

Abstract
The ribosomal protein (RP)-MDM2 interaction is a p53 response pathway critical for preventing oncogenic c-MYC-induced tumorigenesis. To investigate whether the RP-MDM2-p53 pathway is a broad anti-oncogenic mechanism, we crossed mice bearing an MDM2C305F mutation, which disrupts RPL11 binding to MDM2, with mice expressing an oncogenic HrasG12V transgene. Interestingly, the MDM2C305F mutant mice, which are hypersensitive to c-MYC-induced tumorigenesis, are not hypersensitive to oncogenic HrasG12V-induced tumorigenesis. Unlike c-MYC, which induces expression of RPL11, RAS overexpression leads to an increase in RPL23 mRNA and protein while RPL11 expression remains unchanged. The induction of RPL23 involves both MEK and PI3K signaling pathways and requires mTOR function. Increased expression of RPL23, which maintains binding to MDM2C305F mutant, correlates with increased p53 expression in MDM2C305F cells. Furthermore, RAS overexpression can induce p53 in the absence of p19ARF, and the induction can be abolished by down-regulation of RPL23. Thus, while the RPL11-MDM2-p53 pathway coordinates with the p19ARF-MDM2-p53 pathway against oncogenic c-MYC-induced tumorigenesis, the RPL23-MDM2-p53 pathway coordinates with the p19ARF-MDM2-p53 pathway against oncogenic RAS-induced tumorigenesis.
Keywords: MDM2, p53, RPL23, p19ARF, RAS
Introduction
The tumor suppressor gene TP53 is mutated in about 50% of all human cancers (1). As a transcription factor p53 triggers cell cycle arrest, differentiation, apoptosis, and senescence in response to a variety of stresses. Murine double minute 2 (MDM2) is the primary negative regulator of p53, and it accomplishes this by both binding to and inhibiting the transactivation domain of p53 (2), as well as serving as an E3 ubiquitin ligase for p53 degradation (3,4). Meanwhile, p53 enhances MDM2 transcription, forming an auto-regulatory feedback loop (5).
It has been demonstrated that several ribosomal proteins (RPs), such as RPL5, RPL11, and RPL23, interact with MDM2 to inhibit its E3 ligase function, thereby stabilizing and activating p53, suggesting an RP-MDM2-p53 signaling pathway (6). Ribosomal biogenesis is one of the most energy demanding and tightly regulated processes during cell growth and proliferation. Since cancer cells undergo uncontrolled growth and proliferation, they require accelerated ribosomal biogenesis, mandating increased RP production. Using knock-in mice bearing an MDM2C305F point mutation, which prevents binding of RPL5 and RPL11 to MDM2 (7), previous studies have established the physiological significance of the RP-MDM2 interaction in responding to ribosomal stress and demonstrated that the RP-MDM2-p53 pathway is critical in preventing oncogenic c-MYC induced lymphomagenesis in mice (8).
The tumor suppressor p19ARF (p14ARF in human) is uniquely transcribed from an alternative reading frame of the INK4a/ARF gene locus. Similar to RPL11 and RPL5, p19ARF can inhibit MDM2 E3 ligase activity by directly binding to MDM2, stabilizing and activating p53, instituting a p19ARF-MDM2-p53 signaling pathway (9). Previous studies have shown that mice with homozygous deletion of p19ARF (p19Arf−/−) are predisposed to spontaneous tumor development (10). Additionally, oncogenic proteins, such as c-MYC and RAS, can drive tumors by selectively inactivating the p19ARF-MDM2-p53 pathway. Accelerated cancer progression is observed in Eμ-myc;p19Arf−/− transgenic mice, which die of lymphoma within a few weeks of birth (11), and in HrasG12V;p19Arf−/− transgenic mice, which die of melanoma within a few months of birth (12), demonstrating the importance of p19ARF in tumor suppression.
Overexpression of oncogenic RAS induces cell cycle arrest in wild type (WT) murine keratinocytes, which is mediated by increased expression of p19ARF (13). Conversely, oncogenic RAS transforms p19ARF-null mouse embryonic fibroblasts (MEFs) by bypassing p53-mediated checkpoint control (14). The RP-MDM2-p53 signaling pathway responds to deregulated ribosomal biogenesis caused by c-MYC overexpression to activate p53 and prevent tumorigenesis (8). Given that overexpression of RAS promotes growth and proliferation, which like c-MYC overexpression involves enhanced ribosomal biogenesis, we sought to determine whether the RP-MDM2-p53 signaling pathway might also respond to oncogenic RAS overexpression and play a role in tumor suppression.
Materials and Methods
Immunoblotting and immunoprecipitation
For western blotting, MEFs were lysed with 0.5% NP-40 lysis buffer. For mouse tissue protein extraction, tissue from the skin and lymphomas was ground by pestle and mortar with liquid N2, and protein was extracted with 0.5% NP-40 lysis buffer. To assess the half-life of RPL11 and RPL23, low passage MEF cells were treated with cycloheximide (50 μg/ml), chased for the indicated time points, and harvested with SDS lysis buffer (2% SDS, 10% glycerol, 50 mM Tris). Mouse monoclonal anti-Mdm2 (2A10, Calbiochem), mouse monoclonal anti-p53 (NCL-505, Novocastra), goat polyclonal anti-p53 (FL-393, Santa Cruz), mouse monoclonal anti-actin (MAB1501, Chemicon International), rabbit monoclonal anti-β-tubulin (ab179513, Abcam), rabbit monoclonal anti-Ras (ab52939, Abcam), mouse monoclonal anti-Ras (BD610001, BD Biosciences), rat polyclonal anti-p19Arf (Santa Cruz), rabbit polyclonal anti-phospho (Ser473)-AKT (9271S, Cell Signaling), mouse monoclonal anti-phospho-p44/42 (Thr202/204) ERK1/2 (9107S, Cell Signaling), rabbit monoclonal anti-phospho (Ser473)-AKT (4060S, Cell Signaling), rabbit polyclonal anti-AKT (9272S, Cell Signaling), rabbit monoclonal anti-phospho-p44/42 (Thr201/Tyr204) ERK1/2 (4370S, Cell Signaling), rabbit polyclonal anti-ERK1/2 (9102S, Cell Signaling) and anti-GAPDH (RM2002, Ray Antibody Biotechnology, Beijing) antibodies were purchased commercially. Rabbit polyclonal antibodies to p21 were gifts from Dr. Yue Xiong (UNC-Chapel Hill). Rabbit polyclonal antibodies to RPL11 and RPL23 were made in house as previously described (7). Procedures and conditions for immunoprecipitation were performed as previously described (15).
Immunohistochemical Analysis
Antigen retrieval for antibody on formalin-fixed paraffin sections was done by boiling paraffin samples in citrate buffer (pH 6.0) for 15 minutes. Endogenous peroxidase activity was quenched by incubation in 3% H2O2 in methanol for 10 minutes. Ki-67 immunohistochemical staining of mouse spleen samples was used to detect proliferating cells. Antibody detection was done with purified mouse anti-Ki-67 primary antibody (BD Pharmingen, San Diego, CA) and biotin-conjugated anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA). Ki-67 expressing cells were stained brown using a biotin-peroxidase kit (Vectastain Elite, Vector Laboratories). The ratio of positively stained cells to total cells was calculated. Student’s t test (p<0.05 was considered significant) was used to compare the differences in proliferation levels between the different mouse genotypes. Antibodies to p53 (CM5, Leica Biosystems), Bax (#554104, BD Biosciences), and p21 (DCS 60.2, Thermo Scientific) were purchased commercially, while antibodies to RPL23 were made in-house as previously described (7).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 5 Software (Graph-Pad Software, San Diego, CA). Kaplan-Meier survival curves were generated to assess lifespan. Quantitative real time PCR data are represented as mean ± SEM, and were analyzed by Student’s t test.
Results
MDM2C305F mutation partially rescues oncogenic H-RAS induced tumorigenesis
We crossed Mdm2C305F/C305F mice (Mdm2m/m hereafter) with mice expressing an activated melanocyte-specific HrasG12V transgene and examined melanomagenesis in the HrasG12V;Mdm2m/m transgenic mice. Consistent with previous studies (12), HrasG12V;p19Arf−/− mice developed spontaneous melanomas, and the median survival of the transgenic mice was about 6 months (Figure 1A, red line). Unexpectedly, however, the median survival for HrasG12V;Mdm2m/m;p19Arf−/− compound mice was significantly longer than that of Hras;p19Arf−/− mice at more than 12 months (Figure 1A, purple line, p=0.0007). This result indicates that, in contrast to accelerating oncogenic c-MYC-induced tumorigenesis (8), the MDM2C305F mutation partially rescues oncogenic RAS-induced tumorigenesis. Tumors from HrasG12V;Mdm2m/m;p19Arf−/− mice were indistinguishable to those from HrasG12V;p19Arf−/− mice (Figure S1). Although the latency of tumors differed depending on the presence or absence of MDM2C305F mutation, the histological characteristics of established tumors were equivalent between tumors from HrasG12V;Mdm2m/m;p19Arf−/− mice and those from HrasG12V;p19Arf−/− mice, indicating that the MDM2C305F mutation does not ultimately affect the pathophysiological nature of tumors induced upon p19ARF deletion and oncogenic RAS overexpression.
Figure 1. Mdm2C305F mutation partially rescues HRAS induced tumorigenesis.
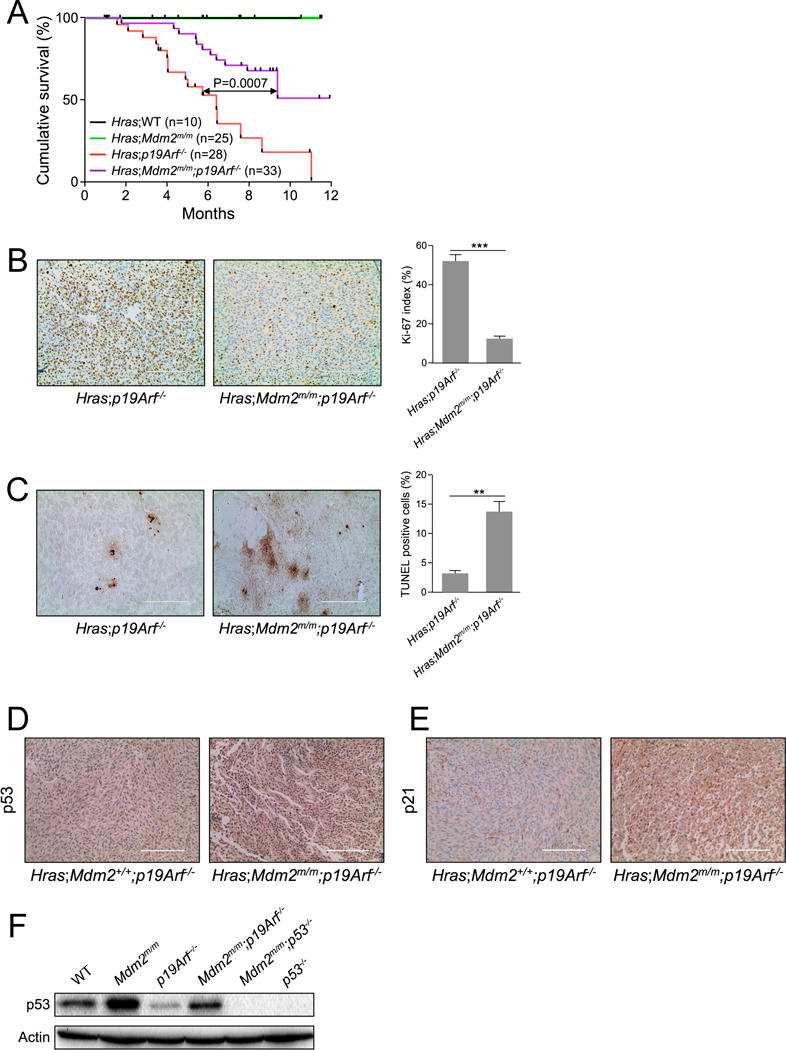
A. Kaplan-Meier survival curves for HrasG12V;WT, HrasG12V;Mdm2m/m, HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− are shown. Median survival time for HrasG12V;p19Arf−/− mice was 6.4 months. There was a significant difference between survival curves for HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− mice (analyzed by log-rank test; p value was 0.0007).
B. Representative Ki-67 staining of skin tumors from 16-week old mice. Brown staining indicates Ki-67-positive proliferating cells. (Scale bar = 200 μm.) The Ki-67 index (calculated as the percentage of Ki-67-positive tumor cells vs. total cells in the view field from at least five randomly chosen fields along the edge of tumors) for the genotypes assayed is indicated in parentheses: HrasG12V;p19Arf−/− mice (51.8), and HrasG12V;Mdm2m/m;p19Arf−/− mice (11.8). Data are represented as mean ± SEM. (*** indicates p value <0.001)
C. Representative TUNEL staining of skin tumors from different genotypes of 16-week old mice. (Scale bar = 200 μm.) The percentage of TUNEL-positive cells in the view field was calculated from at least five randomly chosen fields along the edge of tumors and is indicated in parentheses: HrasG12V;p19Arf−/− mice (3.2%), and HrasG12V;Mdm2m/m;p19Arf−/− mice (13.6%). Data are represented as mean ± SEM. (** indicates p value <0.01)
D. Representative p53 immunohistochemical staining of skin tumors from 16-week old HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− mice. (Scale bar = 200 μm.)
E. Representative p21 immunohistochemical staining of skin tumors from 16-week old HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− mice. (Scale bar = 200 μm.)
F. Early passage WT, Mdm2m/m, p19Arf−/−, Mdm2m/m;p19Arf−/−, Mdm2m/m;p53−/− and p53−/− MEFs were harvested for western blot analysis for p53 expression.
We next determined the proliferative capacities of melanomas from the HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− mice. As shown in Figure 1B, tumors from HrasG12V;p19Arf−/− mice displayed a higher percentage of Ki-67 positive cells (Ki-67 index 51.8) than tumors from HrasG12V;Mdm2m/m;p19Arf−/− mice (Ki-67 index 11.8). TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling) immunohistochemical analysis was performed to measure levels of apoptosis. Tumors isolated from HrasG12V;Mdm2m/m;p19Arf−/− mice displayed a significantly higher percentage of TUNEL-positive cells (13.6%) than those of HrasG12V;p19Arf−/− tumors (3.2%) (Figure 1C). These data suggest that the MDM2C305F mutation decelerates oncogenic RAS-induced tumorigenesis by inhibiting proliferation and inducing apoptosis.
The deceleration of RAS induced tumorigenesis by MDM2C305F mutation was unexpected. Because p53 is the primary target of MDM2, we therefore compared basal levels of p53 in HrasG12V;p19Arf−/− and HrasG12V;Mdm2m/m;p19Arf−/− tumors. As shown in Figure 1D, HrasG12V;Mdm2m/m;p19Arf−/− tumors expressed higher levels of p53. To determine whether p53 activity also correlated with expression in these tumors, we analyzed p21 and found that there was greater p21 staining in HrasG12V;Mdm2m/m;p19Arf−/− tumors compared to HrasG12V;p19Arf−/− tumors (Figure 1E). We then used a tumor-free system to compare whether the MDM2C305F mutation had any effect on basal p53 accumulation. We analyzed p53 levels in WT, Mdm2m/m, p19Arf−/−, and Mdm2m/m;p19Arf−/− mouse embryonic fibroblasts (MEFs). Mdm2m/m MEFs expressed higher levels of p53 than did WT MEFs (Figure 1F). Likewise, Mdm2m/m;p19Arf−/− MEFs expressed higher levels of p53 than did p19Arf−/− MEFs. Thus, the deceleration of RAS-induced tumorigenesis by MDM2C305F mutation correlates with higher levels of p53 expression and activity.
RAS induces RPL23 expression via MEK/PI3K and mTOR pathways
Previous studies have shown that MDM2C305F mutation accelerates oncogenic c-MYC-induced tumorigenesis in mice due to loss of RPL11-MDM2 interaction (8). However, the MDM2C305F mutation does not affect MDM2 binding to RPL23 (7,8); and like RPL11, RPL23 interacts with MDM2 and activates p53 (16,17). To provide further insight for the deceleration of RAS-induced tumorigenesis by the MDM2C305F mutation, we sought to determine whether RAS, like c-MYC (18,19), could also upregulate RP expression. We assessed the levels of RPL11 and RPL23 in RAS-overexpressing mice. Interestingly, the protein levels of RPL23, but not RPL11, were elevated in pre-tumor mouse melanocytes expressing the HrasG12V transgene (Figure 2A, compare lane 1 with lane 3, and lane 2 with lane 4, also Figure S2A, compare lane 1 with lane 3, and lane 2 with lane 4).
Figure 2. RAS induces RPL23 expression.
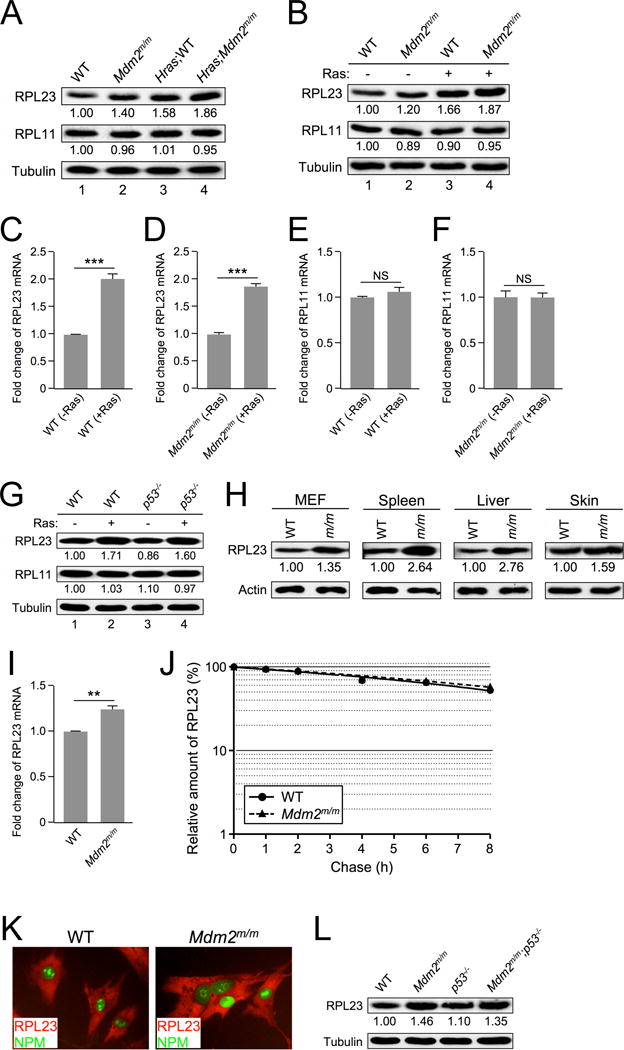
A. Extracts from skin tissue of non-tumor-bearing WT and Mdm2m/m mice and from their Hras transgenic counterparts were analyzed by western blot. The relative expression of RPL23 and RPL11 is shown under the blot (analyzed by ImageJ software, 1.47v).
B. Early passage WT and Mdm2m/m MEFs were infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+), selected in puromycin for three days, then allowed to recover for 48 hours before harvesting for western blot analysis. The relative expression of RPL23 and RPL11 is shown under the blot (analyzed by ImageJ software, 1.47v).
C. WT MEFs were infected with retrovirus expressing either pBabe vector (-Ras) or pBabe-HRASG12V (+Ras) and harvested for qRT-PCR mRNA analysis. Relative RPL23 mRNA expression was calculated using β-GAPDH as an internal control. Data are represented as mean ± SEM, and were analyzed by Student’s t test. (*** indicates p value <0.001)
D. Mdm2m/m MEFs were infected with retrovirus expressing either pBabe vector (−Ras) or pBabe-HRASG12V (+Ras) and harvested for qRT-PCR mRNA analysis. Relative RPL23 mRNA expression was calculated using β-GAPDH as an internal control. (*** indicates p value <0.001)
E. WT MEFs were infected with retrovirus expressing either pBabe vector (−Ras) or pBabe-HRASG12V (+Ras) and harvested for qRT-PCR mRNA analysis. Relative RPL11 mRNA expression was calculated using β-GAPDH as an internal control. (NS indicates no statistically significant difference between samples)
F. Mdm2m/m MEFs were infected with retrovirus expressing either pBabe vector (−Ras) or pBabe-HRASG12V (+Ras) and harvested for qRT-PCR mRNA analysis. Relative RPL11 mRNA expression was calculated using β-GAPDH as an internal control. (NS indicates no statistically significant difference between samples)
G. Early passage WT and p53−/− MEFs were infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+), selected in puromycin for three days, then allowed to recover for 48 hours before harvesting for western blot analysis. The relative expression of RPL23 and RPL11 is shown under the blot (analyzed by ImageJ software, 1.47v).
H. Extracts from WT and Mdm2m/m MEFs and from tissues of 30-week-old WT and Mdm2m/m mice were analyzed by western blot. The relative expression of RPL23 is shown under the blot (analyzed by ImageJ software, 1.47v).
I. Early passage WT and Mdm2m/m MEFs were harvested for qRT-PCR mRNA analysis. Relative RPL23 mRNA expression was calculated using β-GAPDH as an internal control.
J. Half-life assay of RPL23 was carried out using early passage (P1) WT and Mdm2m/m MEFs treated with cycloheximide (50 μg/mL) and harvested with SDS lysis buffer at the indicated time points. The amount of RPL23 was quantified by densitometry, normalized to the level of actin, and plotted.
K. Early passage WT and Mdm2m/m MEFs were fixed and stained with rabbit anti-RPL23 antibody and a fluorescein isothiocyanate-conjugated anti-rabbit secondary antibody (red color), and mouse anti-B23 (NPM) antibody and a fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (green color). Fluorescence images were captured with a cooled charge-coupled device color digital camera (Model 2.2.0, Diagnostic) on an Olympus IX81 inverted microscope equipped with the appropriate fluorescence filters.
L. Extracts from WT, Mdm2m/m, p53−/−, and Mdm2m/m;p53−/− MEFs were analyzed by western blot. The relative expression of RPL23 is shown below the blot (analyzed by ImageJ software, 1.47v).
Furthermore, we infected MEFs with retrovirus expressing pBabe-HRASG12V and found that the expression of RPL23 was induced by ectopic HRASG12V (Figure 2B, compare lane 1 with lane 3, and lane 2 with lane 4). Conversely, the expression of RPL11 was unaffected by either endogenous or ectopic HRASG12V (Figure 2A and 2B). We also observed elevated levels of RPL23 mRNA in HRASG12V retrovirus-infected WT, Mdm2m/m, p19Arf−/−, and Mdm2m/m;p19Arf−/− MEFs (Figure 2C–D, Figure S2B–C). On the other hand, RPL11 mRNA levels remained unchanged in these cells (Figure 2E–F, Figure S2D–E). The induction of RPL23 by oncogenic RAS also appears to be p53-independent, since infection of pBabe-HRASG12V retrovirus resulted in RPL23 overexpression to similar levels in both WT and p53-null MEFs (Figure 2G).
We noticed that the levels of RPL23 were higher in Mdm2m/m mouse skin tissue and Mdm2m/m MEFs compared to their counterparts expressing WT MDM2 (Figure 2A, compare lane 1 with lane 2; Figure 2B, compare lane 1 with lane 2). To analyze this phenomenon, we examined RPL23 expression in multiple tissues, including MEFs, spleen, liver, and skin. We found that RPL23 levels were indeed higher in tissues of Mdm2m/m mice than in those of WT mice (Figure 2H). Furthermore, tumors from HrasG12V;Mdm2m/m;p19Arf−/− mice showed stronger RPL23 staining than those from HrasG12V;p19Arf−/− mice (Figure S2F). To further investigate the MDM2C305F mutation-mediated increase of RPL23 protein level, we analyzed RPL23 mRNA in WT and Mdm2m/m MEFs and observed elevated levels in Mdm2m/m MEFs (Figure 2I). To test whether the elevated levels of RPL23 can also be explained by increased protein stability, we performed a protein half-life assay using early passage MEFs. A normal rate of protein degradation was observed for RPL23 in Mdm2m/m MEFs (Figure 2J), indicating that the stability of RPL23 is not altered by MDM2C205F mutation. An unaltered rate of protein degradation was also observed for RPL11 in in Mdm2m/m MEFs (Figure S2G–I).
We examined RPL23 subcellular localization by immunofluorescence staining, and no difference was observed between WT and Mdm2m/m cells (Figure 2K). Elevated RPL23 expression was also observed in Mdm2m/m cells under a p53-null background (Figure 2L), indicating that it is a p53-independent event. These data indicate that the MDM2C305F mutation increases RPL23 mRNA level and RPL23 protein production. We have yet to explain the mechanism for the increase of RPL23 mRNA expression in cells with the MDM2C305F mutation, but we believe that the increased RPL23 mRNA and protein levels are likely a cause for the increased p53 expression (Figure 1F) and the deceleration of RAS-induced tumorigenesis observed in HrasG12V;Mdm2m/m;p19Arf−/− mice (Figure 1A).
It has been previously shown that regulation of RPL23 can occur at the translational level through mRNA cap binding of eukaryotic translation initiation factor 4E (eIF4e) (20), a well characterized downstream target of the mammalian target of rapamycin (mTOR) pathway. We therefore investigated the mechanism of RAS-induced RPL23 expression at translational control. Because mTOR is a known downstream target of RAS signaling, we hypothesized that RAS induction of RPL23 could be mediated by the mTOR signaling pathway. To test this idea, we infected WT MEFs with pBabe-HRASG12V retrovirus and followed with rapamycin treatment to inhibit mTOR activity. Expression of RPL23, but not RPL11, was induced by RAS overexpression in MEF cells; however, upon treatment with rapamycin RAS-induced RPL23 expression was inhibited (Figure 3A). Consistent with observations made in MEFs, RPL23 expression was elevated in human embryonic kidney (HEK-293T) cells infected with pBabe-HRASG12V retrovirus, and the expression was inhibited by rapamycin (Figure S3A). These data indicate that RAS regulates RPL23 translation through an mTOR-dependent mechanism.
Figure 3. RAS induces RPL23 through the PI3K/MEK and mTOR signaling pathways.
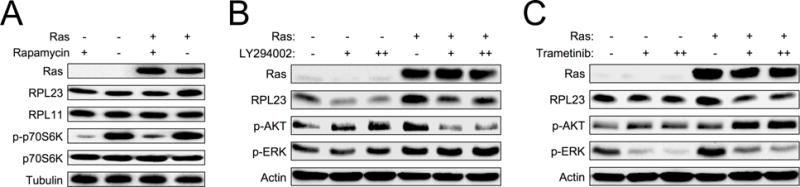
A. Early passage Mdm2m/m MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were treated with 200 nM rapamycin for 18 hours, and then harvested for western blot analysis.
B. Early passage WT MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were treated with PI3K inhibitor LY294002 (1 μM “+” or 5 μM “++”) for 48 hours and then harvested for western blot analysis.
C. Early passage WT MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-H-RasG12V (+) were treated with MEK inhibitor trametinib (1 nM “+” or 2.5 nM “++”) for 48 hours and then harvested for western blot analysis.
RAS signaling to mTOR can occur through phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MEK) pathways. In order to further investigate the signaling pathways through which RAS induces RPL23 expression, we infected WT MEFs and HEK-293T cells with pBabe-HRASG12V retrovirus and treated the cells with small molecule inhibitors of RAS signaling. RPL23 induction by RAS was partially inhibited by treatment with either the MEK inhibitor trametinib or the PI3K inhibitor LY294002, respectively (Figure 3B–C, Figure S3B–C). Together, these data suggest that RAS-mediated induction of RPL23 translation is mTOR dependent and mediated by both PI3K and MEK signaling pathways.
RAS induces p53 expression in the absence of p19ARF
Oncogenic RAS induces p19ARF-dependent activation of p53 (21). To determine whether p19ARF is required for RAS-induced p53 expression in our system, we analyzed pre-tumor skin extracts from p19Arf−/− and Mdm2m/m;p19Arf−/− mice expressing HrasG12V transgene for p53 expression. Interestingly, in the absence of p19ARF, the HrasG12V transgene still induced p53 expression (Figure 4A, compare lane 1 with lane 3), and the induction was further augmented by MDM2C305F mutation (Figure 4A, compare lane 3 with lane 4). A similar conclusion was reached using MEFs infected with pBabe-HRASG12V retrovirus, which resulted in p53 accumulation in p19ARF-null MEFs (Figure 4B, compare lane 1 with lane 3), and this HRASG12V-induced p53 expression was further increased in Mdm2m/m;p19Arf−/− MEFs compared to p19Arf−/− MEFs (Figure 4B, compare lane 3 with lane 4). These results indicate that oncogenic RAS can induce p53 in a p19ARF-independent manner. This is consistent with the observation that HRASG12V induced RPL23 expression in p19ARF-null mice and cells (Figure S2A–S2C, S2F).
Figure 4. RAS induces p53 expression in the absence of p19ARF.
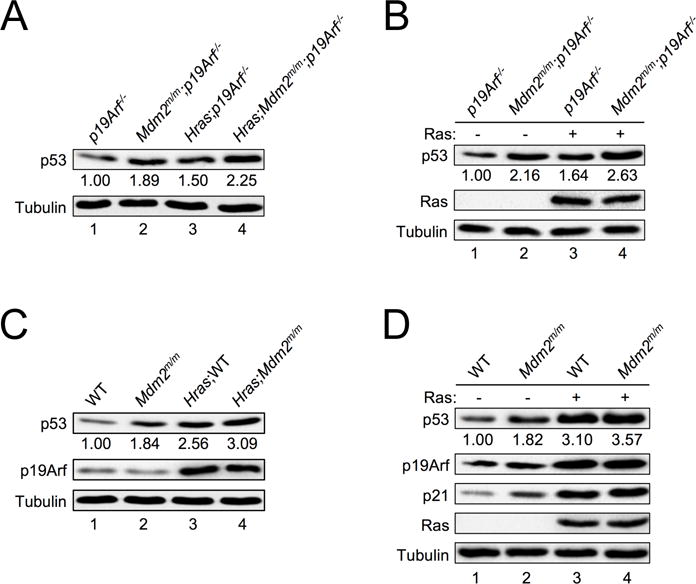
A. Extracts from skin tissues of non-tumor-bearing p19Arf−/− and Mdm2m/m;p19Arf−/− mice and their non-tumor-bearing Hras transgenic counterparts were analyzed by western blot. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
B. Early passage p19Arf−/− and Mdm2m/m;p19Arf−/− MEFs were infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+), selected in puromycin for three days, then allowed to recover for 48 hours before harvesting for western blot analysis. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
C. Extracts from skin tissues of WT and Mdm2m/m mice and their non-tumor-bearing Hras transgenic counterparts were analyzed by western blot. The relative expression of p53 is shown underneath the blot (analyzed by ImageJ software, 1.47v).
D. Early passage WT and Mdm2m/m MEFs were infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+), selected in puromycin for three days, then allowed to recover for 48 hours before harvesting for western blot analysis. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
We noticed that even though oncogenic RAS can induce p53 accumulation in the absence of p19ARF, the levels of the induction were notably reduced compared to those in the presence of p19ARF. In the presence of p19ARF the HrasG12V transgene induced an approximately 2.5-fold increase of p53 in mouse melanocytes (Figure 4C, compare lane 1 with lane 3), whereas in the absence of p19ARF the HrasG12V transgene induced an approximately 1.5-fold increase of p53 (Figure 4A, compare lane 1 with lane 3). Similarly, ectopic expression of pBabe-HRASG12V induced an approximately 3-fold increase of p53 in WT MEFs (Figure 4D, compare lane 1 with lane 3), while the same virus induced an only 1.6-fold increase of p53 in p19ARF-null MEFs (Figure 4B, compare lane 1 with lane 3). This evidence supports the notion that there exists a p19ARF-independent signaling pathway engaged by oncogenic RAS to induce p53 accumulation.
RPL23 is required for RAS induction of p53 in the absence of p19ARF
Given that oncogenic RAS induces expression of RPL23, and that ectopic expression of RPL23 can stabilize p53 by inhibiting MDM2-mediated p53 ubiquitination and degradation (16,17), we wanted to investigate whether RPL23 is necessary for oncogenic RAS-mediated accumulation of p53 in the absence of p19ARF. We knocked down RPL23 by siRNA in p19Arf−/− MEFs, infected the cells with pBabe-HRASG12V retrovirus, and examined p53 levels. Down-regulation of RPL23 significantly attenuated RAS-induced p53 expression in p19Arf−/− MEFs (Figure 5A) as well as in Mdm2m/m;p19Arf−/− compound MEFs (Figure 5B), suggesting that in the absence of p19ARF, RPL23 is a major mediator of p53 expression induced by oncogenic RAS. On the other hand, in the presence of p19ARF down-regulation of RPL23 did not affect RAS induction of p53 (Figures 5C–5D). This suggests that p19ARF is the primary responder to oncogenic RAS expression whereas the RPL23-mediated response might be a fail-safe mechanism that comes into play upon loss of p19ARF.
Figure 5. RPL23 is required for RAS induction of p53 in the absence of p19ARF.
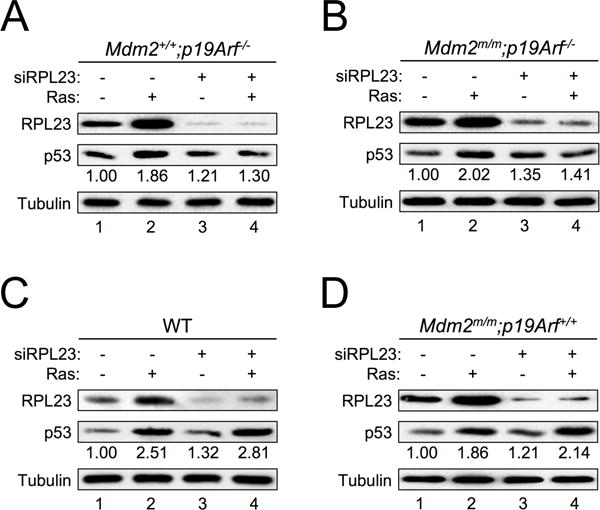
A. p19Arf−/− MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were transfected with a control scrambled RNA duplex (−) or RPL23 siRNA (+) for two days. Cell extracts were collected and analyzed by western blot with the indicated antibodies. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
B. Mdm2m/m;p19Arf−/− MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were transfected with a control scrambled RNA duplex (−) or RPL23 siRNA (+) for two days. Cell extracts were collected and analyzed by western blot with the indicated antibodies. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
C. WT MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were transfected with a control scrambled RNA duplex (−) or RPL23 siRNA (+) for two days. Cell extracts were collected and analyzed by western blot with the indicated antibodies. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
D. Mdm2m/m MEFs infected with retrovirus expressing either pBabe vector (−) or pBabe-HRASG12V (+) were transfected with a control scrambled RNA duplex (−) or RPL23 siRNA (+) for two days. Cell extracts were collected and analyzed by western blot with the indicated antibodies. The relative expression of p53 is shown under the blot (analyzed by ImageJ software, 1.47v).
Discussion
Both RPL23 and p19ARF induce p53 expression in response to oncogenic RAS insult
We have provided evidence that the induction of p53 by oncogenic RAS does not occur solely through p19ARF (Figure 4), and that another pathway to p53 induction exists through RPL23 (Figure 5). There are several possible reasons for this redundancy. First, p19ARF and RPL23 could work together to produce a more rapid and robust p53 response to oncogenic RAS expression than either one alone. We have shown that in the absence of p19ARF, p53 can still be induced by RAS through RPL23, but that this happens to a lesser degree than when p19ARF is present (Figure 4), suggesting that the two pathways could act concurrently (Figure 6A). However, we believe it is more likely that p19ARF, as a canonical tumor suppressor, is the primary responder to RAS overexpression and that RPL23 acts as a backup response to induce p53 activation, particularly when the function of p19ARF is lost. This notion is supported by the observation that knockdown of RPL23 by siRNA does not significantly attenuate p53 activation by RAS when p19ARF is present (Figure 5C–D), but it does so when p19ARF is absent (Figure 5A–B).
Figure 6. A model depicting a RAS-RPL23-MDM2-p53 pathway.
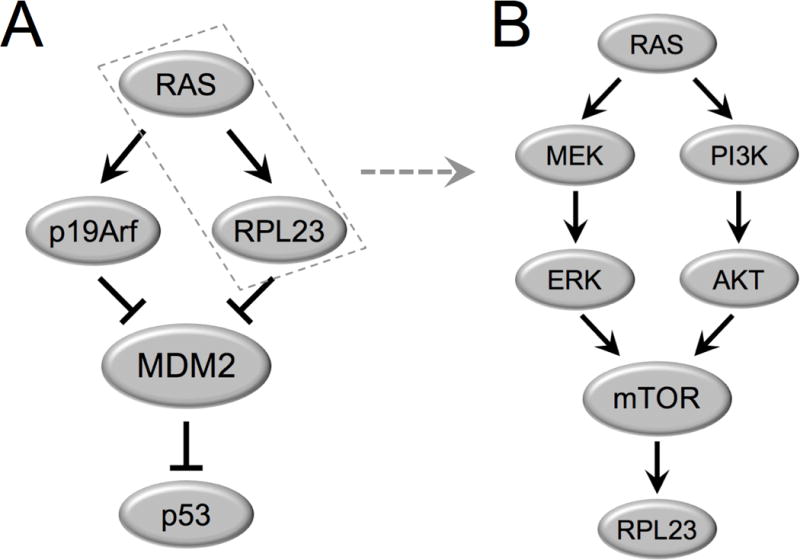
A. Oncogenic RAS induces expression of both RPL23 and p19ARF independently. RPL23 and p19ARF both bind and inhibit MDM2 to stabilize p53.
B. The RAS-MAPK-ERK and RAS-PI3K-AKT signaling cascades induce mTOR dependent expression of RPL23.
The loss of p19ARF in the presence of RAS overexpression can drive tumor progression through inactivation of the p19ARF-MDM2-p53 pathway (12), so the presence of a backup mechanism for p53 activation would be advantageous for tumor prevention. In support of this idea, Mdm2m/m mice, which demonstrate increased levels of RPL23 (Figure S2A) and higher levels of p53 (Figure 4) under a p19ARF deletion background, are more resistant to RAS overexpression-induced tumors as compared to mice with p19ARF deletion alone (Figure 1).
Different ribosomal proteins respond to specific oncogenic stresses to stabilize p53
In general, deregulated oncogenes drive cell growth and proliferation, which requires accelerated ribosomal biogenesis. This current study and studies by others have demonstrated that RAS and c-MYC can drive expression of RPs. Although RP upregulation is generally associated with oncogenic growth, it appears that RPs can also serve as tumor suppressive signaling molecules. It is presently unclear why multiple ribosomal proteins (RPs) interact with MDM2 and appear to have similar functions in p53 stabilization. We propose that the RP-MDM2 interaction represents a system of checkpoints for cell growth, and here we have provided evidence to suggest that the different RPs may respond to distinct oncogenic stimuli to engage the MDM2-p53 pathway. For example, RPL23, but not RPL11, is specifically induced by RAS overexpression (Figure 2), while previous studies have shown that RPL11 responds to oncogenic c-MYC overexpression (8). The MDM2C305F mutation specifically disrupts interaction of RPL11 but not RPL23 with MDM2. Creation of the Mdm2m/m mice thus allowed us to dissect the distinct contributions of RPL11 and RPL23 to p53 induction by oncogenic c-MYC and oncogenic RAS overexpression, respectively. To date at least fourteen RPs have been shown to bind directly to MDM2 and modulate p53 in a similar fashion (22), but the specific signals that these RPs transduce have yet to be elucidated. We postulate that a possible reason that so many RPs bind MDM2 is to confer insult-specific modulation of p53. In this study, we demonstrate that RPL23 can respond to RAS to induce p53. However, although our data demonstrate that RPL23 is essential for RAS-induced p53 expression in the absence of p19ARF, we cannot conclude whether other RPs may have a role in the pathway, given that there are 16 ribosomal proteins have been shown to bind to MDM2 and affect p53. Significant investigation will be required to parse out whether other RPs respond to specific stress signals to induce p53 activation, and which signals are responsible for each potential RP-MDM2 interaction and subsequent p53 activation.
RPL23 expression via the MEK/PI3K and mTOR pathways
We have shown that the increased latency to tumor formation in HrasG12V;Mdm2m/m;p19Arf−/− mice compared to HrasG12V;p19Arf−/− mice is possibly due to oncogenic HRASG12V overexpression combined with MDM2C305F mutation-induced RPL23 expression, leading to increased p53 accumulation and activation. RAS induces RPL23 mRNA expression (Figure 2C–D, Figure S2B–C) and protein production through both MEK and PI3K signaling pathways, which is dependent on mTOR (Figure 3) but is independent of p53 (Figure 2G). It is likely that RAS induction of RPL23 is mediated through both an increase in transcription, as well as an increase in mTOR-dependent translation (Figure 6B). Although our results suggest that RAS can induce p53 through increased inhibition of MDM2 by RPL23, we cannot rule out the possibility that RPL23 induction by RAS can induce p53 through an MDM2-independent mechanism. Previous studies have shown that mTOR can upregulate p53 translation (23), and since mTOR is a downstream target of RAS, it is also possible that upregulation of RPL23 could aid in mTOR-dependent increases in p53 translation.
MDM2C305F mutation facilitates an increase in RPL23 mRNA expression, and we have shown that MDM2C305F mutation does not affect RPL23 protein stability or subcellular localization. Although how MDM2C305F mutation increases RPL23 mRNA transcription is presently unclear, several possibilities can be envisioned. First, RP-MDM2 interactions are thought to occur in response to perturbations of ribosomal biogenesis (6). We speculate that there is some low level of basal RP-MDM2 interaction, mildly inhibiting MDM2, to allow for normal physiological levels of p53 expression. It is possible that upon loss of RPL11-MDM2 binding due to MDM2C305F mutation, the basal level of MDM2 inhibition is decreased and RPL23 could be upregulated as a compensatory mechanism to counteract the decrease in MDM2 inhibition.
Second, ribosomal biogenesis and RP expression is a highly coordinated process, with imbalances in RP ratios often causing p53 dependent cell cycle arrest. In the case of MDM2C305F mutation, the loss of RPL11-MDM2 binding could liberate RPL11, leading to an increase in the relative levels of RPL11 in the ribosomal protein pool and creating imbalances in otherwise tightly regulated RP ratios. In order to combat this imbalance, the cell may increase expression of other ribosomal proteins including, in this case, RPL23.
Finally, a possibility formally remains, for reasons yet unknown, that MDM2C305F mutation could facilitate low but constitutive overexpression of RAS and RAS target genes. The Mdm2m/m mouse, with its elevated expression of endogenous RPL23, serves as a useful in vivo tool for investigating the function and mechanism of a RAS-RPL23-MDM2-p53 pathway, without the effects of RPL11-Mdm2 binding, and sets the stage for further investigations of RP-MDM2-p53 pathway activation.
Supplementary Material
Acknowledgments
We thank Aiwen Jin and Yong Liu for their helpful advice and technical assistance. We are in debt to Yue Xiong, Guillermina Lozano, Gerard Evan, Charles Sherr, and Norman Sharpless for their generosity in sharing mouse strains and reagents.
Financial Support
This research was supported by grants from the National Institutes of Health (CA155235, CA127770, CA 100302 and CA167637), and Natural Science Foundation of China (NSFC) from China to Y. Zhang, the National S&T Major Project for Infectious Diseases of China (NO.2012ZX10002-017) to J. Dong, the NSFC from China (NO.81502376) to X. Meng, and the National Institute of General Medical Sciences (5T32 GM007092) to N.R. Tackmann.
Footnotes
Conflict of Interest
The authors declare that they have no competing financial interests.
Additional methods are included in the supplemental materials
References
- 1.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358(6381):15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 3.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS letters. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 4.Geyer RK, Yu ZK, Maki CG. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol. 2000;2(9):569–73. doi: 10.1038/35023507. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7(7A):1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16(5):369–77. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol. 2007;27(3):1056–68. doi: 10.1128/MCB.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS, et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell. 2010;18(3):231–43. doi: 10.1016/j.ccr.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6(9):663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 10.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59(9):2217–22. [PubMed] [Google Scholar]
- 11.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11(21):2822–34. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci U S A. 2001;98(9):5025–30. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 15.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12(5):1151–64. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 16.Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24(17):7669–80. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24(17):7654–68. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci U S A. 2000;97(21):11198–202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, et al. Identification of c-myc responsive genes using rat cDNA microarray. Cancer research. 2000;60(21):5922–8. [PubMed] [Google Scholar]
- 20.Mamane Y, Petroulakis E, Martineau Y, Sato T-A, Larsson O, Rajasekhar VK, et al. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS One. 2007;2(2):e242–e42. doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 22.Kim TH, Leslie P, Zhang Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget. 2014;5(4):860–71. doi: 10.18632/oncotarget.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2012;31(15):1949–62. doi: 10.1038/onc.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.