven NAFLD (353 with simple steatosis and 182 with NASH). The clinical characteristics and serum metabolomics of this cohort has been published.18 Silhouette cluster analysis revealed that this signature subclassified patients with NAFLD into two main clusters, one of them showing a metabolic profile similar to that identified in the 0.1MCD mouse model (Fig. 2B). For validation, we used the developed procedure,18 which consists of carrying out a random partition of samples (50/50) in two cohorts (estimation and validation), and executed the cluster analysis described above to select those patients with a metabolic profile resembling that of the 0.1MCD‐fed mice. After a 1,000‐fold repetition of this random partition, patients showing a metabolic profile similar to that identified in the 0.1MCD mouse model ≥700 times were selected. This metabocentric analysis showed that 47% (n = 250) of patients with NAFLD displayed a metabolic phenotype similar to the 0.1MCD‐fed mice, 27% (n = 147) were classified ≥700 times as non‐0.1MCD, and the remaining 26% (n = 138) were indeterminate (classified <700 times as either 0.1MCD or non‐0.1MCD) (Fig. 2C).
Figure 2.
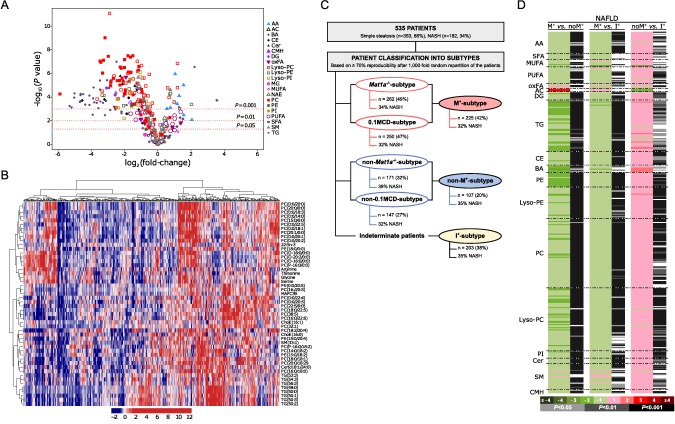
Identification of a subset of patients with NAFLD showing a serum metabolomic profile similar to that of the 0.1MCD mouse model. (A) Volcano plot representation indicating the –log10(P value) and log2(fold change) of individual serum metabolic ion features of mice fed a 0.1MCD diet compared to mice fed a normal diet. (B) Heatmap representation of the serum metabolomic profile from 535 patients with biopsy‐confirmed NAFLD. The scale indicates (vertical axis) the relative ion abundance of each metabolite in the serum extract of a given subject with respect to that found in the rest of the study population, 0 being the mean value. Metabolite selection is based on the top 50 serum metabolites that more significantly differentiated between mice fed a 0.1MCD diet and mice fed a normal diet. The hierarchical clustering is based on optimum average silhouette width obtaining two main clusters; the first (left‐hand cluster) resembles the serum metabolomic profile observed in mice fed a 0.1MCD diet (0.1MCD subtype), while the second cluster (right‐hand cluster) shows a different metabolomic profile (non‐0.1MCD subtype). (C) Schematic representation summarizing the results obtained from the metabocentric analysis. A 1,000‐fold repetition of a random partition of samples (50/50) in two cohorts (estimation and validation) and subsequent cluster analysis was performed based on the 50 serum metabolites selected. Patients were classified into 0.1MCD subtype and non‐0.1MCD subtype depending on the frequency distribution. Following the criteria based on 70% reproducibility, 250 patients showed a metabolic phenotype similar to 0.1MCD‐fed mice and 147 were classified as non‐0.1MCD subtype. The remaining 138 patients showed a reproducibility of less than 70% (indeterminate group). Similarities were found between patients showing a 0.1MCD metabolic signature and patients showing a Mat1a –/– metabolic signature.18 Of the patients with NAFLD classified as Mat1a–/– subtype, 90% were also classified as 0.1MCD subtype and called M+ subtype; 73% of the patients classified as non‐M subtype were also classified as non‐0.1MCD subtype and called non‐M+ subtype. The remaining patients (203) were considered indeterminate and called I+ subtype. (D) Heatmap representing the comparisons of the serum metabolic profiles of validated M+, non‐M+, and I+ groups, displaying the log2(fold change) and unpaired Student t test. Abbreviations: AA, amino acids; AC, acyl carnitines; BA, bile acids; CE, cholesteryl esters; Cer, ceramides; CMH, monohexosylceramides; oxFA, oxidized fatty acids; MG, monoglycerides; NAE, N‐acylethanolamines; SFA, saturated fatty acids; SM, sphingomyelins.
Using this same approach, we recently demonstrated that 49% of this same cohort of patients (n = 262) showed a metabolomic profile similar to methionine adenosyltransferase‐1a–/– (Mat1a–/–) mice.18 Because Mat1a encodes the main gene involved in hepatic SAMe synthesis and Mat1a–/– mice show a reduction in liver SAMe, GSH, and PC‐PUFAs as well as impaired VLDL export and spontaneously developed NASH,18 we hypothesized that patients with NAFLD showing an 0.1MCD metabolomic signature and those with a Mat1a–/– signature would coincide. We observed that 90% (n = 225) of patients classified as 0.1MCD subtype (0.1MCD signature) were also classified as M subtype (Mat1a–/– signature). We called this group M+ subtype (Fig. 2C). These results strengthen the concept that an impaired synthesis of hepatic SAMe may be a frequent feature in patients with NAFLD.18 Similarly, of the 147 patients classified as non‐0.1MCD‐fed mice, 107 (73%) had previously been classified as non‐M subtype using the Mat1a –/– mouse model. We called this group non‐M+ subtype (Fig. 2C). The remaining 203 patients were classified as indeterminate. We called this group I+ subtype (Fig. 2C).
Comparison of the serum metabolomic profile of the validated M+ and non‐M+ subtypes revealed that 93.6% of all lipids and amino acids analyzed (n = 328) were significantly reduced in the M+ subtype (Fig. 2D). The list of lipids analyzed included saturated FAs, MUFAs, PUFAs, DGs, TGs, choline, and ethanolamine glycerophospholipids (diacyl, plasmalogens, and lyso phospholipids (Lyso) forms); sphingolipids (ceramides and sphingomyelins); cholesteryl esters; and bile acids. These results suggest that, as in the Mat1a –/– and 0.1MCD models, VLDL export and perhaps the secretion of other lipid vesicles, such as exosomes, are impaired in M+ subtype patients with NAFLD compared to the non‐M+ subtype. Consistent with this hypothesis, the serum contents of the four amino acids that feed one‐carbon metabolism to generate SAMe (threonine, serine, glycine, and methionine) and the levels of taurine, an output of SAMe catabolism, were reduced in M+ subtype patients with NAFLD compared to the non‐M+ subtype group (Fig. 2D). The I+ subtype group showed an intermediate metabolomic phenotype, exhibiting higher content of serum lipids and amino acids than the M+ subtype but lower than the non‐M+ subtype (Fig. 2D).
HEPATIC LIPID METABOLISM IN 0.1MCD‐FED MICE
Feeding with the 0.1MCD diet associated with hepatic accumulation of FAs, DGs, TGs, and cholesteryl esters as well as with the accumulation of oxidized FAs, including oxidized derivatives of linoleic acid (oxLA), a human NASH biomarker26 (Fig. 3A,B). Proteomics data analysis searching for proteins differentially expressed in 0.1MCD‐fed mice involved in lipid metabolism revealed that the content of fatty acid translocase (CD36), an FA transporter with an overexpression that correlates with TG accumulation in human NAFLD,27 was augmented whereas the content of SCD1 was reduced (Fig. 3A,C). This reduction in SCD1 activity was confirmed by measuring the hepatic levels of MUFAs and the FA(16:1)/(16:0) ratio, which were decreased (Fig. 3B). The FA(16:1)/(16:0) ratio was also decreased in the serum of 0.1MCD‐fed mice compared to animals fed a control diet (Fig. 3B). The content of several key enzymes involved in de novo lipogenesis (adenosine triphosphate citrate lyase, acetyl‐CoA carboxylase 1 [ACC1], and fatty acid synthase) was slightly reduced or unaltered, while the protein content of acylglycerol‐3‐phosphate O‐acyltransferase 3, a key enzyme involved in TG synthesis, was increased (Fig. 3A,C). Our results also showed an increase in the content of carnitine palmitoyltransferase 1A (CPT1A), the rate‐limiting step in mitochondrial β‐oxidation, and normal levels of 3‐hydroxyacyl‐CoA dehydrogenase A and B, the α and β subunits of the trifunctional enzyme that catalyzes the three last steps of mitochondrial β‐oxidation (Fig. 3A,C). In agreement with previous findings,24, 28 the liver of 0.1MCD‐fed mice exhibited an increase in the content of mitochondrial uncoupling protein 2 and microsomal cytochrome P450 family 4, subfamily a, polypeptide 14 (Fig. 3C). The protein content of ACC2 (Fig. 3A,C), the enzyme that synthesizes mitochondrial malonyl‐CoA, an inhibitor of CPT1A, was reduced (Fig. 3A,C). These results agree with previous studies showing that MCD‐mediated reduction of SCD1 stimulates FA oxidation through the β‐oxidation pathway.24
Figure 3.
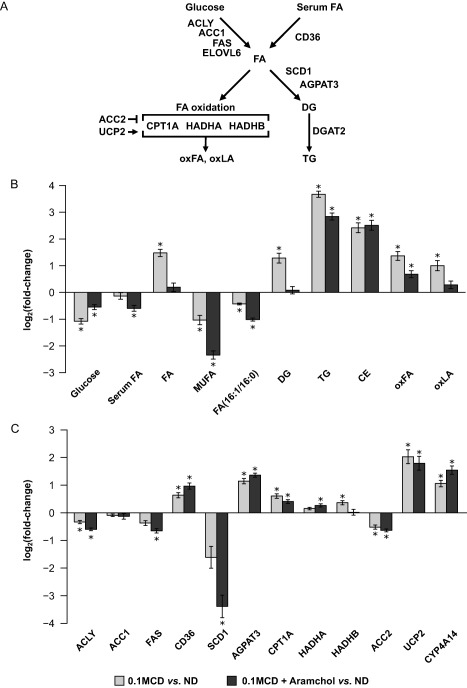
Hepatic lipid metabolism in 0.1MCD‐fed mice treated with vehicle or with Aramchol. (A) Schematic representation of hepatic lipid metabolism. Hepatic FAs originate from serum and through de novo lipogenesis. FAs can either be oxidized in the mitochondria or esterified to form TGs. The rate‐limiting step in mitochondrial β‐oxidation is CPT1A. The accumulation of FAs in the cytoplasm increases their oxidation and generates ROS, which induce GSH depletion and produce oxFA, such as oxLA, which can lead to fibrosis and cell death. (B) Relative fold change (log2) in the hepatic content of the main metabolites involved in lipid metabolism in mice fed a 0.1MCD diet and treated with vehicle or with Aramchol compared to mice fed a normal diet. A 0.1MCD diet led to reduced levels of glucose, serum FAs, and MUFAs and to a reduction in the FA(16:1/16:0) ratio. In contrast, feeding the mice a 0.1MCD diet led to increased levels of FAs, DGs, TGs, CEs, oxFA, and oxLA. (C) Relative fold change (log2) in the content of proteins involved in liver lipid metabolism in mice fed a 0.1MCD diet and treated with vehicle or with Aramchol compared to mice fed a normal diet. Feeding the mice a 0.1MCD diet resulted in reduced levels of ACLY, ACC1, ACC2, FAS, and SCD1. The 0.1MCD mice also showed increased levels of CD36, AGPAT3, CPT1A, HADHA, HADHB, UCP2, and CYP4A14. *P < 0.05. Abbreviations: ACLY, adenosine triphosphate citrate lyase; AGPAT3, 1‐acylglycerol‐3‐phosphate O‐acyltransferase 3; CYP4A14, cytochrome P450, family 4, subfamily a, polypeptide 14; FAS, fatty acid synthase; HADHA, HADHB, 3‐hydroxyacyl‐CoA dehydrogenase A and B; LA, linoleic acid; oxFA, oxidized FA; ND, normal diet; UCP2, uncoupling protein 2. Data were represented as mean ± SEM.
ARAMCHOL REDUCED FEATURES OF STEATOHEPATITIS IN 0.1MCD‐FED MICE
Mice given Aramchol displayed an improvement of liver histology, as indicated by hematoxylin and eosin histology (Fig. 4A). This was confirmed by Sudan III (1.5‐fold improvement compared to 0.1MCD‐fed mice administered Aramchol's vehicle; P < 0.05), F4/80 (3.2‐fold improvement compared to 0.1MCD‐fed mice administered Aramchol's vehicle; P < 0.05), CD64 (5‐fold improvement compared to 0.1MCD‐fed mice administered Aramchol's vehicle; P < 0.05), and Sirius Red (2.3‐fold improvement compared to 0.1MCD‐fed mice administered Aramchol's vehicle; P < 0.05) staining and measurement of COL1A1 protein by western blot (Fig. 4A,B,D). The content of COL1A1 in Aramchol‐treated mice decreased and did not differ significantly compared to mice fed a control diet (1.2‐fold 0.1MCD + Aramchol versus control diet). Feeding mice the 0.1MCD diet for 4 weeks caused a marked increase in serum liver enzymes (ALT and AST) (Fig. 4C). The trend to increase ALT and AST observed in the 0.1MCD‐fed mice treated with Aramchol was not statistically significant (Fig. 4C). Likewise, the low TG levels observed in the 0.1MCD‐fed mice were not significantly affected by Aramchol treatment (Fig. 4C).
Figure 4.
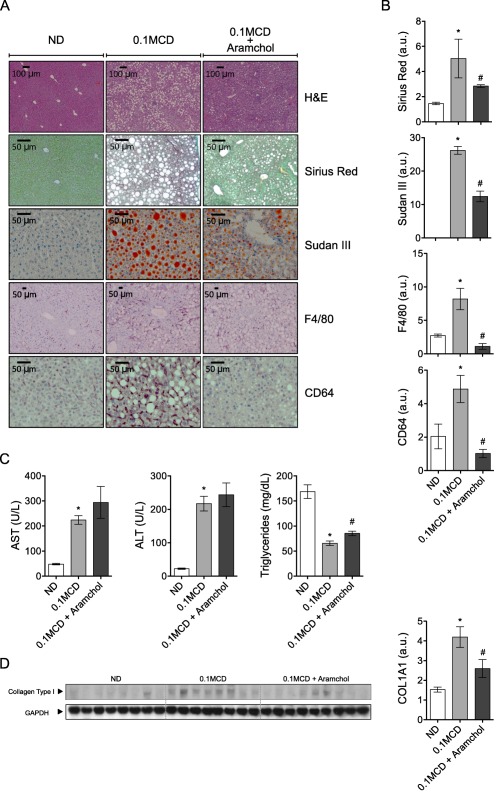
Aramchol attenuated steatohepatitis in 0.1MCD‐fed mice. Livers from mice fed with normal or 0.1MCD diet treated either with Aramchol or vehicle were extracted and the following analyses performed: (A,B) hematoxilin and eosin staining, Sirius Red, and Sudan III staining and quantification of positive areas; F4/80 and CD64 immunostaining and quantification of positive areas. (C) Serum levels of ALT, AST, and TGs. (D) Collagen type I protein content in the liver of mice was assessed by immunoblotting using GAPDH as the loading control. *P < 0.05 versus control; #P < 0.05 versus MCD. Data were represented as mean ± SEM. Abbreviations: ND, normal diet; a.u., arbitrary units.
Consistent with the decrease in inflammation and fibrosis, we observed that the 0.1MCD‐fed animals given Aramchol had a normal content of FAs and oxidized FAs, including oxLA, which are lipotoxic (Fig. 3B). The concentration of cystathionine, GSH, and the GSH/GSSG ratio was also normalized in the 0.1MCD‐fed mice given Aramchol (Fig. 1B). Mammals have three major systems to maintain redox homeostasis, two of these are nicotinamide dinucleotide reduced phosphate dependent (TXNRD1 and GSR) and the third is made by GCL and GS, the two enzymes in GSH synthesis.29 Analysis of protein content data showed that the 0.1MCD‐fed mice given Aramchol showed no significant effect in the content of the main enzymes involved in GSH synthesis and redox regulation (CBS, GCL catalytic subunit, GCL modifier subunit, GS, GSR, GPX1, GSTM1‐3, and TXNRD1) compared to animals treated with vehicle alone (Fig. 1C). However, the marked increase (3‐fold) exerted by Aramchol on the liver content of 2‐aminobutyric acid (Fig. 1B), a biomarker of the flux through the transsulfuration pathway,30 indicates that animals given Aramchol have developed mechanisms that compensate for the loss of GSH induced by the 0.1MCD diet and are effective in preventing oxidative stress while increasing FA expenditure.
We also analyzed the effect of Aramchol administration to 0.1MCD‐fed mice on the content of key enzymes involved in de novo lipogenesis, FA uptake, TG synthesis, and mitochondrial β‐oxidation. These mice showed a further reduction in SCD1 function as evaluated by protein content and by measuring the FA(16:1)/(16:0) ratio and the content of MUFAs (Fig. 3B,C). With the exception of SCD1, the protein content of other enzymes involved in de novo lipogenesis, FA uptake, TG synthesis, and FA oxidation was similar in 0.1MCD‐fed mice treated with Aramchol or vehicle alone (Fig. 3C). Based on previous findings showing increased FA oxidation in SCD1‐deficient mice,8 these results indicate that the antisteatotic effect of Aramchol is largely the result of a reduction in TG synthesis and increased β‐oxidation triggered by SCD1 suppression.
IDENTIFICATION OF SERUM BIOMARKERS ASSOCIATED WITH ARAMCHOL TREATMENT
As there is an urgent need to identify reliable biomarkers reflecting treatment response in NASH, we compared the serum metabolome of 0.1MCD‐fed mice treated with Aramchol with that of animals fed the 0.1MCD diet treated with vehicle alone (Supporting Table S2). Volcano plot analysis of the metabolomic data revealed the existence of 91 metabolites that significantly distinguished between both conditions (Fig. 5A; Supporting Table S2). Among them, we identified 30 metabolites that were also significantly different when the livers of both groups of mice were compared (Fig. 5B). This list included 2‐aminobutyric acid, which increased after Aramchol treatment, and three MUFAs, 10 PUFAs, four Lyso‐PE, nine Lyso‐PC, and three Lyso‐PI that were all reduced in 0.1MCD‐fed animals treated with Aramchol (Fig. 5B). The dose response for these biomarkers was investigated in serum and liver by treating 0.1MCD‐fed mice with 1 mg/kg/day Aramchol, a dose that did not improve liver histology (not shown). As shown in Fig. 5B, treatment of 0.1MCD‐fed mice with 1 mg/kg/day Aramchol only modified the content of two Lyso‐PC in serum and three Lyso‐PC and two lysophosphatidylinositols in liver.
Figure 5.
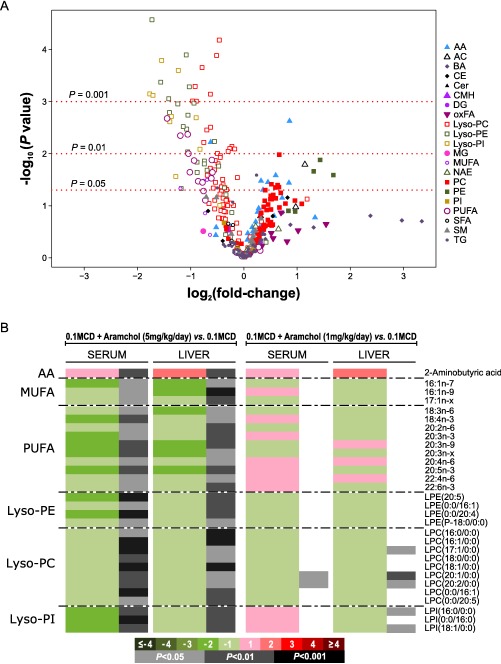
Identification of serum biomarkers associated with Aramchol treatment. (A) Volcano plot representation indicating the –log10(P value) and log2(fold change) of individual serum metabolic ion features of mice fed a 0.1MCD diet and treated with Aramchol (5 mg/kg/day) compared to untreated mice fed a 0.1MCD diet. (B) Heatmap of the 30 serum metabolites found to reflect the effect of Aramchol in the liver. This graph shows the log2(fold change) together with the unpaired Student t test of each metabolite. For the comparisons, log‐transformed ion abundance ratios are depicted as represented by the scale. Darker green and red colors indicate higher drops and elevations of the metabolite levels, respectively, in every comparison. Gray lines correspond to significant fold changes of individual metabolites; darker gray colors have been used to highlight higher significances (P < 0.05, P < 0.01, P < 0.001). Metabolites have been ordered in the heatmap according to their carbon number and unsaturation degree of their acyl chains. Abbreviations: AA, amino acids; AC, acyl carnitines; BA, bile acids; CE, cholesteryl esters; Cer, ceramides; CMH, monohexosylceramides; LPE, Lyso‐PE; LPC, Lyso‐PC; LPI, Lyso‐PI; oxFA, oxidized fatty acids; MG, monoglycerides; NAE, N‐acylethanolamines; SFA, saturated fatty acids; SM, sphingomyelins.
ARAMCHOL IMPROVED STEATOSIS AND OXIDATIVE STRESS IN HEPATOCYTES EXPOSED TO MCD MEDIUM AND REDUCED COL1A1 mRNA IN HUMAN STELLATE CELLS
We analyzed the effect of Aramchol (10 μM) in mouse hepatocytes incubated in MCD culture medium with regard to the development of steatosis and oxidative stress. We observed that incubation of hepatocytes with MCD medium resulted in cellular neutral lipids accumulation as determined by BODIPY staining and that this effect was prevented in Aramchol‐treated cells (Fig. 6A). We also observed that incubation of hepatocytes with MCD medium increased total hepatocyte ROS 4.5‐fold, but this was significantly reduced by the addition of Aramchol (Fig. 6B). Finally, we found that incubation of LX2 human stellate cells with medium containing Aramchol (10 μM) was associated with a decrease in COL1A1 mRNA content and an increase in PPARγ transcript (Fig. 6C). PPARγ is a negative regulator of type I collagen expression.31
Figure 6.
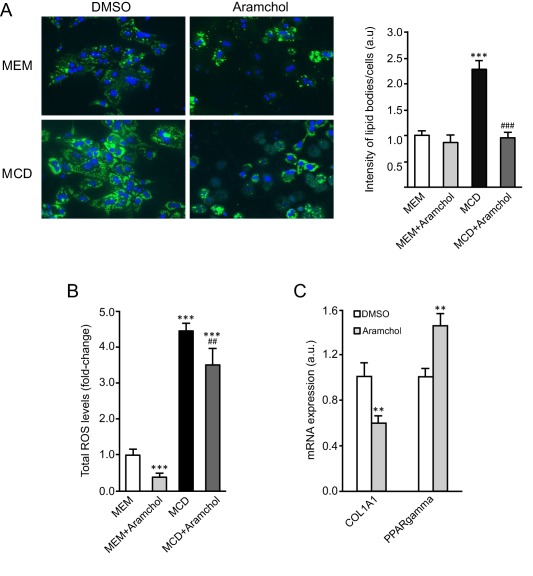
Aramchol attenuated lipid accumulation and ROS production in hepatocytes and collagen production in LX‐2 cells. Hepatocytes were cultured during 48 hours in serum‐free MEM or MCD medium, with or without Aramchol (10 μM). (A) BODIPY staining and quantification of the intensity of lipid bodies per cell. (B) CellROX Deep Green Reagent loading and total ROS levels quantification by FACS. (C) LX‐2 cells were treated with Aramchol (10 μM) for 24 hours, and the mRNA expression of COL1A1 and PPARY was assessed by quantitative PCR. **P < 0.01 versus control; ***P < 0.001 versus control; ##P < 0.01 versus MCD; ###P < 0.001 versus MCD. Data were represented as mean ± SEM. Abbreviation: FACS, fluorescence‐activated cell sorting.
Discussion
The MCD diet model is a widely used murine model of NASH.10 MCD diets produce the most severe phenotype in the shortest time period. In mice, MCD diets induce steatosis, inflammation, and fibrosis within 2 weeks, and these conditions continue to progress. The major disadvantage of this model is that animals fed MCD diets lose weight rapidly (up to 50% compared to control mice in 10 weeks), have low serum TG levels, and do not become insulin resistant. MCD‐fed mice also exhibit higher serum ALT and AST levels compared to patients with NASH. Comparison of gene expression patterns among various murine models of NASH and liver tissues from patients with NAFLD has shown little overlap.32 These models included a high‐fat/high‐carbohydrate diet, high‐fat/high‐carbohydrate/streptozocin diet, Western‐type diet, Pten –/–, and MCD diet. This lack of correlation between human and murine samples indicates that in this gene‐centered analysis none of the models replicate all aspects of the molecular changes in the human disease. The overlap between human samples and the MCD‐fed animals was not inferior to that of any other mouse model.32 Here, we demonstrated that despite the phenotype of the 0.1MCD mouse model being quite different from the majority of human subjects with NAFLD who have metabolic syndrome and obesity, around 50% of NAFLD subjects show a serum metabolomic phenotype that resembles the metabolism observed in this dietary‐induced mouse model of NASH. This finding supports the conclussion that, from the perspective of the regulation of liver metabolism, the 0.1MCD diet is a good model to study NASH and the mechanism of action of potential therapeutic agents.
Moreover, these findings confirm the existence of human NAFLD subtypes with marked differences in liver lipid metabolism. In particular, these results validate the existence of an M+ subtype which, based on its similarities with the Mat1a –/– and 0.1MCD mouse models, would be characterized by reduced synthesis of SAMe, low GSH content, impaired export of VLDLs, and conceivably impaired export of other intracellular vesicles, such as exosomes. This would lead to TG accumulation, a major rearrangement of lipid metabolism, and eventually progression to NASH. These results agree with the observation that impaired SAMe synthesis and a lower rate of transmethylation is a frequent feature in human NAFLD.33, 34
The 0.1MCD diet model may be considered as a modification of the CD diet model where the content of methionine has been reduced to 0.1%. In the control diet, the content of methionine was 0.3%. A CD diet also reliably induces fatty liver in mice, but unlike the MCD model, liver fat accumulation in the CD model is not accompanied with weight loss or steatohepatitis.35 Here, we used a modification of the canonical MCD model by adding 0.1% methionine to the diet to minimize the effect of weight loss on liver TG accumulation without affecting the development of steatohepatitis. The 0.1MCD diet induced steatosis, inflammation, and fibrosis with a small loss of body weight, which stabilized after the first 2 or 3 days compared to the control group. Importantly, when Aramchol treatment was initiated, weight loss had stabilized at least a week before. As in the canonical MCD model,24, 28, 30, 36 0.1MCD feeding impaired the synthesis of PC‐PUFAs through the PE N‐methyltransferase pathway, which is an essential component of the outer phospholipid coat of lipoproteins. This led to reduced secretion of hepatic TGs as VLDLs, resulting in the accumulation of hepatic TGs. The increase in acylglycerol‐3‐phosphate O‐acyltransferase 3 content, a critical enzyme in glycerol esterification, may also contribute to the development of steatosis in the 0.1MCD model. As in the canonical MCD model,24, 28, 30, 36 de novo lipogenesis in 0.1MCD‐fed mice remained normal or slightly reduced with the exception of SCD1. Feeding with 0.1MCD markedly reduced liver SCD1 mRNA and protein content, although these did not disappear as in the classical MCD diet model.24, 28, 30, 36 Consistent with this, the content of MUFAs and the FA(16:1/16:0) ratio, a marker of SCD1 activity, decreased in 0.1MCD‐fed mice compared to the control group which, together with the increase in CD36, would likely contribute to hepatic FA accumulation, lipotoxicity, and increased FA oxidation, as suggested by the abnormal protein content of CPT1A, uncoupling protein 2, and ACC2.
These alterations may be at the base of increased ROS production and the buildup of oxidized species of FAs, including oxLA, a biomarker of human NASH,37 inflammation, and fibrosis. Accordingly, the liver content of GSH and the GSH/GSSG ratio were reduced in 0.1MCD‐fed mice compared to animals fed a control diet. To maintain redox homeostasis, the liver relies mainly on two mechanisms: the GSH system, which comprises both de novo synthesis of GSH and the regeneration of GSH from GSSG; and the TXNRD1 system, which scavenges ROS and maintains intracellular redox status.29 In 0.1MCD‐fed mice, the protein content of GSR, the enzyme that catalyzes the regeneration of GSH from GSSG, was normal compared to control mice, whereas GCL and GS, the last two steps in GSH synthesis, and TXNRD1 were augmented. The protein content of CBS, the enzyme that removes homocysteine from the methionine cycle and moves it toward the transsulfuration pathway, was diminished. Conversely, in 0.1MCD‐fed mice, the protein content of numerous enzymes that consume GSH, including GPX1 and GSTM1‐3, was increased compared to control mice.
The oral administration of Aramchol to 0.1MCD‐fed mice further suppressed SCD1 expression and activity. However, this additional reduction of SCD1 was not associated with an exacerbation of the lipotoxicity and inflammatory response induced by the 0.1MCD diet, as may have been expected,38, 39 but reduced these responses in addition to reducing fibrosis. Consistent with this anti‐inflammatory activity, Aramchol normalized hepatic GSH content and the GSH/GSSG ratio as well as the levels of FAs and oxidized FAs, including the NASH biomarker oxLA, compared to control mice. Aramchol also induced a marked increase in the liver content of 2‐aminobutyric acid, a biomarker of the transsulfuration pathway.30 Viewed together, these results indicate that in 0.1MCD‐fed mice, flux through the transsulfuration pathway increased in response to Aramchol, leading to decreased oxidative stress and less GSH/GSSG turnover, which in turn would reduce the inflammatory and fibrotic response induced by 0.1MCD feeding. Despite the existence of fundamental differences between the in cellulo and in vivo experiments, with regard to the development of steatosis and oxidative stress induced by MCD, Aramchol's reduction of cellular lipid accumulation and the diminution of cellular ROS in hepatocytes incubated in MCD medium recapitulate the in vivo findings.
As oxidative stress is a major trigger of liver inflammation and fibrosis, Aramchol‐treated 0.1MCD‐fed mice showed less inflammation (determined histologically by F4/80 and CD64 staining) and fibrosis (measured both by Sirius Red staining, and COL1A1 and COL6A1 protein content) compared to control 0.1MCD‐fed mice administered vehicle alone. These studies also indicate that Aramchol negatively regulates collagen production in LX2 human stellate cells. In summary, these two disparate consequences of Aramchol treatment, the reduction of SCD1 and the increased flux through the transsulfuration pathway, may be the main targets through which Aramchol exerts its antisteatotic, anti‐inflammatory, and antifibrotic effects, although it is possible that there may be other targets not yet identified. In the 0.1MCD mouse model, fibrosis is slight. If treatment with Aramchol is also beneficial in more advanced stages of NASH when hepatic fibrosis is widespread is not known. Improvement of fibrosis and resolution of NASH are the secondary histologic endpoints of the ongoing Aramchol phase IIb clinical trial.
Aramchol administration failed to decrease the elevation of serum ALT and AST induced by the 0.1MCD diet. It is important to note, however, that the trend to increase the levels of ALT and AST observed in the group of 0.1MCD‐fed mice treated with Aramchol compared with the 0.1MCD animals receiving vehicle alone was not statistically significant. Moreover, in the published phase IIa study,4 there was no signal for toxicity (as determined by the serum levels of ALT, AST, and alkaline phosphatase) after 3 months treatment with Aramchol.
Finally, we also identified a group of serum and liver lipid biomarkers associated with Aramchol treatment in 0.1MCD‐fed mice that were dose dependent. These lipids included several MUFAs, PUFAs, as well as different Lyso‐PE, Lyso‐PC, and Lyso‐PI species, which had decreased content in response to Aramchol. The reduction of MUFAs agrees with Aramchol's mechanism of action, whereas the increase in lysophospholipid species has been associated with lipotoxicity and NASH progression.40, 41 Although it is at present not known if these noninvasive biomarkers of Aramchol's administration could be translated to determine the response of patients with NASH to treatment with this molecule, these results demonstrate that metabolomics is a most promising technique to identify noninvasive response biomarkers in NASH.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1107/full.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.
Supported by National Institutes of Health grants R01AT001576 (S.C.L. and J.M.M.) and R01DK092407 (S.C.L.), the Agencia Estatal de Investigación of MINECO SAF 2014‐52097R (J.M.M.) and SAF 2014‐54658R (M.L.M.‐C.), MINECO‐ISCiii PIE14/00031 (M.L.M.‐C. and J.M.M.), CIBERehd‐ISCiii (J.M.M., M.L.M.‐C. and A.M.A.), Basque Government Health Department 2013111114 and Spanish‐AECC (M.L.M.‐C.). We thank MINECO for the Severo Ochoa Excellence Accreditation (SEV‐2016‐0644). Mice studies were supported by Galmed Pharmaceuticals (Tel Aviv, Israel).
*These authors contributed equally to this work.
REFERENCES
- 1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Angulo P. Long‐term mortality in nonalcoholic fatty liver disease: is liver histology of any prognostic significance? Hepatology 2010;51:373‐375. Erratum in: Hepatology 2010;52:1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cassidy S, Syed BA. Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov 2016;15:745‐746. [DOI] [PubMed] [Google Scholar]
- 4. Safadi R, Konikoff FM, Mahamid M, Zelber‐Sagi S, Halpern M, Gilat T, et al.; FLORA Group . The fatty acid‐bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2014;12:2085‐2091. [DOI] [PubMed] [Google Scholar]
- 5. Leikin‐Frenkel A, Gonen A, Shaish A, Goldiner I, Leikin‐Gobbi D, Konikoff FM, et al. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non‐atherogenic. Arch Med Res 2010;41:397‐404. [DOI] [PubMed] [Google Scholar]
- 6. Enoch HG, Catalá A, Strittmatter P. Mechanism of rat liver microsomal stearyl‐CoA desaturase. Studies of the substrate specificity, enzyme‐substrate interactions, and the function of lipid. J Biol Chem 1976;251:5095‐5103. [PubMed] [Google Scholar]
- 7. Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl‐CoA desaturase‐1 function protects mice against adiposity. Proc Natl Acad Sci U S A 2002;99:11482‐11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flowers MT, Ntambi JM. Role of stearoyl‐coenzyme A desaturase in regulating lipid metabolism. Curr Opin Lipidol 2008;19:248‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, et al. Hepatic stearoyl‐CoA desaturase‐1 deficiency protects mice from carbohydrate‐induced adiposity and hepatic steatosis. Cell Metab 2007;6:484‐496. [DOI] [PubMed] [Google Scholar]
- 10. Anstee QM, Goldin RD. Mouse models in non‐alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 2006;87:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leffert HL, Koch KS, Moran T, Williams M. Liver cells. Methods Enzymol 1979;58:536‐544 . [DOI] [PubMed] [Google Scholar]
- 12. Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX‐1 and LX‐2: new tools for analysis of hepatic fibrosis. Gut 2005;54:142‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gold ES, Ramsey SA, Sartain MJ, Selinummi J, Podolsky I, Rodriguez DJ, et al. ATF3 protects against atherosclerosis by suppressing 25‐hydroxycholesterol‐induced lipid body formation. J Exp Med 2012;209:807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barr J, Caballería J, Martínez‐Arranz I, Domínguez‐Díez A, Alonso C, Muntané J, et al. Obesity‐dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res 2012;11:2521‐2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martínez‐Uña M, Varela‐Rey M, Cano A, Fernández‐Ares L, Beraza N, Aurrekoetxea I, et al. Excess S‐adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology 2013;58:1296‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martínez‐Arranz I, Mayo R, Pérez‐Cormenzana M, Mincholé I, Salazar L, Alonso C, et al. Enhancing metabolomics research through data mining. J Proteomics 2015;127:275‐288. [DOI] [PubMed] [Google Scholar]
- 17. van Liempd S, Cabrera D, Mato JM, Falcon‐Perez JM. A fast method for the quantitation of key metabolites of the methionine pathway in liver tissue by high‐resolution mass spectrometry and hydrophilic interaction ultra‐performance liquid chromatography. Anal Bioanal Chem 2013;405:5301‐5310. [DOI] [PubMed] [Google Scholar]
- 18. Alonso C, Fernández‐Ramos D, Varela‐Rey M, Martínez‐Arranz I, Navasa N, van Liempd SM, et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology 2017;152:1449‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911‐917. [DOI] [PubMed] [Google Scholar]
- 20. Ruiz JI, Ochoa B. Quantification in the subnanomolar range of phospholipids and neutral lipids by monodimensional thin‐layer chromatography and image analysis. J Lipid Res 1997;38:1482‐1489. [PubMed] [Google Scholar]
- 21. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6:359‐362. [DOI] [PubMed] [Google Scholar]
- 22. Gomez‐Sanchez JA, Carty L, Iruarrizaga‐Lejarreta M, Palomo‐Irigoyen M, Varela‐Rey M, Griffith M, et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 2015;210:153‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shields DJ, Altarejos JY, Wang X, Agellon LB, Vance DE. Molecular dissection of the S‐adenosylmethionine‐binding site of phosphatidylethanolamine N‐methyltransferase. J Biol Chem 2003;278:35826‐35836. [DOI] [PubMed] [Google Scholar]
- 24. Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, et al. Mice fed a lipogenic methionine‐choline‐deficient diet develop hypermetabolism coincident with hepatic suppression of SCD‐1. J Lipid Res 2006;47:2280‐2290. [DOI] [PubMed] [Google Scholar]
- 25. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med 2009;30:42‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, et al. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res 2010;51:3046‐3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol 2008;294:G1281‐G1287. [DOI] [PubMed] [Google Scholar]
- 28. Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PloS One 2015;10:e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eriksson S, Prigge JR, Talago EA, Arnér ES, Schmidt EE. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat Commun 2015;6:6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Medici V, Peerson JM, Stabler SP, French SW, Gregory JF 3rd, Virata MC, et al. Impaired homocysteine transsulfuration is an indicator of alcoholic liver disease. J Hepatol 2010;53:551‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARγ agonists prevent TGFβ1/Smad3‐signaling in human hepatic stellate cells. Biochem Biophys Res Commun 2006;350:385‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 2016;151:513‐525. [DOI] [PubMed] [Google Scholar]
- 33. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalhan SC, Edmison J, Marczewski S, Dasarathy S, Gruca LL, Bennett C, et al. Methionine and protein metabolism in non‐alcoholic steatohepatitis: evidence for lower rate of transmethylation of methionine. Clin Sci (Lond) 2011;121:179‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raubenheimer PJ, Nyirenda MJ, Walker BR. A choline‐deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high‐fat diet. Diabetes 2006;55:2015‐2020. [DOI] [PubMed] [Google Scholar]
- 36. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res 2008; 49:1068‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Santoro N, Caprio S, Feldstein AE. Oxidized metabolites of linoleic acid as biomarkers of liver injury in nonalcoholic steatohepatitis. Clin Lipidol 2013;8:411‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, et al. Metabolomics reveals that hepatic stearoyl‐CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab 2008;7:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl‐CoA desaturase. J Biol Chem 2009;284:5637‐5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee JH, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res 2008;49:84‐97. [DOI] [PubMed] [Google Scholar]
- 41. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1107/full.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.