

Abstract
Substantial evidence shows that activation of glial cells in the spinal cord may promote central sensitization and pain. Descending facilitation from the rostroventromedial medulla (RVM) is a critical component in the maintenance of chronic pain states, although the precise mechanisms through which facilitation maintains pain are unclear. Here, we investigated the possibility that glial activation in the RVM could promote descending facilitation from the RVM in states of inflammatory pain. Peripheral inflammation was induced with carrageenan injected into the hindpaws of male Sprague–Dawley rats, and behavioral responses to noxious thermal and light tactile stimuli were determined. Microinjection of the glial inhibitors minocycline or fluorocitrate, or of the p38 mitogen-activated protein kinase (MAPK) inhibitor SB 203580, produced a significant and time-related attenuation of behavioral hypersensitivity resulting from hindpaw inflammation. Carrageenan-induced inflammation increased immunolabeling for microglia and astrocytes in the RVM, as well as for phosphorylated p38 MAPK. Phosphorylated p38 MAPK was found in microglia and neurons of the RVM. Inflammation-induced microglial and astrocytic activation in the RVM were attenuated by RVM microinjection of the glial inhibitors. The data show that inflammatory pain is associated with glial activation in the RVM that probably participates in driving descending pain facilitation. These findings reveal a novel site of glial modulation of inflammatory pain.
Keywords: astrocytes, brainstem, chronic pain, microglia, p38 MAPK
Introduction
Chronic pain is maintained in part by neuroplastic changes at spinal and supraspinal sites. Increased activity of sensitized peripheral nerves can cause spinal sensitization, which results in enhancement of sensory information arriving at supraspinal centers (Woolf & Salter, 2000). The enhancement of nociceptive inputs can evoke neuroplastic changes, including activation of descending pain facilitatory systems from the rostroventromedial medulla (RVM) (Urban & Gebhart, 1999; Porreca et al., 2001, 2002; Gebhart, 2004). Activation of descending facilitation further promotes the release of pronociceptive excitatory neurotransmitters from primary afferent terminals, thus maintaining a state of enhanced pain (Pertovaara et al., 1998; Urban & Gebhart, 1999; Terayama et al., 2000; Porreca et al., 2002). Blocking descending facilitation abolishes enhanced pain (Pertovaara et al., 1996; Porreca et al., 2001, 2002; Burgess et al., 2002; Gardell et al., 2003).
Activation of spinal glia contributes to the sensitization of spinal neurons to nociceptive inputs. Chronic constriction injury of the sciatic nerve results in activation of spinal astrocytes in a distribution that overlaps with central terminals of the sciatic nerve, and is correlative with hyperalgesic intensity (Garrison et al., 1991). Animal models of inflammation, peripheral nerve injury, bone cancer pain and spinal cord injury have all shown activation of spinal glia (Fu et al., 1999, 2000; Watkins et al., 2001; Watkins & Maier, 2003; Raghavendra et al., 2004). Activated glial cells release proinflammatory substances, including cytokines, prostaglandins and excitatory amino acids, that sensitize neurons and also activate other glial cells (Kreutzberg, 1996; Ridet et al., 1997). Accordingly, inhibition of spinal glial activation or of pronociceptive mediators secreted by the activated glia attenuates behavioral signs of enhanced pain in animal models (Meller et al., 1994; Watkins et al., 1994; Milligan et al., 2000, 2003; Sweitzer et al., 2001; Raghavendra et al., 2003; Chacur et al., 2004). Peripheral inflammation also elicits phosphorylation of p38 mitogen-activated protein kinase (MAPK), which is a promoter of proinflammatory cytokines, in spinal neurons and microglia (Milligan et al., 2001; Svensson et al., 2003). Conversely, inhibition of the activity of phosphorylated p38 MAPK (p-p38 MAPK) attenuates hyperalgesia (Milligan et al., 2001; Svensson et al., 2003).
Most of the studies investigating the role of glial activation in spinal sensitization and enhancement of nociception have focused on changes occurring within the spinal cord. More recently, however, several studies have demonstrated that microglial activation and phosphorylation of neuronal and glial p38 MAPK occur at supraspinal sites after peripheral inflammation, nerve injury, or spinal cord injury (Raghavendra et al., 2004; Imbe et al., 2007; Zhao et al., 2007). Microglia and astrocytes in the RVM were activated within 2 days of constriction injury of the infraorbital nerve in rats (Guo et al., 2007; Wei et al., 2008). Because of the considerable mechanistic differences between nerve injury and peripheral inflammation, we undertook the present investigation in order to determine whether inflammation induced by carrageenan injection into the hindpaw is capable of eliciting glial activation in the RVM, and to determine whether this glial activation contributes to descending facilitation.
Materials and methods
Animals
Male Sprague–Dawley rats (250–300 g; Harlan Labs) were housed in a climate-controlled room on a 12-h light/dark cycle (lights on at 07:00 h and off at 19:00 h) and were allowed food and water ad libitum. All testing was performed under a protocol approved by the Institutional Animal Care and Use Committee of the University of Arizona and was in accordance with the policies and recommendations of the guidelines laid down by the NIH in the USA. Animals were kept three to a cage until they were implanted with a paired guide cannula, and then they were housed separately. The animal rooms housed only rats.
Surgery and injections
RVM cannulation
Rats were anesthetized with a ketamine-HCl/xylazine (80 mg/kg ketamine and 12 mg/kg xylazine) solution (100 mg/kg, intraperitoneal; Sigma, St Louis, MO, USA) and placed into a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). A 2 cm incision was made in the scalp, and the underlying connective tissue was retracted with hemostats to expose the skull. Paired guide cannulae 1.2 mm apart (26GA, #C235G-1.2 mm; Plastics One Inc., Roanoke, VA, USA) were directed towards the RVM (10.8 mm caudal to bregma, 0.6 mm to each side of the sagittal suture, and 7.0 mm ventral to the dura mater surface). Bone wax was used to seal the hole around the cannula. The paired guide cannula was secured to the skull using stainless steel screws and dental acrylic. A dummy cannula (#C235DC; Plastics One Inc.) was inserted to prevent contaminants from entering the RVM guide cannula. Rats were given an injection of antibiotic (amikacin C, 5 mg/kg, intraperitoneal) and allowed to recover for 5 days.
RVM microinjections
Microinjection of drugs was performed through an injection cannula (33GA, #C235G; Plastics One Inc.) that extended 2 mm beyond the tip of the paired guide cannula and into fresh brain tissue. The drug solutions were slowly expelled with a 10 μL Hamilton syringe connected to the injection cannula with tygone tubing (#CP95607-14; Cole-Parmer Instrument Company, Vernon Hills, IL, USA). Microinjections were made in a volume of 0.5 μL administered into the paired guide cannula. Drug doses are reported as the total amount administered bilaterally.
At the termination of the experiments, microinjection sites in the RVM were verified histologically for proper placement; only data from rats with correctly placed injections were included in the analysis. Injection sites were visualized by the microinjection of 0.5 μL of India ink into the paired guide cannula, and medullary sections were counterstained with Nissl stain (Fig. 1).
Fig. 1.
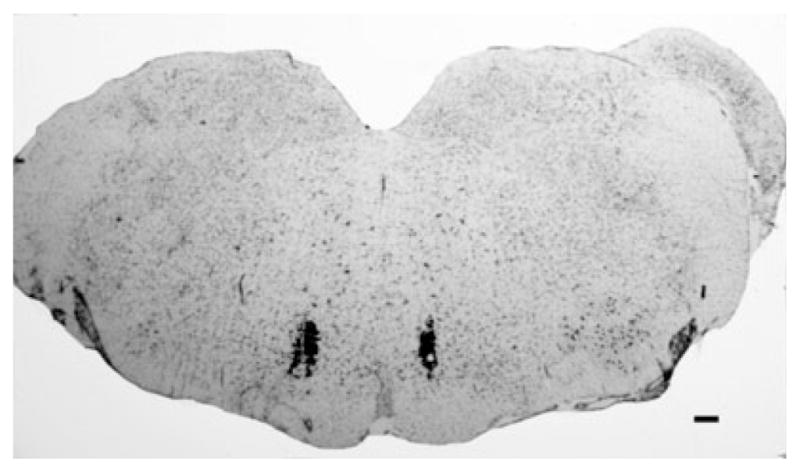
A representative photomicrograph of a medullary section at the level of the rostroventromedial medulla (RVM) counterstained with Nissl stain. The microinjection sites in the RVM were visualized with the microinjection of India ink to verify proper cannula placement. Scale bar: 100 μm.
Carrageenan injections
Rats were briefly anesthetized with isofluorane (4% in 95% O2/5% CO2 at a flow rate of 2–3 L/min), and subcutaneous injections of 100 μL of a 3% suspension of λ-carrageenan (Sigma) were made into the plantar aspect of the hindpaw. Control injections were made as above with 0.9% saline solution. Paw volumes were measured before and 3 h after carrageenan or saline injections with a plethysmomter (Ugo Basile) and expressed in milliliters.
Inhibitors
Minocycline hydrochloride (Sigma) was freshly dissolved daily in distilled H2O and heated briefly in a water bath until completely dissolved and the solution was clear. Fluorocitrate (DL-fluorocitric acid barium salt; Sigma) was first dissolved in 1 N HCl and then diluted in 0.9% sterile, isotonic saline. The p38 MAPK inhibitor SB 203580 hydrochloride (EMD Biosciences, La Jolla, CA, USA) was dissolved in distilled H2O.
Behavioral tests
Thermal hyperalgesia
Rats were placed on a hot-plate at 52°C and latency to lifting, stomping or licking of the hindpaw was measured. Hot-plate latencies were determined prior to carrageenan-induced inflammation (baseline), 3 h after carrageenan or vehicle injection, and at several time points after microinjection. Thermal hyperalgesia was indicated by a significant (P = 0.05) reduction in hot-plate latency from the baseline response latency. In the studies where a reversal of the thermal hyperalgesia was produced, the reversal was indicated by a significant (P < 0.05) increase in the hot-plate latency from the post-carrageenan value that indicated thermal hyperalgesia.
Tactile allodynia
Rats were placed in elevated plexiglass boxes (Plastics Plus, Tucson, AZ, USA) on wire-mesh. The paw withdrawal thresholds were determined by probing the hindpaw with a series of eight von Frey filaments (Stoelting) calibrated in logarithmically spaced increments from 0.41 to 15 g. Each filament was held against the plantar surface of the hindpaw until it buckled. The mean paw withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength, and analysed by the Dixon non-parametric test (Dixon, 1980; Chaplan et al., 1994). Paw withdrawal thresholds were determined prior to carrageenan-induced inflammation (baseline), 3 h after carrageenan or vehicle injection, and at several time points after microinjection. A significant (P = 0.05) reduction in paw withdrawal threshold from baseline indicated tactile allodynia, and a significant (P < 0.05) increase from the post-carrageenan baseline indicated attenuation of tactile allodynia.
Immunofluorescent labeling and imaging
At the termination of the experiment, the rats were killed by suffocation with CO2 and decapitated. The brainstems were removed and fixed overnight in 10% formalin (Sigma). This method of fixation was employed because of sensitive immunolabeling of OX-42. Comparisons between perfusion with 4% paraformaldehyde, perfusion with 10% formalin and fresh tissue immersion in 10% formalin were made, and no noticeable difference between the groups was observed, with the exception that the tissue immersed in 10% formalin showed more consistent labeling of the tissue sections. The fixation method did not affect any of the other antibodies used in this study. The tissue was then cryoprotected for 48 h in sterile 30% sucrose–Tris-buffered saline solution. Medullary sections 20 μm thick were cut in a −20°C cryostat and transferred serially to multiwell tissue culture plates containing 0.1 M Tris-buffered saline (TBS). Free-floating sections were incubated in TBS containing 0.1% Triton X-100 (Sigma) and 8% bovine serum albumin (BSA; Sigma) at room temperature for 1 h. The following primary antibodies were used: antibodies against CD11b/c (clone OX-42, purified mouse monoclonal antibody, 1 : 500; BMA Biomedicals, BMA Biomedicals, Switzerland; #T-3103), glial fibrillary acidic protein (GFAP; mouse monoclonal antibody, 1 : 1000; Sigma; #G 3893), neuronal nuclei (NeuN; purified mouse monoclonal antibody, 1 : 1000; Chemicon, Temecula, CA, USA; #MAB377), and p-p38 MAPK (purified rabbit polyclonal antibody, 1 : 500; Cell Signaling Technology, Danvers, MA, USA; #9211). All incubations with primary antibodies were performed in TBS with 3% BSA overnight at room temperature. Control sections with omitted primary antibody were examined to verify that there was no non-specific labeling with the secondary antibody. These primary antibodies have also been used to show glial staining in the RVM (Imbe et al., 2007). Sections were then washed with TBS (three times for 10 min each) and incubated with secondary antibody for 2 h at room temperature. Cy3-conjugated donkey anti-rabbit antibody (purified, 1 : 1000; Jackson ImmunoResearch, West Grove, PA, USA; #711-165-152) was used as the secondary antibody for p-p38 MAPK staining. Fluorescein isothiocyanate-conjugated donkey anti-mouse antibody (purified, 1 : 1000; Jackson ImmunoResearch; #715-095-150) was used as the secondary antibody for OX-42, GFAP and NeuN staining. After incubation with secondary antibody, sections were washed (three times for 10 min each) and mounted onto slides, air-dried, and coverslipped using Vectashield Hard Set Mounting Medium (Vector Laboratories, Burlingame, CA, USA). The fluorescent signals were detected using a Nikon E800 fluorescence microscope (Plan Apo 20 ×; numerical aperture, 0.75) and images (1344 × 1024 pixels shown at a resolution of 300 pixels/in.) were acquired with a Hamamatsu C5810 color CCD camera and its proprietary Image Processor software (Hamamatsu Photonic Systems, Bridgewater, NJ, USA).
The presence of activated microglia and astrocytes was suggested by changes in morphology, as described by DeLeo and coworkers (Colburn et al., 1997; Raghavendra et al., 2004). Resting microglia appear as small profiles with long, thin and wispy ramifications, and astrocytes show long and thin projections. The glial cells appear to be fairly well spaced apart. Activated microglia are indicated by larger, thicker cell bodies with shortened and thickened projections, and astrocytes appear more rounded, with decreased ramifications (Colburn et al., 1997; Raghavendra et al., 2004). Activated microglia and astrocytes show vivid, intense immunolabeling for OX-42 or GFAP, respectively, and the populations of the glia appear much denser.
Western blots
At the termination of the experiment, the animals were killed by suffocation with CO2 and decapitated, and the brainstem tissue was removed. A 1 mm coronal slice containing the RVM was obtained, and a tissue punch 1 mm in diameter was used to isolate the RVM area, including the nucleus raphe magnus, gigantocellularis, and gigantocelularis pars alpha. The RVM tissue was immediately frozen in liquid nitrogen and stored at −80°C until processing. The tissue was sonicated in cold Tris buffer (pH 7.4) containing 10 mM EDTA, 1% Triton X-100, 1% protease inhibitor, and 1% phosphatase inhibitor cocktail (Sigma), and centrifuged at 6500 r.p.m. for 15 min at 4°C. Protein concentrations were determined, and samples were prepared for electrophoresis. Proteins (30 μg) were separated using Bis-Tris gel electrophoresis (4–12% Bis-Tris; Invitrogen, Carlsbad, CA, USA), and electrophoretically transferred to poly(vinylidene fluoride) membranes (Invitrogen). Membranes were blocked with 5% non-fat dry milk in TBS containing 1% Triton X-100 (TBS-T) for 3 h at room temperature. The following primary antibodies were used: antibodies against GFAP (mouse monoclonal antibody, 1 : 2500; Santa Cruz, Santa Cruz, CA, USA; #sc-33673) and ionized calcium-binding adaptor molecule 1 (Iba1; 1 : 1000; Wako Chemicals USA, Richmond, VA, USA; #016-20001). All incubations with primary antibodies were performed in TBS-T with 5% BSA (Fraction V; Sigma) overnight at 4°C. Membranes were washed in TBS-T and incubated with appropriate secondary antibodies conjugated with horseradish peroxidase (goat anti-rabbit or goat anti-mouse, 1 : 5000; Thermo Scientific, Rockford, IL, USA; #31460 and #31430) at room temperature for 1 h. The membranes were washed, and immuno-reactive proteins were detected by enhanced chemiluminescence (Thermo Scientific) and visualized by exposure to X-ray film. The membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific) and re-blotted (as above) with primary antibody α-tubulin horseradish peroxidase (1 : 8000; Abcam, Cambridge, MA, USA; #ab40742) to serve as a loading control. For the quantification of western signals, the densities of specific GFAP, Iba1 and α-tubulin bands were measured with a computer-assisted imaging analysis system (ImageJ; NIH, Bethesda, MD). Target protein levels were normalized against the corresponding loading control levels, and the relative protein levels are presented as the mean ± standard error of the mean. No primary control membranes were examined to verify that there was no non-specific labeling with the secondary antibody, and membranes were examined after stripping with chemiluminescence to verify complete stripping of the previous antibody.
Statistical analysis
Reversal of hyperalgesia or allodynia was indicated by significant increases from the post-carrageenan means. Changes in means within each group over time were detected by one-factor ANOVA followed by Fisher’s least significant difference post hoc test. Significant differences between treatment groups over time were determined by two-factor ANOVA. Significance was set at P < 0.05. Treatment groups for the behavioral studies consisted of at least six rats per group. Power analyses (G*Power; Universitat Kiel, Germany) indicated that this group size was sufficient to achieve an acceptable power level (i.e. > 0.80 for α = 0.05). A total of 148 rats were prepared with a paired guide cannula and, over the course of the study, 14 were eliminated because of adverse behavioral effects, blocked cannulas, or incorrect cannula placement.
Results
Carrageenan-induced glial activation in the RVM
Peripheral inflammation was induced by injecting 100 μL of 3% carrageenan suspension into the plantar aspect of the hindpaw. Control animals received saline injections. The rats were killed 3 h after carrageenan or saline injection, and brainstem tissue was prepared for immunodetection of the microglial marker OX-42 or the astrocyte marker GFAP. Carrageenan-induced inflammation produced increased immunofluorescent labeling for both OX-42 and GFAP in the RVM, suggesting activation of both microglia and astrocytes (Fig. 2). These images are representative of multiple sections examined between −10.8 and −11.4 mm from bregma (Fig. 2E), and show that activated microglia and astrocytes exhibited hypertrophied cell bodies as compared with tissue from rats treated with saline (Fig. 2B and D).
Fig. 2.

Medullary sections that include the rostroventromedial medulla (RVM) obtained from rats treated with saline (A and C) or subjected to carrageenan-induced inflammation (B and D) and immunolabeled for OX-42 (microglia; A and B) and glial fibrillary acidic protein (GFAP) (astrocytes; C and D). Rats with inflammation indicate an intensification in labeling for OX-42 (B) and GFAP (D) along with changes in morphology indicative of activation of glia. Scale bar: 100 μm. Insets show magnified microglia cells (A and C) and astrocytes (B and D). Scale bar: 25 μm. (E) All RVM images are representative of multiple sections examined between −10.8 and −11.4 mm from bregma (Paxinos & Watson, 1998). The drawing represents the RVM location, including the nucleus raphe magnus and gigantocellularis pars alpha (green area). The shaded rectangle represents the location of the photomicrograph.
Carrageenan-induced hypersensitivity is reversed by microglia inhibitor
In order to determine whether the observed activation of microglia contributes to the development of behavioral signs of enhanced pain produced by inflammation, the specific microglia inhibitor minocycline was microinjected into the RVM. Intraplantar injections of carrageenan produced behavioral signs of thermal hyperalgesia and tactile allodynia within 3 h, as indicated by significant reductions in 52°C hot-plate latencies to a mean of 8.4 ± 0.3 s from a baseline value of 16.8 ± 0.4 s (F1,46 = 84.09, P < 0.001; n = 70) and in paw withdrawal thresholds to 4.4 ± 0.4 g from a baseline mean of 12.6 ± 0.5 g (F1,34 = 215.83, P < 0.001; n = 58) (Fig. 3). Rats treated with vehicle showed no significant difference from baseline. RVM microinjections of vehicle or minocycline at various doses (10, 25, or 50 μg) were administered, and hot-plate latencies were determined at 10 min intervals for 30 min (data not shown). Minocycline produced a dose-dependent reversal of thermal hyperalgesia after microinjection of either 25 or 50 μg, as indicated by significant increases in hot-plate latencies. Microinjection of 10 μg did not produce any significant changes in behavioral responses. Peak reversal of thermal hyperalgesia occurred 20 min after microinjection of either 25 or 50 μg of minocycline (data not shown). As there were no significant differences in responses to either 25 or 50 μg of minocycline, the lower dose was employed in all subsequent experiments.
Fig. 3.

Microinjection of the microglia inhibitor minocycline (25 μg) into the rostroventromedial medulla (RVM) produced a time-dependent attenuation of carrageenan-induced thermal hyperalgesia (A) and tactile allodynia (B). *Significant (P < 0.05) increases in responses relative to the post-carrageenan values. BL, mean baseline responses; Carr., mean post-carrageenan responses. (C) Paw volumes (mL) were measured 30 min after microinjection into the RVM of minocycline or vehicle in rats treated with carrageenan or saline. No significant difference was observed after the microinjection of minocycline into the RVM.
Microinjection of minocycline (25 μg) produced a significant (F5,35 = 7.93, P < 0.001) attenuation of carrageenan-induced thermal hyperalgesia 15 min (14.1 ± 1.2 s; n = 10) after administration, with a return to post-carrageenan levels at the 60 min time point (7.8 ± 1.0 s), indicating a reversible effect of minocycline (Fig. 3A). Minocycline also reversed tactile allodynia in a time-dependent manner, with a peak anti-allodynic effect being observed 30 min after injection and indicated by a mean paw withdrawal threshold of 9.8 ± 1.3 g (F4,55 = 7.41, P < 0.001; n = 13). Withdrawal thresholds returned to post-carrageenan levels 60 min after minocycline administration, as indicated by a mean withdrawal threshold of 4.9 ± 0.8 g (Fig. 3B). The injection of minocycline into the RVM of rats without hindpaw inflammation did not produce any significant changes in behavioral responses relative to baseline values (data not shown). Importantly, the injection of minocycline into the RVM did not alter carrageenan-induced hindpaw edema, as increased paw volume was not reduced by minocycline microinjection (Fig. 3C). These observations indicate that the attenuation of behavioral signs of inflammation-induced thermal and tactile hypersensitivity by inhibition of RVM microglia is not due to reversal of peripheral inflammation.
In order to examine whether the reversal of behavioral signs of inflammatory hyperesthesias by minocycline was related to inhibition of microglial activation in the RVM, medullary brain sections were obtained from control rats and rats with carrageenan-induced inflammation between 15 and 30 min after microinjection of minocycline or vehicle into the RVM. The sections were prepared for immunolabeling of the microglia marker OX-42. Carrageenan-induced inflammation increased OX-42 immunofluorescence relative to the control section (Fig. 4B). Importantly, tissue obtained from rats treated with RVM minocycline after carrageenan-induced inflammation showed decreased immunofluorescent labeling for OX-42, indicative of reduced microglial activation (Fig. 4C).
Fig. 4.

Medullary sections obtained from rats treated with hindpaw injections of saline (A) or subjected to carrageenan-induced inflammation and receiving microinjections of vehicle (B) or 25 μg of minocycline (C) in the rostroventromedial medulla (RVM) were labeled with OX-42 for immunofluorescent visualization of microglia. Microinjection of minocycline into the RVM 3 h after hindpaw injection of carrageenan produced a reduction in immunofluorescence intensity for OX-42 along with morphological changes suggesting reduced microglial activation. Scale bar: 100 μm. (D) The top blot shows examples of the bands immunoreactive against anti-ionized calcium-binding adaptor molecule 1 (Iba1). The bottom blot shows bands immunoreactive against anti-tubulin after stripping and reprobing of the same membrane. The bar graph shows the mean levels of Iba1 normalized to tubulin. Carrageenan administration produced significantly increased levels of Iba1 as compared with saline-treated animals. Protein levels of Iba1 from animals treated with minocycline in the RVM were not significantly different from those in saline-treated animals. *P < 0.05, relative to saline/saline.
Activated microglia upregulate a variety of cell surface molecules, including OX-42 and Iba1, that can serve as a measure of microglial activation (Ito et al., 1998; Salter, 2005; Romero-Sandoval et al., 2008). Western blot analysis performed with RVM tissue identified a 17 kDa band that corresponded to the molecular mass of Iba1 and that showed significantly increased levels (0.84 ± 0.15, F1,19 = 4.47, P = 0.048; n = 5) with carrageenan administration (Fig. 4D) as compared with saline-treated animals (0.46 ± 0.12; n = 5). Microinjection of minocycline into the RVM of animals with carrageenan-induced inflammation resulted in Iba1 protein levels that were not significantly (F2,25 = 2.73, P = 0.08) different from those of the vehicle-treated control animals (0.55 ± 0.12; n = 5) (Fig. 4D), indicating a decrease in microglia activation. These results suggest that minocycline diminishes carrageenan-induced microglia activation in the RVM, and that a decrease in microglia activation in the RVM correlates with an attenuation of inflammation-induced thermal and tactile hypersensitivity. The reversibility of the behavioral effects of minocycline, along with an absence of observable changes in immunofluorescent imaging or protein levels in control groups receiving minocycline, suggests that the observations made are probably due to an effect of the drug microinjection, and not neuronal or glial cell death.
Fluorocitrate in the RVM reverses carrageenan-induced hypersensitivity
To determine whether astrocytes as well as microglia play a role in inflammation-induced hypersensitivity, the general glial metabolic inhibitor fluorocitrate was microinjected into the RVM. Baseline responses to thermal and tactile stimuli were determined, and carrageenan-induced inflammation was confirmed by significant reductions in hot-plate latencies and paw withdrawal thresholds, as described above. Either vehicle or fluorocitrate (1 μg) was micro-injected into the RVM. The microinjection of fluorocitrate into the RVM blocked carrageenan-induced thermal hyperalgesia at the 60 min time point, as indicated by a significant increase in hot-plate latency to 15.2 ± 2.3 s from a post-carrageenan mean of 8.4 ± 0.3 s (F6,29 = 46.01, P = 0.014; n = 6) (Fig. 5A). Tactile allodynia was attenuated at the 30 min time point of fluorocitrate administration, as indicated by a significant increase in paw withdrawal threshold to 9.6 ± 1.7 g from a post-carrageenan mean of 4.4 ± 0.4 g (F7,68 = 2.28, P = 0.037; n = 10) (Fig. 5B). Hot-plate latencies and von Frey filament withdrawal thresholds returned to post-carrageenan levels by the 120 and 180 min time points, as indicated by mean response values of 10.6 ± 0.3 s and 5.7 ± 0.3 g, respectively, indicating reversibility of the effect of fluorocitrate (Fig. 5). Fluorocitrate was given to rats with hindpaw injections of vehicle, and no significant difference from baseline was observed (data not shown). Fluorocitrate in the RVM did not alter carrageenan-induced hindpaw edema, as indicated by non-significant changes in paw volume (Fig. 5C).
Fig. 5.

Microinjection of the astrocyte inhibitor fluorocitrate (1 μg) into the rostroventromedial medulla (RVM) produced a time-dependent attenuation of carrageenan-induced thermal hyperalgesia (A) and tactile allodynia (B). *Significant (P < 0.05) increases in responses relative to the post-carrageenan values. BL, mean baseline responses; Carr., mean post-carrageenan responses. (C) Paw volumes (mL) were measured 60 min after microinjection into the RVM of fluorocitrate or vehicle in rats treated with carrageenan or saline.
Separate groups of rats received RVM microinjections of fluorocitrate 3 h after the injection of vehicle or carrageenan. At the time of peak effect of the inhibitor (60 min after microinjection), the rats were killed, and brainstem tissue was prepared for immunofluorescent detection of the astrocyte marker GFAP. Carrageenan-induced inflammation produced a visible intensification of immunofluorescent labeling for GFAP, along with the morphological changes indicative of astrocytic activation (Fig. 6B). The microinjection of fluorocitrate into the RVM resulted in decreased immunolabeling for GFAP indicative of a reduction in activated astrocytes (Fig. 6C).
Fig. 6.

Medullary sections obtained from rats treated with hindpaw injections of saline (A) or subjected to carrageenan-induced inflammation and receiving microinjections of vehicle (B) or 1 μg of fluorocitrate (C) in the rostroventromedial medulla (RVM) were labeled with glial fibrillary acidic protein (GFAP) for immunofluorescent visualization of astrocytes. Microinjection of fluorocitrate into the RVM 3 h after hindpaw injection of carrageenan produced a reduction in immunofluorescence intensity for GFAP along with morphological changes suggesting reduced astrocyte activation. Scale bar: 100 μm. (D) The top blot shows examples of the bands immunoreactive against anti-GFAP. The bottom blot shows the bands immunoreactive against anti-tubulin after stripping and reprobing of the same membrane. The bar graph shows the mean levels of GFAP normalized to tubulin. Carrageenan administration produced significantly increased protein levels of GFAP as compared with saline-treated animals. Protein levels of GFAP from animals treated with fluorocitrate in the RVM were not significantly different from those in saline-treated animals. *P < 0.05, relative to saline/saline.
Activated astrocytes upregulate GFAP expression (Romero-Sandoval et al., 2008), and increased protein levels can be used as a measure of activation. RVM western blot analysis identified a 50 kDa band that corresponded to the molecular mass of GFAP that showed significantly increased levels (0.97 ± 0.26, F1,18 = 4.37, P = 0.047; n = 5) with carrageenan administration (Fig. 6D) as compared with saline-treated animals (0.35 ± 0.12; n = 5). After RVM microinjection of fluorocitrate in animals with carrageenan-induced inflammation, GFAP protein levels were not significantly different from those of the saline-treated control animals (0.60 ± 0.03, F2,25 = 2.99, P = 0.068; n = 5) (Fig. 6D), indicating a decrease in astrocyte activation. These results suggest that carrageenan-induced inflammation causes astrocyte activation and that application of fluorocitrate diminishes this activation in the RVM.
Inhibition of p38 MAPK in the RVM attenuates inflammation-induced hypersensitivity
To investigate whether p38 MAPK activation occurs within the RVM following inflammatory injury and whether p-p38 MAPK is located in glia and/or neurons of the RVM, tissue of rats treated with vehicle or carrageenan was collected 3 h after the hindpaw injection and prepared for immunohistochemical analysis. The sections were labeled with fluorescent marker for p-p38 MAPK and co-labeled for OX-42 (microglial marker), GFAP (astrocytic marker), or NeuN (neuronal marker). Immunofluorescent label for p-p38 MAPK was found in labeled microglia and neurons of the RVM, but was not present in astrocytes (Fig. 7). In addition, there was an increase in co-labeled microglia after carrageenan-induced inflammation, which is consistent with microglial activation in the RVM (Imbe et al., 2007).
Fig. 7.

Medullary sections were obtained from saline-treated control rats (A, C and E) and rats with carrageenan-induced inflammation (B, D and F) 3 h after injection. Sections were immunolabeled for phosphorylated p38 mitogen-activated protein kinase (MAPK) (red), and co-labeled (green) with OX-42 (microglia; A and B), glial fibrillary acidic protein (GFAP) (astrocytes; C and D) and neuronal nuclei (NeuN) (neurons; E and F). The label for phosphorylated p38 MAPK is co-localized with microglia and neurons, but not with astrocytes. Additionally, there is a visible increase in expression of microglia co-expressing phosphorylated p38 MAPK after carrageenan-induced inflammation, whereas there is no observable change in neurons expressing this marker. Scale bar: 100 μm. White arrows indicate examples of co-localization.
Baseline responses to thermal and tactile stimuli were determined and carrageenan-induced inflammation was confirmed by significant reductions in hot-plate latencies and paw withdrawal thresholds, as described above. Either vehicle or SB 203580, a p38 MAPK inhibitor, was microinjected into the RVM at various doses (1, 3, or 10 μg), and behavioral responses to a 52°C hot-plate were measured every 10 min for 30 min (data not shown). Only 10 μg SB 203580 produced a significant attenuation of behavioral hypersensitivity (data not shown), and this dose of SB 203580 was used for the remainder of this study.
The microinjection of SB 203580 (10 μg) into the RVM blocked carrageenan-induced thermal hyperalgesia at 30 min after administration, as indicated by a significant increase in hot-plate latency to 13.0 ± 1.0 s (F5,84 = 10.02, P < 0.001) from a post-carrageenan mean of 8.4 ± 0.3 s (n = 13) (Fig. 8A). Thermal responses returned to post-carrageenan levels (10.4 ± 0.8 s) by the 90 min time point after microinjection (Fig. 8A). Tactile allodynia was also attenuated at 30 min after administration of SB 203580, as indicated by a significant increase in paw withdrawal threshold to 9.2 ± 1.0 g (F5,79 = 2.66, P = 0.028; n = 18) (Fig. 8B). Paw withdrawal thresholds returned to post-carrageenan levels (5.5 ± 1.4 g) by 90 min after administration (Fig. 8B). Administration of SB 203580 to rats with hindpaw injections of saline did not produce any significant changes in behavioral responses (data not shown).
Fig. 8.

Microinjection of the p38 mitogen-activated protein kinase (MAPK) inhibitor SB 203580 (10 μg) into the rostroventromedial medulla (RVM) produced a time-dependent attenuation of carrageenan-induced thermal hyperalgesia (A) and tactile allodynia (B). *Significant (P < 0.05) increases in responses relative to the post-carrageenan values. BL, mean baseline responses; Carr., mean post-carrageenan responses. *P = 0.05 as compared with saline. Medullary sections obtained from rats treated with hindpaw injections of saline (C) or with carrageenan-induced inflammation and receiving microinjections of vehicle (D) or 10 μg of SB 203580 (E) in the RVM were labeled with OX-42 for immunofluorescent visualization of microglia. Microinjection of SB 203580 into the RVM 3 h after hindpaw injection of carrageenan produced a reduction in immunofluorescence intensity for OX-42 along with morphological changes suggesting reduced microglial activation. Scale bar: 100 μm.
Additional groups of rats received saline injections or carrageenan-induced inflammation of the hindpaw. Thermal hyperalgesia was confirmed 3 h after carrageenan administration, and either vehicle or SB 203580 was injected into the RVM. At the time of peak behavioral effect following the microinjection of SB 203580 (30 min), the rats were killed, and medullary sections were obtained and immunofluorescently labeled for OX-42, in order to examine changes in microglial activation. Carrageenan-induced inflammation produced increased intensity of immunofluorescence for OX-42 relative to the control sections, along with morphological features indicative of microglial activation (Fig. 8D). Tissue obtained from rats treated with SB 203580 in the RVM after carrageenan-induced inflammation showed reduced intensity of immunofluorescent labeling for OX-42, as well as a reduction in the morphological signs associated with microglial activation (Fig. 8E). The microinjection of SB 203580 did not appear to reduce immunofluorescent labeling for GFAP in tissue from rats treated with carrageenan (data not shown).
Discussion
The present study demonstrated that peripheral inflammation leads to activation of microglia and astrocytes in the RVM. Microinjection of the selective microglial inhibitor minocycline into the RVM attenuated behavioral signs of hyperalgesia and tactile allodynia, and inhibited the activation of microglia in the RVM. Likewise, the non-specific glial inhibitor fluorocitrate microinjected into the RVM also reversed the behavioral signs of inflammatory pain and attenuated the immunohistological evidence of activation of astrocytes, as indicated by the decrease in immunolabeling of GFAP. Additionally, phosphorylation of p38 MAPK mediated, at least in part, behavioral signs of inflammatory pain, as microinjection of the p38 MAPK inhibitor SB 203580 into the RVM attenuated both carrageenan-induced thermal hyperalgesia and tactile allodynia. Activation of microglial p38 MAPK may be partly responsible for the behavioral expression of hyperesthesia due to inflammation, as the microinjection of SB 203580 into the RVM decreased OX-42 labeling of RVM tissue. The results of the present investigation indicate that peripheral inflammation induces glial activation in the RVM, and that activated glia of the RVM may contribute to enhanced pain, probably through activation of descending facilitation from the RVM.
Converging lines of evidence indicate that the RVM is a central component of descending facilitation of nociceptive inputs at the level of the spinal cord (Urban & Gebhart, 1999; Porreca et al., 2002; Gebhart, 2004). Considerable evidence has been generated to show that descending facilitatory influences from the RVM can contribute to chronic pain states (Urban & Gebhart, 1999; Fields, 2000) and such contributions are important for the maintenance of thermal and tactile hypersensitivity. Administration of lidocaine into the RVM and lesions of the dorsolateral funiculus (DLF) have been shown to block behavioral signs of neuropathic and inflammatory nociception (Pertovaara et al., 1996, 1998; Burgess et al., 2002; Xie et al., 2005). Selective ablation of RVM neurons expressing μ-opioid receptors, which are probably pain facilitatory cells, by microinjection of the cytotoxin saporin conjugated to dermorphin either prevented or reversed behavioral signs of enhanced pain (Porreca et al., 2001; Burgess et al., 2002). Finally, pharmacological and surgical manipulations that block descending facilitation from the RVM have also blocked enhanced capsaicin-evoked release of excitatory transmitters from primary afferent terminals and the upregulation of spinal dynorphin to pathologically elevated pronociceptive levels (Porreca et al., 2001; Burgess et al., 2002). Whereas activation of descending facilitation has been considered to be a neuronal phenomenon, we now present evidence that activation of glial cells in the RVM contributes to descending facilitation of inflammatory pain.
Although the neuronal processes that contribute to central sensitization in the spinal cord have been examined extensively, the potential contribution of activated glial cells to this process has only recently been explored. It is now appreciated that peripheral nerve injury, as well as inflammation, results in the activation of spinal glia (Garrison et al., 1991; Sweitzer et al., 1999; Svensson et al., 2005). Several studies have now shown that sensitization of peripheral nociceptors by subcutaneous injection of irritants, peripheral inflammation, or nerve injury, as well as intrathecal injection of sensitizing agents such as N-methyl-D-aspartate (NMDA), results in the activation of microglia and astrocytes in the spinal cord, and that glial activation correlates with enhanced nociceptive responses (Fu et al., 1999, 2000; Sweitzer et al., 1999; Raghavendra et al., 2004; Tanga et al., 2005; Scholz & Woolf, 2007). Studies performed with genetically modified mice or by selective knockdown of receptors and peptides with antisense oligodeoxynucleotides further revealed that activated spinal microglia promote enhanced pain caused by nerve injury and inflammation (Abbadie et al., 2003; Tsuda et al., 2003, 2005; Tanga et al., 2005). Inhibition of spinal microglial or astrocytic activity with intrathecal glial inhibitors attenuated behavioral signs of hyperalgesia and allodynia induced by formalin, carrageenan, and spinal injection of NMDA (Raghavendra et al., 2003; Hua et al., 2005; Ledeboer et al., 2005; Tsuda et al., 2005). Whereas the preponderance of such studies have focused on the role of spinal glia in enhanced pain states, at present there is little work that examines changes in glial function at supraspinal sites. Thus, the results of the present investigation extend the role of glial activation after peripheral inflammation to a supraspinal site, specifically the RVM. Recent studies demonstrated that peripheral nerve injury resulted in microglial activation of the RVM (Guo et al., 2007; Wei et al., 2008). An earlier study had indicated that Complete Freund’s Adjuvant-induced inflammation caused supraspinal glial activation, as indicated by increased expression of glial markers in the medulla, pons, midbrain, and thalamus (Raghavendra et al., 2004). However, in that study, there was no attempt to attenuate supraspinal microglial or astrocytic activation and to correlate such inhibition of glial activation with changes in inflammatory pain (Raghavendra et al., 2004). In the present investigation, we have extended these observations to include the RVM and to correlate glial activation with inflammatory pain.
Carrageenan-induced inflammation led to evidence of activation of microglia and astrocytes in the RVM. Inhibitors of activation of microglia and astrocytes injected into the RVM reduced carrageenan-induced activation of these glial cells, and attenuated tactile allodynia and thermal hyperalgesia. Upon activation, microglia and astrocytes are known to upregulate the expression of OX-42, Iba1 and GFAP in the central nervous system (Romero-Sandoval et al., 2008). Immuno-histochemistry has been used extensively to monitor the morphological and biological transformation of activated glia in the spinal cord after injury, but western blot analysis was employed when quantification of glial activation was attempted (Ledeboer et al., 2005; Guo et al., 2007; Romero-Sandoval et al., 2008). In the present investigation, carrageenan-induced inflammation increased Iba1 and GFAP protein levels, indicating microglia and astrocyte activation, respectively, and the microinjection of glial inhibitors into the RVM resulted in Iba1 and GFAP protein levels that were not different from those in saline-treated animals. The effects of minocycline and fluorocitrate on glial activation and behavioral hypersensitivity are caused by direct administration into the RVM and not a decrease in peripheral inflammation, as the administration of the glial inhibitors had no effect on paw edema. Whereas these results are in agreement with those of a recent study that examined glial activation in the RVM occurring within 2 days after a trigeminal nerve injury (Wei et al., 2008), the present investigation extends these observations to events occurring within 3 h after induction of peripheral inflammation. Moreover, the present study shows that modulation of glial activation in the RVM by the microinjection of inhibitors attenuated behavioral hypersensitivity within 30 min.
As activated microglia in spinal and supraspinal sites increases the expression of proinflammatory cytokines, it is possible that the activated glia in the RVM can enhance the activity of adjacent neurons, thus increasing descending pain facilitation from the RVM. Accordingly, disruption of this glial activation reverses behavioral hyperalgesia and allodynia. Glial activation leads to the phosphorylation of p38 MAPK, which, in turn, promotes the production of inflammatory mediators such as prostaglandin E2 and cytokines (Obata et al., 2000; Shi & Gaestel, 2002; Takeda & Ichijo, 2002). It has been shown that peripheral inflammation and nerve injury provoke phosphorylation of p38 MAPK in dorsal root ganglion neurons and in both microglia and neurons in the dorsal horn of the spinal cord (Kim et al., 2002). Importantly, however, whereas spinal microglia demonstrate phosphorylation of p38 MAPK after nerve injury, peripheral inflammation, subcutaneous injection of algogenic substances, or the spinal injection of NMDA or substance P, increased phosphorylation of neuronal p38 MAPK is inconsistent and is not detected after peripheral injection of formalin or intrathecal injection of substance P (Hua et al., 2005; Tsuda et al., 2005; Svensson et al., 2007). It was also found that the p38α isoform of p38 MAPK is found predominantly in neurons, whereas the p38β isoform is found in microglia (Svensson et al., 2005). Antisense experiments that produced selective knockdown of either isoform of p38 MAPK showed that the p38β isoform, and not the p38α isoform, mediates hyperalgesia due to intraplantar formalin or spinal administration of substance P (Svensson et al., 2005). These observations indicate that, at least in the spinal cord, microglial, but not neuronal, p38 MAPK contributes to enhanced sensitivity in chronic pain states. In the present investigation, p-p38 MAPK was localized in both microglia and neurons in the RVM in the resting state, with increased phosphorylation of p38 MAPK being seen in microglia after carrageenan-induced inflammation. The relative contribution of neuronal p38 MAPK to the expression of enhanced pain requires further investigation.
Whereas inhibitors of p38 MAPK activation do not alter basal responses to acute noxious stimuli (Hua et al., 2005), pretreatment with intrathecal injections of p38 MAPK inhibitors has been shown to block hyperalgesia induced by formalin, capsaicin or carrageenan injected into the hindpaw, peripheral nerve injury, or intrathecal injections of inflammatory mediators such as interleukin-6β (Sweitzer et al., 1999; Mizushima et al., 2005; Svensson et al., 2005, 2007). In addition, p38 MAPK inhibitors blocked substance P-induced evoked release of prostaglandin E2 into the cerebrospinal fluid (Svensson et al., 2005). One of the most commonly used compounds among various p38 MAPK inhibitors is SB 203580. We report that microinjection of SB 203580 into the RVM attenuated behavioral hyperalgesia and allodynia as well as immunofluorescent evidence of activated microglia in the RVM. Interestingly, the time course of action of both SB 203580 and minocycline on thermal hyperalgesia and tactile allodynia was approximately 30 min, suggesting that the activation of p38 MAPK correlates with the activation of microglia. Others reported that peripheral inflammation induced by injection of Complete Freund’s Adjuvant into the hindpaw resulted in phosphorylation of p38 MAPK that was predominant in RVM neurons but also present in some microglia of the RVM within 30 min of the injection (Imbe et al., 2007). However, in that study, the functional significance of p-p38 MAPK in non-neuronal cells was not investigated (Imbe et al., 2007). The phosphorylation of p38 MAPK diminished within 7 h, corresponding with increased activation of extracellular signal-related kinase, which itself might be transcriptionally related to the activation of p38 MAPK (Imbe et al., 2007). In that study, it was also found that approximately 40% of neurons expressing p-p38 MAPK were serotonergic, and it was hypothesized that p38 MAPK activation could lead to neuroplastic changes in descending pain modulation, in this case by increasing the transcription of tryptophan hydroxylase and promoting serotonergic transmission from the brainstem (Imbe et al., 2007). Transcriptional events produced by activated p38 MAPK could also lead to increased expression of pronociceptive mediators in the RVM; it is known that p38 MAPK activates basic fibroblast growth factor-mediated transcription of cholecystokinin (Hansen et al., 1999). Previous reports indicated that increased cholecystokinin levels in the RVM mediate, in part, enhanced descending facilitation (Xie et al., 2005).
In the present investigation, we demonstrated that peripheral inflammation induced with carrageenan activates microglia and astrocytes in the RVM as well as phosphorylation of p38 MAPK in neurons and microglia in the RVM. Immunofluorescence examination of medullary sections shows an intensification in labeling for OX-42, GFAP and p-p38 MAPK in RVM tissue obtained from rats with carrageenan-induced inflammation. Microinjection of inhibitors of glial activation or of p38 MAPK reverses both carrageenan-induced thermal hyperalgesia and tactile allodynia. Minocycline, fluorocitrate and SB 203580 also attenuate the activation of microglia and astrocytes in the RVM, as observed by immunodetection and western blot. As disruption of descending facilitation from the RVM inhibits enhanced pain conditions (Gebhart, 2004; Xie et al., 2005; Ossipov & Porreca, 2006), it seems likely that blockade of glial activation associated with this system may also attenuate pain facilitation. The results of this study indicate a mechanism that may represent a novel target for developmental efforts aimed at the control of chronic pain states.
Abbreviations
- BSA
bovine serum albumin
- GFAP
glial fibrillary acidic protein
- Iba1
ionized calcium-binding adaptor molecule 1
- MAPK
mitogen-activated protein kinase
- NeuN
neuronal nuclei
- NMDA
N-methyl-D-aspartate
- p-p38 MAPK
phos-phorylated p38 mitogen-activated protein kinase
- RVM
rostroventromedial medulla
- TBS
Tris-buffered saline
- TBS-T
Tris-buffered saline containing 1% Triton X-100
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–5136. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacur M, Gutierrez JM, Milligan ED, Wieseler-Frank J, Britto LR, Maier SF, Watkins LR, Cury Y. Snake venom components enhance pain upon subcutaneous injection: an initial examination of spinal cord mediators. Pain. 2004;111:65–76. doi: 10.1016/j.pain.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–175. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fields HL. Pain modulation: expectation, opioid analgesia and virtual pain. Prog Brain Res. 2000;122:245–253. doi: 10.1016/s0079-6123(08)62143-3. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825:59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Maixner W. Relationship between nociceptor activity, peripheral edema, spinal microglial activation and long-term hyperalgesia induced by formalin. Neuroscience. 2000;101:1127–1135. doi: 10.1016/s0306-4522(00)00376-6. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Vanderah TW, Gardell SE, Wang R, Ossipov MH, Lai J, Porreca F. Enhanced evoked excitatory transmitter release in experimental neuropathy requires descending facilitation. J Neurosci. 2003;23:8370–8379. doi: 10.1523/JNEUROSCI.23-23-08370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27:729–737. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial–cytokine–neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TV, Rehfeld JF, Nielsen FC. Mitogen-activated protein kinase and protein kinase A signaling pathways stimulate cholecystokinin transcription via activation of cyclic adenosine 3′,5′-monophosphate response element-binding protein. Mol Endocrinol. 1999;13:466–475. doi: 10.1210/mend.13.3.0257. [DOI] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Imbe H, Okamoto K, Aikawa F, Kimura A, Donishi T, Tamai Y, Iwai-Liao Y, Senba E. Effects of peripheral inflammation on activation of p38 mitogen-activated protein kinase in the rostral ventromedial medulla. Brain Res. 2007;1134:131–139. doi: 10.1016/j.brainres.2006.11.091. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Kim SY, Bae JC, Kim JY, Lee HL, Lee KM, Kim DS, Cho HJ. Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. Neuroreport. 2002;13:2483–2486. doi: 10.1097/00001756-200212200-00021. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiencyvirus-1(HIV-1)envelopeglycoprotein,gp120. BrainRes. 2000;861:105–116. doi: 10.1016/s0006-8993(00)02050-3. [DOI] [PubMed] [Google Scholar]
- Milligan ED, O’Connor KA, Armstrong CB, Hansen MK, Martin D, Tracey KJ, Maier SF, Watkins LR. Systemic administration of CNI-1493, a p38 mitogen-activated protein kinase inhibitor, blocks intrathecal human immunodeficiency virus-1 gp120-induced enhanced pain states in rats. J Pain. 2001;2:326–333. doi: 10.1054/jpai.2001.26174. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima T, Obata K, Yamanaka H, Dai Y, Fukuoka T, Tokunaga A, Mashimo T, Noguchi K. Activation of p38 MAPK in primary afferent neurons by noxious stimulation and its involvement in the development of thermal hyperalgesia. Pain. 2005;113:51–60. doi: 10.1016/j.pain.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med. 2000;28:N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Porreca F. Ascending and descending facilitatory circuits in neuropathic pain states. In: Campbell JN, Basbaum AI, Dray A, Dubner R, Dworkin RH, Sang CN, editors. Emerging Strategies for the Treatment of Neuropathic Pain. IASP Press; Seattle: 2006. pp. 211–238. [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1998. [Google Scholar]
- Pertovaara A, Wei H, Hamalainen MM. Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci Lett. 1996;218:127–130. doi: 10.1016/s0304-3940(96)13136-0. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Hamalainen MM, Kauppila T, Panula P. Carrageenan-induced changes in spinal nociception and its modulation by the brain stem. Neuroreport. 1998;9:351–355. doi: 10.1097/00001756-199801260-00032. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Chai N, Nutile-McMenemy N, DeLeo JA. A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res. 2008;1219:116–126. doi: 10.1016/j.brainres.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW. Cellular signalling pathways of spinal pain neuroplasticity as targets for analgesic development. Curr Top Med Chem. 2005;5:557–567. doi: 10.2174/1568026054367638. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Shi Y, Gaestel M. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol Chem. 2002;383:1519–1536. doi: 10.1515/BC.2002.173. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38beta isoform mediates tissue injury-induced hyper-algesia and spinal sensitization. J Neurochem. 2005;92:1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Yaksh TL, Sorkin LS. The role of p38 in microglial regulation of spinal pain processing. In: DeLeo JA, Sorkin LS, Watkins LR, editors. Immune and Glial Regulation of Pain. IASP Press; Seattle: 2007. pp. 297–317. [Google Scholar]
- Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA. Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res. 1999;829:209–221. doi: 10.1016/s0006-8993(99)01326-8. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- Takeda K, Ichijo H. Neuronal p38 MAPK signalling: an emerging regulator of cell fate and function in the nervous system. Genes Cells. 2002;7:1099–1111. doi: 10.1046/j.1365-2443.2002.00591.x. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terayama R, Guan Y, Dubner R, Ren K. Activity-induced plasticity in brain stem pain modulatory circuitry after inflammation. Neuroreport. 2000;11:1915–1919. doi: 10.1097/00001756-200006260-00022. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in ‘small’ glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci USA. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Furness LE, Maier SF. Illness-induced hyperalgesia is mediated by spinal neuropeptides and excitatory amino acids. Brain Res. 1994;664:17–24. doi: 10.1016/0006-8993(94)91948-8. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial–neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28:10482–10495. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xie JY, Herman DS, Stiller CO, Gardell LR, Ossipov MH, Lai J, Porreca F, Vanderah TW. Cholecystokinin in the rostral ventromedial medulla mediates opioid-induced hyperalgesia and antinociceptive tolerance. J Neurosci. 2005;25:409–416. doi: 10.1523/JNEUROSCI.4054-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007;27:8893–8902. doi: 10.1523/JNEUROSCI.2209-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]