

Abstract
Morphine is a well-characterized and effective analgesic commonly used to provide pain relief to patients suffering from both acute and chronic pain conditions. Despite its widespread use and effectiveness, one of the major drawbacks of morphine is its relatively short half-life of approximately 4 h. This short half-life often necessitates multiple administrations of the drug each day, which may contribute to both dependence and tolerance to morphine. Here, we tested the analgesic properties of a new polymer form of morphine known as PolyMorphine. This polymer has monomeric units of morphine incorporated into a poly(anhydride-ester) backbone that has been shown to hydrolyze into free morphine in vitro. Using an animal model of chronic pain, the spared nerve injury surgery, we showed that PolyMorphine is able to block spared nerve injury-induced hypersensitivity in mice for up to 24-h post-administration. Free morphine was shown to only block spared nerve injury-induced hypersensitivity for up to 2-h post-injection. PolyMorphine was also shown to act through the mu opioid receptor due to the ability of naloxone (a mu opioid receptor antagonist) to block PolyMorphine-induced analgesia in spared nerve injury animals pretreated with PolyMorphine. Additionally, we observed that PolyMorphine causes similar locomotor and constipation side effects as free morphine. Finally, we investigated if PolyMorphine had any effects in a non-evoked pain assay, conditioned place preference. Pretreatment of spared nerve injury mice with PolyMorphine blocked the development of conditioned place preference for 2-methyl-6-(phenylethynyl)pyridine (MPEP), a short-lasting mGluR5 antagonist with analgesic-like properties. Free morphine does not block the development of preference for MPEP, suggesting that PolyMorphine has longer lasting analgesic effects compared to free morphine. Together, these data show that PolyMorphine has the potential to provide analgesia for significantly longer than free morphine while likely working through the same receptor.
Keywords: PolyMorphine, morphine polymers, spared nerve injury, von Frey testing, conditioned place preference
Introduction
Morphine is one of the most commonly used and most well-known analgesics in both humans and animals.1 Morphine is an agonist of the mu opioid receptor (MOR) and binds to this receptor 3.4 and 10 times stronger than the other opioid receptor subtypes, the kappa and delta opioid receptors, respectively.2 With the activation of the MOR, morphine inhibits the release of neurotransmitter by decreasing calcium entry at presynaptic nerve endings and induces hyperpolarization in the post-synaptic cell at a number of CNS sites and on primary afferent neurons.3 These mechanisms are believed to cause the analgesic effects of morphine. Yet, despite its success in blocking pain, a major drawback to morphine use is its relatively short half-life of approximately 4 h.4 In vivo, morphine is quickly distributed5 and subsequently metabolized, mainly in the liver, into inactive morphine-3-glucaronide and the analgesic morphine-6-glucaronide.6 These metabolites are then excreted via the kidneys.7 This fast metabolism leads to the need for repeated administration of morphine, can result in low compliance, and can contribute to the development of tolerance and dependence over time.8 While several capsule forms9–11 have been developed to circumvent some of these problems, these tablets often lead to abuse because they can be crushed into a powder. Crushing these tablets causes a large amount of morphine to be available at once, rather than slowly releasing the drug over time, leading to greater psychotropic effects and risk for abuse. Further, these extended release tablets only modestly extend the analgesic effects of morphine.12–14
Recently, we developed a new formulation of morphine, called PolyMorphine, that could circumvent these issues.15 PolyMorphine is efficiently synthesized by chemically incorporating morphine in each repeat unit through a poly(anhydride ester) backbone in a three-step reaction.15 PolyMorphine was shown to be analgesic for three days in naïve mice using the tail-flick assay, which is 20 times longer than free morphine.15 What remains unclear is the extent to which PolyMorphine works as an analgesic in chronic pain models, how long its effects may last in these other models, if it acts on the MOR in a manner similar to free morphine, and what side effects may be associated with PolyMorphine treatment. In this study, PolyMorphine was evaluated for prolonged analgesia potential in the spared nerve injury (SNI) model of chronic pain.16 The SNI model has been shown to induce long-lasting hypersensitivity in the ipsilateral (injured) paw for up to nine weeks post-surgery.17,18 Herein, this chronic pain model was combined with several measures of pain-like behaviors including von Frey mechanosensory testing and the conditioned place preference (CPP) assay. Based on the previously shown extended analgesic effects of PolyMorphine, we hypothesized that this polymer could block SNI-induced hypersensitivity as measured by von Frey mechanosensory testing for up to three days post-administration. We also hypothesized that, if PolyMorphine works through the activation of the MOR, then pretreatment with naloxone, an MOR antagonist, would block the analgesic-like effects of PolyMorphine. Since the effects of PolyMorphine are ultimately due to the release of free morphine monomers, we also predicted that PolyMorphine would cause side effects similar to free morphine such as hyperactivity19,20and constipation.21 Finally, we hypothesized that the long-lasting analgesic effects of PolyMorphine would have the ability to block the development of preference for a short-lasting analgesic in the analgesic CPP assay.22–24
Materials and methods
PolyMorphine
The polymer was prepared according to previously published procedures.15 Briefly, Morphine (1 in Figure 1, 1.00 g, 1 eq) was dissolved in anhydrous pyridine under argon and stirred for 5 min. Glutaric anhydride (2, 3.97 g, 10 eq) was slowly added manually. The reaction mixture was heated to 60℃ and stirred overnight. Pyridine was azeotropically removed using toluene. The brown paste obtained was washed 10 × 50 mL with dichloromethane (DCM) to remove the excess glutaric acid. The final product was dried under vacuum at room temperature. Morphine-based diacid (3, 0.18 g) was acetylated by reacting with an excess of acetic anhydride (36 mL, Fisher, Fair Lawn, NJ). The reaction mixture was stirred overnight at room temperature. The excess acetic anhydride was removed under reduced pressure. Yield: 0.16 g (89 %), orange paste. Morphine-based monomer (4, 1.00 g) was polymerized by melt-condensation polymerization at 170℃ under constant vacuum (<2 mmHg) and constant stirring (100 r/min) using an overhead mechanical stirrer (T-line laboratory stirrer, Talboys Engineering Corp., Montrose, PA). Polymerization continued until the mixture solidified (∼30 min). The product was cooled to room temperature and dissolved in DCM (2 mL). The polymer was precipitated dropwise over excess diethyl ether (50 mL) and isolated by vacuum filtration. The product was dried under vacuum at room temperature overnight. Yield: 0.70 g (70 %), tan solid.
Figure 1.

Synthesis of PolyMorphine (5). PolyMorphine 5 was synthesized from the reaction of morphine 1 and glutaric anhydride 2 via ring opening followed by the acetylation of the diacid 3 and polymerization of the monomer 4 by melt condensation.
Animals
All mouse protocols were in accordance with the guidelines of National Institutes of Health and approved by the Animal Care and Use Committee of Duquesne University (Pittsburgh, PA). All behavioral experiments were performed on male C57Bl/6J mice. All mice were 8–10 weeks old when surgery and behavioral experimentation took place. Animals were individually housed after SNI surgery and kept on a 12-h light/dark cycle from 7 a.m. to 7 p.m. with ad libitum access to both food and water. Experiments were conducted during the light cycle and performed by an experimenter who was blinded to treatment and surgery type.
Surgery
SNI was performed as described previously.16 Mice were anesthetized with a 10:1 mixture of ketamine/xylazine at a dose of 10 µL/g. The hair overlying the sciatic nerve was shaved and a 1 cm incision was made parallel to the nerve. The sciatic nerve of the left hind paw was exposed and two of the three branches (tibial and common peroneal) were ligated. The third branch of the sciatic nerve, the sural branch, was not manipulated. Mice were allowed to recover on heating pads following surgery and a 2% lidocaine cream was placed over the suture on the skin for acute pain relief.
Drugs
Free morphine (morphine sulfate, Sigma) and PolyMorphine were separately dissolved in a 5% Cremophor EL saline solution at a dose of 10 mg/kg and 200 mg/kg, respectively. Dosages of these compounds were determined based on previously published results.15,24 Naloxone (naloxone hydrochloride dihydrate, Sigma, St Louis, MO) was dissolved in 0.9% saline at a dose of 1 or 10 mg/kg. These doses were based on previous publications.25,26 2-Methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP hydrochloride, Enzo Life Sciences) was dissolved in 0.9% saline on the first day of drug pairing (day 2 of CPP) at a dosage rate of 30 mg/kg. The dose of MPEP was determined using previously published CPP results with MPEP in the SNI model.24
Drug administration
Morphine and PolyMorphine were both administered intraperitoneally (IP) (10 mg/kg and 200 mg/kg, respectively) in a volume of 100 µL. The vehicle control for both morphine and PolyMorphine was 5% Cremophor EL saline in a volume of 100 µL. In the naloxone experiments, naloxone (1 or 10 mg/kg) was administered subcutaneously in a volume of 10 µL/g. The vehicle control for naloxone was 0.9% saline in a volume of 10 µL/g. In the CPP experiments, MPEP (30 mg/kg) was administered IP in a volume of 20 µL 5 min prior to behavioral testing. The vehicle control for MPEP was 0.9% saline in a volume of 20 µL. In von Frey testing, morphine, PolyMorphine, or vehicle was injected once following von Frey testing, which was completed one week after surgery. In the naloxone von Frey experiment, PolyMorphine was injected once following von Frey testing one-week post-SNI and naloxone was administered 2 h prior to the beginning of each time point measured. In the formalin test, naloxone (or saline) and morphine (10 mg/kg or saline) were administered 20 min before the injection of formalin (4% in a volume of 10 µL). Spontaneous behavior (described below) was then analyzed for 60 min. For the side effect experiments, morphine, PolyMorphine, or vehicle was injected once immediately prior to starting testing. In the CPP test, morphine, PolyMorphine, or vehicle was injected in the evening on day 1 of CPP (several hours after baseline day 1 times were measured). MPEP and vehicle were given to mice once a day for three consecutive days (on days 2, 3 and 4 of CPP, see below).
Behavioral testing
For mechanosensory studies with von Frey filaments, mice underwent baseline testing to determine 50% withdrawal thresholds using the up/down method.27,28 Animals were placed in Plexiglas boxes on a wire mesh surface for 2 h prior to testing. Withdrawals were defined as a full lifting of the foot off of the wire-mesh surface when a filament was applied to the lateral edge of the paw (area innervated by the sural branch of sciatic nerve). Filaments ranging from 0.20 mN to 25.1 mN (0.02–2.56 g) were used to determine the 50% withdrawal thresholds. After baseline testing, mice underwent SNI. One week after the surgery, mice were subjected to von Frey testing again to observe withdrawal thresholds. All animals were then given an injection of PolyMorphine (200 mg/kg), free morphine (10 mg/kg), or vehicle. von Frey testing was performed at 2 h and 4 h, as well as 1, 2, 3, 4, and 7 days after injection (corresponding to days 7, 8, 9, 10, and 14 days after surgery).This same procedure was repeated for the mechanosensory testing with PolyMorphine (200 mg/kg) and naloxone (10 mg/kg) studies, with naloxone or vehicle injections occurring 2 h before each von Frey time point.
For spontaneous formalin behavior, animals were placed in Plexiglas boxes on a clear Plexiglas surface for 2 h prior to testing. After drug and formalin administration (see above), spontaneous pain-like behaviors were recorded with a web-camera from beneath the animals for 60 min and scored offline. Pain-like behaviors were defined as the animal lifting, flinching, or biting the injected paw.
For side effect behaviors, animals were injected with PolyMorphine (200 mg/kg), free morphine (10 mg/kg), or vehicle and individually housed for 3-h post-injection. During the 3-h time period, animals were subjected to sensory motor battery tests (platform test, ledge test, pole test, and 60/90/inverted screen tests29) at 30-min and 2-h post-injection. Also at the 2-h time point, animals were placed in an open field box to measure total distance traveled in a 10-min time period. Activity was monitored with an overhead web camera (Logitec Webcam Pro 9000) using AnyMaze software (Stoelting Co., version 4.98). Total food and water consumed after injection at the end of the 3h time point were also measured. Finally, body temperature (rectal) and total fecal boli deposited over the time period were also measured. On days 1 to 3 post-injection, mice underwent sensory motor battery and open field again; food and water consumed and body temperature/fecal boli were also measured over these days as described for the injection day.
For spontaneous pain-like behavior, the analgesic CPP (aCPP) assay was used as previously described.24,30 Mice first underwent SNI surgery. One week after the surgery, mice were subjected to the five-day aCPP assay, with the addition of a pretreatment that allowed for the testing of long-lasting analgesic compounds. In the aCPP assay, positive reinforcement is used to detect non-evoked spontaneous pain where animals learn to associate contextual cues (e.g. visual stripes on walls of box) with the positive effects of a drug.22 The assay uses a box with three different chambers: two outer chambers that differ only in the visual patterns on the wall (horizontal versus vertical black and white stripes) and a small neutral chamber connecting the two outer chambers. On the first day of the aCPP experiment (known as day 1 or “pre-conditioning”), the mice are placed in the neutral chamber and allowed to roam free between all three chambers for 30 min. No drugs are given to the mice during this time. The total time that the animals spend in each chamber is recorded with an overhead camera. Six hours following baseline testing on day 1 animals are given a pretreatment injection (100 µL) of PolyMorphine (200 mg/kg), free morphine (10 mg/kg) or vehicle. On the next three days of the experiment (days 2–4 or conditioning days), the mice receive a control vehicle injection (20 µL of saline) in the morning and are restricted to one of the outer chambers for 30 min. In the afternoon, the mice then receive an injection of the short-term analgesic drug (20 µL of 30 mg/kg MPEP) and are placed in the opposite outer chamber for 30 min. Finally, on the last day of the experiment (day 5 or post-conditioning), the mice are placed with no injection in the neutral chamber and allowed to roam free across the entire apparatus for 30 min, this exposure being a repeat of the testing from day 1. The total time spent in each chamber is recorded using two overhead web cameras (Logitec Webcam Pro 9000 and Canon ZR420) and AnyMaze software. Total times recorded on day 1 are compared to those of day 5 to measure whether a preference for the chambers developed. All behavioral testing was done blinded to treatment and surgery.
Statistical analysis
Prism (version 6.0, GraphPad) was used for all behavioral analysis. All data are shown as mean ± SEM. Statistical significance was determined using t-tests or two-way analysis of variance (ANOVA) followed by a post hoc test. When analyzing von Frey, open field, and constipation data, withdrawal thresholds, distance traveled, and total fecal boli deposited were analyzed using a two-way ANOVA followed by Bonferroni post hoc tests, respectively. When analyzing formalin data, total amount of time exhibiting pain-like behaviors was analyzed using a one-way ANOVA followed by a Bonferroni post hoc test. When analyzing time difference data from the CPP assay, a paired t-test was used. Statistical significance was determined with a 95% confidence interval (P < 0.05).
Results
PolyMorphine synthesis and degradation into free morphine
Morphine diacid was synthesized via ring-opening reaction between glutaric anhydride and morphine, followed by the acetylation of the diacid and polymerization of the monomer by melt condensation (Figure 1). PolyMorphine was shown to hydrolytically degrade in vitro at physiological conditions (37℃ and pH 7.4) to free morphine in the following stages: hydrolytic cleavage of the anhydride bonds to morphine-diacid 3 followed by hydrolysis of the ester bonds to yield free morphine (Figure 2).
Figure 2.

Hydrolytic degradation of PolyMorphine 5.The anhydride linkages of polymer 5 are hydrolyzed first to generate diacid intermediates (3), which are then further hydrolyzed to the final products, morphine (1) and adipic acid (7).
PolyMorphine blocks SNI-induced hypersensitivity
The ability of PolyMorphine to block SNI-induced mechanical hypersensitivity was measured. Using von Frey testing to determine 50% withdrawal thresholds, a pretreatment of PolyMorphine (n = 8) reversed SNI-induced hypersensitivity for up to 24-h post-injection compared to vehicle-treated SNI animals (n = 8). The reversal of SNI-induced hypersensitivity in mice treated with regular morphine (n = 8) was gone after 2 h (Figure 3). These data demonstrate a significant increase in PolyMorphine analgesia over time compared to free morphine.
Figure 3.

PolyMorphine blocks SNI-induced hypersensitivity longer than free morphine. SNI mice treated with 200 mg/kg of PolyMorphine (n = 8) show reduced hypersensitivity compared to control SNI mice (n = 8) for up to 24 h after administration. SNI mice treated with 10 mg/kg of free morphine (n = 8) show reduced hypersensitivity compared to control SNI mice for only 2 h after administration. Two-way ANOVA, overall main effect of treatment, *P = 0.0153; overall main effect of time, ***P < 0.0001; Bonferroni post hoc tests compared to vehicle (saline), *P < 0.05 and ***P < 0.0001. SNI: spared nerve injury.
Naloxone blocks the analgesic-like effects of PolyMorphine
To determine if PolyMorphine works through the same molecular mechanism as free morphine, an MOR antagonist (naloxone) was used to potentially inhibit the analgesic effects of PolyMorphine. First, we determined a dose of naloxone sufficient to block free morphine effects using the spontaneous formalin test. Animals received a pretreatment of either vehicle or morphine (10 mg/kg) IP and vehicle or naloxone (1 or 10 mg/kg) prior to formalin injection. Using this assay, a dose of 10 mg/kg of naloxone was found to be effective in fully blocking the analgesic effects of free morphine (Figure 4(a)). Once this dosage was determined, the ability of this dose of naloxone to block the analgesic effects of PolyMorphine was measured. Using von Frey testing to determine 50% withdrawal thresholds, pretreatments of naloxone prior to each experimental time point blocked the analgesic effects of PolyMorphine in SNI animals with PolyMorphine (Figure 4(b)). Animals that received pretreatments of vehicle showed the normal reversal of SNI-induced hypersensitivity of up to 24-h post-PolyMorphine injection (Figure 4(b)). These data show that PolyMorphine likely works through the MOR.
Figure 4.
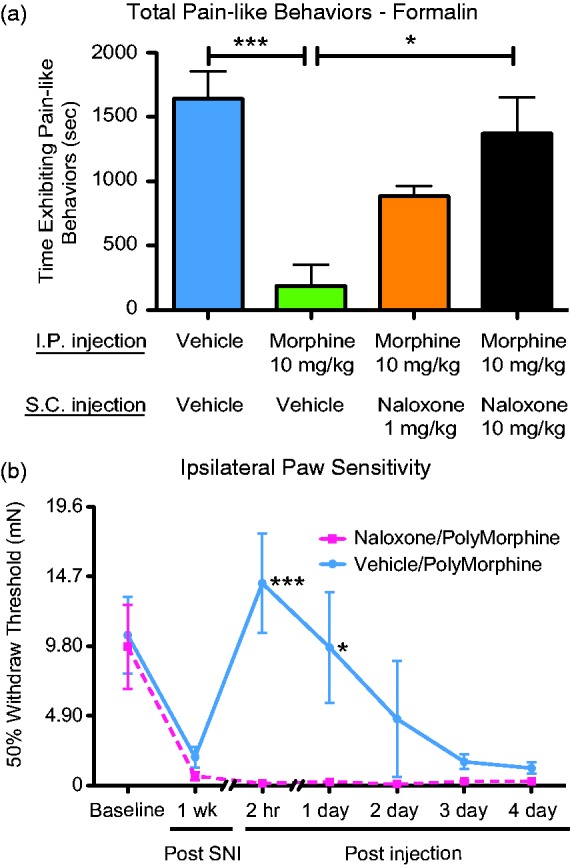
Naloxone blocks both free morphine and PolyMorphine-induced analgesia. (a) Mice treated with 10 mg/kg of free morphine and 10 mg/kg of naloxone (n = 6) show equal amounts of pain-like behaviors compared to control mice (n = 12). Mice treated with only 10 mg/kg of morphine (n = 5) show significantly less pain-like behaviors compared to the control and morphine/naloxone (10 mg/kg) groups. Naloxone (1 mg/kg, n = 6) was not a sufficient dose to block the analgesic effects of morphine. (b) SNI mice treated with 200 mg/kg of PolyMorphine and 10 mg/kg of naloxone (n = 6) prior to each post-injection time point remained hypersensitive compared to SNI mice (n = 6) that received PolyMorphine with vehicle injections. Formalin test—one-way ANOVA, overall main effect, ***P = 0.0007; Bonferroni post hoc tests, *P < 0.05 and ***P < 0.0001. von Frey test—two-way ANOVA, overall main effect of treatment, *P = 0.0211; overall main effect of time, ***P < 0.0001; Bonferroni post hoc tests, *P < 0.05 and ***P < 0.0001. SNI: spared nerve injury.
PolyMorphine causes constipation and increased motor activity
Next, the side effects of free morphine and PolyMorphine were compared. Animals were pretreated with free morphine, PolyMorphine, or vehicle, and motor deficiencies, constipation, hyperactivity, body weight, body temperature, and food/water consumed for up to three days post-injection were measured. No differences were found between the groups in terms of body weight, body temperature, or food/water consumed at any of the time points measured (data not shown). For constipation, animals treated with both free morphine and PolyMorphine showed constipation for the first 3-h post-injection (Figure 5(a)). This effect was not seen on days 1 to 3 post-injection. Finally, hyperactivity was measured using the open field test. Animals treated with free morphine showed hyperactivity at 3-h post-injection, while no hyperactivity was observed with PolyMorphine at this time point (Figure 5(b)). However, by one-day post-injection, PolyMorphine pretreated animals now showed hyperactivity whereas free morphine pretreated animals did not. Overall, these data show that PolyMorphine has some of the same side effects as free morphine with subtle but potentially important differences.
Figure 5.

PolyMorphine and free morphine induce constipation and hyperactivity. (a) Mice treated with 200 mg/kg of PolyMorphine (n = 6) or 10 mg/kg of free morphine (n = 8) show constipation effects at 3-h post-injection compared to control animals (n = 6), with no effects one to three days post-injection. (b) Mice treated with 200 mg/kg of PolyMorphine (n = 6) show hyperactivity at one-day post-injection with no effects 3-h or two to three days post-injection compared to vehicle-treated animals (n = 6). Mice treated with 10 mg/kg of free morphine (n = 8) show hyperactivity at 3-h post-injection with no effects one to three days post-injection compared to vehicle-treated animals. Constipation—two-way ANOVA, overall main effect of treatment *P = 0.0385, overall main effect of time, ***P < 0.0001; Bonferroni post hoc tests compared to vehicle, **P < 0.01. Hyperactivity—two-way ANOVA, overall main effect of treatment, P = 0.1275; overall main effect of time, ***P < 0.0001; Bonferroni post hoc tests compared to vehicle, *P < 0.05 and ***P < 0.0001.
PolyMorphine blocks the development of preference for MPEP in the analgesic CPP assay
For spontaneous pain-like behaviors, the analgesic CPP assay was used. Typically in the analgesic CPP assay, a short-lasting analgesic is repeatedly paired with a physical space to induce a conditioned preference response in a mouse with on-going spontaneous pain.31 As PolyMorphine is hypothesized to have a long-lasting analgesic effect, PolyMorphine could not be used as the normal pairing drug during the pairing days. Instead, animals were given a pretreatment of the PolyMorphine to determine if pretreatment blocked the preference-inducing effect of a short-lasting analgesic, MPEP. Pretreatment with PolyMorphine was compared to pretreatment with free morphine or vehicle. Neither free morphine nor vehicle pretreatment were hypothesized to block MPEP CPP in SNI mice. Here, pretreatments with saline caused mice to develop normal MPEP-induced CPP (Figure 6(a)); SNI mice pretreated with regular morphine also developed MPEP-induced CPP (Figure 6(b)). However, pretreatment with PolyMorphine blocked MPEP-induced CPP in SNI mice (Figure 6(c)). Overall, these results support the hypothesis that PolyMorphine causes analgesic-like behavioral effects in both reflexive and spontaneous pain-like behavior assays.
Figure 6.

PolyMorphine blocks MPEP-induced CPP in male SNI mice. (a) SNI mice pretreated with vehicle (n = 8) develop normal preference for the MPEP-paired chamber. (b) SNI mice pretreated with 10 mg/kg of free morphine (n = 8) also develop preference for the MPEP-paired chamber. (c) SNI mice pretreated with PolyMorphine (n = 8) do not develop preference for the MPEP-paired chamber. Paired t-tests,*P < 0.05. MPEP: 2-methyl-6-(phenylethynyl)pyridine.
Discussion
Morphine and other mu opioid agonists have a long history in being used to treat both acute and chronic pain conditions. There is an active push in the United States to prepare compounds that could avoid the negative side effects and short half-lives of most opioids. A majority of the newer treatments using morphine have specifically focused on trying to circumvent the problem of rapid metabolism of the compound by focusing on extended-release formulations of the drug.32 Here, for the first time, we show the extended analgesic-like effects of a polymer form of morphine, PolyMorphine, in a preclinical model of chronic pain combined with behavioral testing in mice. Using von Frey mechanosensory testing, PolyMorphine was able to block SNI-induced hypersensitivity for up to 24-h post-administration. This effect is in contrast to the short-lived one of free morphine, wherein the SNI-induced hypersensitivity is suppressed by morphine for only 2-h post-administration. Mechanistically, naloxone antagonism of MOR receptor blocked the analgesic-like effects of PolyMorphine in SNI animals. Additionally, PolyMorphine caused comparable side effects of free morphine, such as constipation and hyperactivity. Finally, in a non-evoked behavioral pain test, the aCPP assay, PolyMorphine blocked the effects of the short-term analgesic MPEP. This is again in contrast to free morphine wherein normal MPEP-induced CPP developed with free morphine pretreatment.
The use of opioids to treat pain has substantially increased over the past few years, especially in the treatment of non-cancer pain.33 This increase in use has coincided with the development of extended release forms of morphine that allow patients to be administered the compounds less frequently. Some of the more commonly used formulations that are available are Embeda®, which contains pellets of morphine sulfate with sequestered naltrexone34 and Kadian®, which contains polymer coated morphine sulfate pellets.32 Morphine has also been incorporated into nanogels35 and liposomes36 in order to increase the duration of its analgesic effects. The main difference between these formulations and PolyMorphine is that in these compounds, free morphine is sequestered in a polymer coating, whereas in PolyMorphine, free morphine is directly incorporated into a polymer form. Despite these differences in design, Embeda®, Kadian®, and Polymorphine have all been found to release free morphine over time.37 Other approaches have also been taken that attempt to limit abuse liability by slowing the rate at which compounds cross the blood–brain barrier (BBB) instead of extending the release of drugs over time. One notable example is NKTR-181, a compound that contains a five-ring morphine-like scaffold bonded to a short-chain poly(ethylene glycol).38,39 This functional group has previously been shown to reduce oral bioavailability and transmission rate across the BBB.40,41 Through this mechanism, NKTR-181 provides analgesia in humans and is currently in Phase III clinical trials. However, this type of formulation only circumvents potential abuse problems and does not extend the analgesic effects of the compound for longer than free opioids. Other attempts, such as polymer formulations to extend analgesia and prevent abuse, have tried using a polyurethane backbone, but these formulations are problematic because they lead to poor bioavailability due to their inability to degrade in vivo.42 The poly(anhydride-ester) backbone used in PolyMorphine can circumvent many of the problems associated with other extended release morphine formulations and has already been used to make polymer formulations of NSAIDs,43 antiseptics,44 and antioxidants.45 This specific polymer-type formulation may be the key behind extending analgesia while reducing abuse liability.
Additionally, our results show that PolyMorphine has the potential to be effective in providing extended analgesia in animal models. Original testing with PolyMorphine using the tail-flick assay showed that the drug provided extended analgesic relief in mice, for up to three days (20 times longer than free morphine).15 While these results were promising, the tests were only done in naïve mice and only in one “experimenter-induced” pain assay. Our results expand upon these data, showing that PolyMorphine is effective in a classic model of chronic neuropathic pain, SNI, providing similar reduction in mechanical hypersensitivity compared to free morphine initially along with extended relief up to 24 h after delivery. These extended analgesic results are similar to those obtained in other rodent studies that looked at liposome-encapsulated morphine in vivo. Liposome-encapsulated morphine was shown to block the hypersensitivity associated with male rats that had spinal nerve ligation in thermal sensory testing36 and extend analgesic-like effects in naïve animals using the tail-flick test.46 Other extended release formulas of morphine, however, such as those produced using nanogels, only resulted in the extension of analgesic effects for approximately 1 h longer than that of free morphine when tested using the hot-plate test in naïve mice.35 Similarly, NKTR-181, the five-ring morphine-like scaffold bonded to a short-chain PEG produced analgesic-like effects for only up to 4-h post-administration in the hot-plate test in mice, this being similar to the effects of free opioids.39
In addition to this 24-h block in hypersensitivity with PolyMorphine administration, we also showed that PolyMorphine is effective in altering spontaneous pain-like behaviors. In the classic aCPP assay, animals receive a pain-inducing injury prior to testing. Following injury, animals are conditioned to learn the effects of a short-lasting analgesic agent. If animals receive relief from their ongoing nociception, they will develop a preference for the drug-paired chamber. We had previously shown that three days of pairing with MPEP, an mGluR5 antagonist, alone induces preference in SNI but not sham-operated mice.24 In our experiments here, we used a pretreatment injection to determine if the extended analgesic effects of PolyMorphine could block the short-lasting analgesic effects of MPEP, such that animals do not develop preference for the MPEP-paired chamber. The MPEP results from vehicle-pretreated mice were consistent with our previous data with MPEP in SNI mice,24 wherein MPEP-induced CPP. In a similar matter, pretreatment with free morphine also resulted in mice developing a preference for MPEP. This is because the short-term effects of the morphine wear off before the pairing with MPEP occurs (e.g., approximately 20 h after morphine administration). Conversely, animals pretreated with PolyMorphine did not develop any preference for the MPEP-paired chamber. This suggests that unlike free morphine, PolyMorphine provided relief from the animal’s ongoing nociception, such that when MPEP was administered, the animals did not receive any additional relief during the three days of pairing. These aCPP results, which suggest PolyMorphine induces analgesic-like effects over a three-day time period are in slight contrast to our von Frey data that show the effects of PolyMorphine wearing off by the two-day time point. One possible reason for this difference could be the fundamentally different types of pain that are being measured in each assay (i.e. experimenter-induced pain measured with von Frey testing compared to spontaneous non-evoked pain measured with aCPP). Our data suggest that PolyMorphine is able to provide continuous ongoing relief to injured animals with spontaneous non-evoked pain for a significantly longer period of time. The lack of preference for MPEP in the PolyMorphine pretreatment group also shows that there is no additive effect of the two drugs. This observation is in contrast to studies that have shown that coadministration of mGluR5 antagonists and acute MOR agonists can generate enhanced effects.47,48 If there had been some type of additive effects where animals received greater relief from the combined effects of MPEP and PolyMorphine, we would have predicted preference to still develop after PolyMorphine pretreatment.
Our results, with both the experimenter-induced and spontaneous pain-like behavior assays, suggest that PolyMorphine has the potential to work significantly better than other slow-release morphine formulations. Further investigation with PolyMorphine is required to determine if its extended analgesic-like effects are observed when using female animals since all of the testing done thus far has focused on males. However, since female animals are known to respond to morphine similar to males,24 we expect PolyMorphine to work in females as well. Additional work investigating the potential addictive nature of PolyMorphine will also need to be completed. To our knowledge, no studies have investigated the self-administration rates of polymers of opioids. However, since the morphine is sequestered into a polymer that slowly releases free morphine, animals would not be predicted to self-administer.
Mechanistically, we demonstrated that PolyMorphine most likely works through the same pathway as free morphine. Since PolyMorphine was previously shown to generate free morphine in vivo,15 it was expected that the free morphine generated would also work through MOR activation. Here, we were able to reverse PolyMorphine-induced analgesia with a pretreatment of the drug naloxone. Naloxone is a selective antagonist of the MOR and has been extremely valuable for investigating the opioid actions of compounds preclinically. Naloxone has 9 times greater affinity for mu compared to the kappa opioid receptor and 60 times greater affinity for mu compared to the delta opioid receptor.49 Unlike other selective antagonists (such as β-FNA and norBNI) whose actions can last for weeks after the elimination of the drug, naloxone acts as a simple competitive antagonist, whose effects are much shorter and reversible.50 Thus, our results show that PolyMorphine, just like free morphine, likely works through MOR activation.
Finally, PolyMorphine was shown to have a comparable side effect profile to free morphine. With the doses of PolyMorphine (200 mg/kg) and free morphine (10 mg/kg) used, no motor deficiencies, ataxia, or hypothermia were observed. This result is not surprising however, since this dose of morphine may be too low to observe these particular effects.19 It also suggests that when PolyMorphine is metabolized into free morphine in vivo, the amount of free morphine in the animal’s bloodstream remains at or below the amount when free morphine is administered at the rate of 10 mg/kg. However, further testing of blood concentrations of free morphine in animals treated with PolyMorphine would be needed to confirm this hypothesis. Other side effects of morphine and related compounds include opioid-induced constipation21,51 due to the presence of the MOR in the colon. Animals treated with both PolyMorphine and free morphine develop this acute side effect 3 h after administration. Similarly, opioids are known to stimulate motor activity.19,20 Hyperactivity was seen at different times post-administration between free morphine (3 h) and PolyMorphine (24 h). This lack of increase in the magnitude of side effects with an extension in analgesic-like properties has been shown in other animal models with liposome deliveries of morphine.52,53
The varying times at which each of the different side effects are seen may provide some insight into how PolyMorphine is distributed and metabolized after injection. Our data suggest that when PolyMorphine is first administered, some of the first free morphine generated in the bloodstream from polymer breakdown remains in the periphery and immediately binds with the MORs in the gastrointestinal tract54 causing constipation. The delay in locomotor hyperactivity with PolyMorphine shows that there may not be enough free morphine present in the CNS until 24 h after injection to stimulate the dopaminergic locomotor system.55 On the other hand, PolyMorphine induces both acute (2 h and 4 h) and extended (24 h) analgesic effects. These data suggest that enough free morphine is cleaved off the polymer quickly and for an extended period of time, where it can act at analgesic targets in the periphery and CNS. With regular morphine, since no breakdown of a polymer is needed to generate free morphine, sufficient drug is immediately available to induce analgesia and all of the side effects immediately after administration, and none of the effects persist longer than 3 h after injection. These data suggest the potential for slightly altered release kinetics of PolyMorphine after injection on side effect profiles.
Overall, mice that receive an injection of PolyMorphine have a reversal of SNI-induced hypersensitivity for up to 24-h post-administration and this effect is blocked by naloxone. PolyMorphine also has a similar side effect profile to free opioids. Finally, SNI animals do not develop preference for the short-term analgesic MPEP when given a PolyMorphine pretreatment, presumably because of PolyMorphine’s long-lasting effects. These effects are in contrast to those of free morphine, whose effects are no longer observed after a couple of hours. Our results demonstrate that the poly(anhydride-ester) backbone in the synthesis of an extended-release morphine may have a promising future in the treatment of chronic pain conditions. A valuable goal is to find an opioid that can be administered in the clinic and last for weeks or longer to treat persistent acute pain (e.g., following major trauma), thus preventing the need for distribution and potential abuse. While PolyMorphine does not yet achieve this level of efficacy, future formulations may represent a step in the right direction for battling the current opioid epidemic.
Acknowledgments
KEU gratefully acknowledges a gift from Dr. Phil Cox (Noramco).
Author Contributions
NCL, SRL, and BJK performed behavioral experiments including data analysis and interpretation. NCL, BJK, and LY designed behavioral experiments. RC and KEU synthesized PolyMorphine. NCL, BJK, RC, KEU, and LY wrote and edited the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: RC gratefully acknowledges funding and support by the US Fulbright Scholar program (Australian-American Fulbright Commission). BJK gratefully acknowledges funding and support from the National Institutes of Health National Center for Complementary and Integrative Health (NCCIH R15AT008060).
References
- 1.Mohammed W, Alhaddad H, Marie N, et al. Comparison of tolerance to morphine-induced respiratory and analgesic effects in mice. Toxicol Lett 2013; 217: 251–259. [DOI] [PubMed] [Google Scholar]
- 2.Kilpatrick GJ, Smith TW. Morphine-6-glucuronide: actions and mechanisms. Med Res Rev 2005; 25: 521–544. [DOI] [PubMed] [Google Scholar]
- 3.Chahl LA. Experimental and clinical pharmacology: opioids-mechanisms of action. Austr Prescr 1996; 19: 63–65. [Google Scholar]
- 4.Sverrisdottir E, Lund TM, Olesen AE, et al. A review of morphine and morphine-6-glucuronide's pharmacokinetic-pharmacodynamic relationships in experimental and clinical pain. Eur J Pharm Sci 2015; 74: 45–62. [DOI] [PubMed] [Google Scholar]
- 5.Spector S, Vesell ES. Disposition of morphine in man. Science 1971; 174: 421–422. [DOI] [PubMed] [Google Scholar]
- 6.Christrup LL. Morphine metabolites. Acta Anaesthesiol Scand 1997; 41: 116–122. [DOI] [PubMed] [Google Scholar]
- 7.Yeh SY. Urinary excretion of morphine and its metabolites in morphine-dependent subjects. J Pharmacol Exp Ther 1975; 192: 201–210. [PubMed] [Google Scholar]
- 8.Akbarali HI, Inkisar A, Dewey WL. Site and mechanism of morphine tolerance in the gastrointestinal tract. Neurogastroenterol Motil 2014; 26: 1361–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross EL, Hahn K. KADIAN (morphine sulfate extended-release) capsules for treatment of chronic, moderate-to-severe, nonmalignant pain. Int J Clin Pract 2008; 62: 471–479. [DOI] [PubMed] [Google Scholar]
- 10.King CR, Khabazian A. Avinza (morphine sulfate extended-release capsules). Clinical J Oncol Nurs 2003; 7: 458–460, 78. [DOI] [PubMed] [Google Scholar]
- 11.Hagen NA, Thirlwell M, Eisenhoffer J, et al. Efficacy, safety, and steady-state pharmacokinetics of once-a-day controlled-release morphine (MS Contin XL) in cancer pain. J Pain Symptom Manage 2005; 29: 80–90. [DOI] [PubMed] [Google Scholar]
- 12.Setnik B, Sommerville K, Goli V, et al. Assessment of pharmacodynamic effects following oral administration of crushed morphine sulfate and naltrexone hydrochloride extended-release capsules compared with crushed morphine sulfate controlled-release tablets and placebo in nondependent recreational opioid users. Pain Med 2013; 14: 1173–1186. [DOI] [PubMed] [Google Scholar]
- 13.Katz N, Dart RC, Bailey E, et al. Tampering with prescription opioids: nature and extent of the problem, health consequences, and solutions. Am J Drug Alcohol Abuse 2011; 37: 205–217. [DOI] [PubMed] [Google Scholar]
- 14.Butler SF, Black RA, Cassidy TA, et al. Abuse risks and routes of administration of different prescription opioid compounds and formulations. Harm Reduct J 2011; 8: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosario-Melendez R, Harris CL, Delgado-Rivera R, et al. PolyMorphine: an innovative biodegradable polymer drug for extended pain relief. J Control Release 2012; 162: 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000; 87: 149–158. [DOI] [PubMed] [Google Scholar]
- 17.Richner M, Bjerrum OJ, Nykjaer A, et al. The spared nerve injury (SNI) model of induced mechanical allodynia in mice. J Vis Exp 2011; 54: e3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bourquin A-F, Süveges M, Pertin M, et al. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 2006; 122: e1–e14. [DOI] [PubMed] [Google Scholar]
- 19.Koek W, France CP, Javors MA. Morphine-induced motor stimulation, motor incoordination, and hypothermia in adolescent and adult mice. Psychopharmacology 2012; 219: 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koek W. Effects of repeated exposure to morphine in adolescent and adult male C57BL/6J mice: age-dependent differences in locomotor stimulation, sensitization, and body weight loss. Psychopharmacology 2014; 231: 1517–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. Pharmacol Exp Ther 2005; 314: 1195–1201. [DOI] [PubMed] [Google Scholar]
- 22.Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 1998; 56: 613–672. [DOI] [PubMed] [Google Scholar]
- 23.He Y, Tian X, Hu X, et al. Negative reinforcement reveals non-evoked ongoing pain in mice with tissue or nerve injury. J Pain 2012; 13: 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lax NC, George DC, Ignatz C, Kolber BJ. The mGluR5 antagonist fenobam induces analgesic conditioned place preference in mice with spared nerve injury. PloS One 2014; 9: e103524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cendan CM, Pujalte JM, Portillo-Salido E, et al. Antinociceptive effects of haloperidol and its metabolites in the formalin test in mice. Psychopharmacology 2005; 182: 485–493. [DOI] [PubMed] [Google Scholar]
- 26.Gupta R, Gupta LK, Bhattacharya SK. Naloxone blocks the beneficial effects of aqueous extract of Murraya koenigii (L.) Spreng leaves in models of pain. Eur Rev Med Pharmacol Sci 2013; 17: 1748–1751. [PubMed] [Google Scholar]
- 27.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980; 20: 441–462. [DOI] [PubMed] [Google Scholar]
- 28.Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53: 55–63. [DOI] [PubMed] [Google Scholar]
- 29.Kolber BJ, Boyle MP, Wieczorek L, et al. Transient early-life forebrain corticotropin-releasing hormone elevation causes long-lasting anxiogenic and despair-like changes in mice. J Neurosci 2010; 30: 2571–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Felice M, Eyde N, Dodick D, et al. Capturing the aversive state of cephalic pain preclinically. Ann Neurol 2013; 74: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Refsgaard LK, Hoffmann-Petersen J, Sahlholt M, et al. Modelling affective pain in mice: effects of inflammatory hypersensitivity on place escape/avoidance behaviour, anxiety and hedonic state. J Neurosci Methods 2016; 262: 85–92. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson B. Morphine sulfate extended-release capsules for the treatment of chronic, moderate-to-severe pain. Expert Opin Pharmacother 2008; 9: 1585–1594. [DOI] [PubMed] [Google Scholar]
- 33.Chou R, Fanciullo GJ, Fine PG, et al. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10: 113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badalamenti VC, Buckley JW, Smith ET. Safety of EMBEda (morphine sulfate and naltrexone hydrochloride) extended-release capsules: review of postmarketing adverse events during the first year. J Opioid Manag 2012; 8: 115–125. [DOI] [PubMed] [Google Scholar]
- 35.Hassanzadeh M, Ghaemy M, Ahmadi S. Extending time profile of morphine-induced analgesia using a chitosan-based molecular imprinted polymer nanogel. Macromol Biosci 2016; 16: 1515–1523. [DOI] [PubMed] [Google Scholar]
- 36.Smith LJ, Krugner-Higby L, Clark M, et al. A single dose of liposome-encapsulated oxymorphone or morphine provides long-term analgesia in an animal model of neuropathic pain. Comp Med 2003; 53: 280–287. [PubMed] [Google Scholar]
- 37.Johnson FK, Ciric S, Boudriau S, et al. The relative bioavailability of morphine sulfate and naltrexone hydrochloride extended release capsules (EMBEDA(R)) and an extended release morphine sulfate capsule formulation (KADIAN(R)) in healthy adults under fasting conditions. Am J Ther 2011; 18: 2–8. [DOI] [PubMed] [Google Scholar]
- 38.Gonçalves L, Silva R, Pinto-Ribeiro F, et al. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp Neurol 2008; 213: 48–56. [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki T, Choi IY, Rubas W, et al. NKTR-181: a novel mu-opioid analgesic with inherently low abuse potential. The J Pharmacol Exp Ther 2017; 363: 104–113. [DOI] [PubMed] [Google Scholar]
- 40.Corsetti M, Tack J. Naloxegol: the first orally administered, peripherally acting, mu opioid receptor antagonist, approved for the treatment of opioid-induced constipation. Drugs Today (Barc) 2015; 51: 479–489. [DOI] [PubMed] [Google Scholar]
- 41.Bui K, She F, Zhou D, et al. The effect of quinidine, a strong P-glycoprotein inhibitor, on the pharmacokinetics and central nervous system distribution of naloxegol. J Clin Pharmacol 2016; 56: 497–505. [DOI] [PubMed] [Google Scholar]
- 42.Mahkam M, Sharifi-Sanjani N. Preparation of new biodegradable polyurethanes as a therapeutic agent. Polym Degrad Stab 2003; 80: 199–202. [Google Scholar]
- 43.Schmeltzer RC, Anastasiou TJ, Uhrich KE. Optimized synthesis of salicylate-based poly(anhydride-esters). Polym Bull (Berl) 2013; 49: 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmeltzer RC, Uhrich KE. Synthesis and characterization of antiseptic-based poly(anhydride-esters). Polym Bull (Berl) 2006; 57: 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anastasiou TJ, Uhrich KE. Aminosalicylate-based biodegradable polymers: syntheses and in vitro characterization of poly(anhydride-ester)s and poly(anhydride-amide)s. J Polym Sci A Polym Chem 2003; 41: 3667–3679. [Google Scholar]
- 46.Grant GJ, Vermeulen K, Zakowski MI, et al. Prolonged analgesia and decreased toxicity with liposomal morphine in a mouse model. Anesth Analg 1994; 79: 706–709. [DOI] [PubMed] [Google Scholar]
- 47.Kozela E, Pilc A, Popik P. Inhibitory effects of MPEP, an mGluR5 antagonist, and memantine, an N-methyl-D-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology (Berl) 2003; 165: 245–251. [DOI] [PubMed] [Google Scholar]
- 48.Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur J Pharmacol 2014; 743: 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Codd EE, Shank RP, Schupsky JJ, et al. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 1995; 274: 1263–1270. [PubMed] [Google Scholar]
- 50.Pasternak GW, Pan Y-X. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev 2013; 65: 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wheeler M, Oderda GM, Ashburn MA, et al. Adverse events associated with postoperative opioid analgesia: a systematic review. J Pain 2002; 3: 159–180. [DOI] [PubMed] [Google Scholar]
- 52.Kim T, Murdande S, Gruber A, et al. Sustained-release morphine for epidural analgesia in rats. Anesthesiology 1996; 85: 331–338. [DOI] [PubMed] [Google Scholar]
- 53.Yaksh TL, Provencher JC, Rathbun ML, et al. Pharmacokinetics and efficacy of epidurally delivered sustained-release encapsulated morphine in dogs. Anesthesiology 1999; 90: 1402–1412. [DOI] [PubMed] [Google Scholar]
- 54.Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg 2001; 182: 11s–18s. [DOI] [PubMed] [Google Scholar]
- 55.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci 1992; 12: 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]