

Abstract
Formyl peptide receptor-2 (FPR2) is a G-protein coupled receptor that plays critical roles in inflammatory reactions. FPR2-specific interaction can be possibly used to facilitate the resolution of pathological inflammatory responses by enhancing endogenous anti-inflammation systems. Starting from our lead agonist 5, we designed new ureidopropanamides derivatives able to activate FPR2 in transfected cells and human neutrophils. The new FPR2 agonists showed good stability towards oxidative metabolism in vitro. Moreover, selected compounds showed anti-inflammatory properties in LPS-stimulated rat primary microglial cells. (S)-3-(4-Cyanophenyl)-N-[[1-(3-chloro-4-fluorophenyl)cyclopropyl]methyl]-2-[3-(4-fluorophenyl)ureido]propanamide ((S)-17) emerged as prospective pharmacological tool to study the effects of FPR2 activation in the central nervous system (CNS) being able to reduce IL-1β and TNF-α levels in LPS-stimulated microglial cells and showing good permeation rate in hCMEC/D3 cells, an in vitro model of blood brain barrier. These results are very promising and can open new therapeutic perspectives in the treatment of CNS disorders characterized by neuroinflammation.
Keywords: Formyl peptide receptor 2, neuroinflammation, ureidopropanamides, microglia, metabolic stability, blood-brain barrier permeation
Graphical Abstract
Introduction
Neuroinflammation is a complex multicellular process that plays a central role in a variety of neurological diseases, including ischemia, neurodegenerative diseases, psychiatric and immune-mediated disorders [1,2]. Increasing evidence suggests that microglia, the resident immune cells of the central nervous system (CNS), play a pivotal role in inflammation-associated disorders. Under normal conditions, activation of microglia cells exerts a protective role by regulating the response to pathogens and promoting tissue repair through the release of anti-inflammatory and neurotrophic factors [3]. On the other hand, chronic activation of immune responses can lead to the functional switch of microglia from regulatory to neurotoxic leading to the excessive release of pro-inflammatory cytokines, such as IL-1β, IL-18, and TNF-α, as well as of neurotoxic mediators like nitric oxide (NO), prostaglandin E2, and superoxide anion [4]. Microglial activation is a phenotypically and functionally dynamic process, which may progress differently depending on aging, stage of disease or on the status of the brain environment [5,6].
In general, inflammation requires tight regulation and control: pro-inflammatory mediators operate in a parallel and serial fashion to provoke the classical signs of inflammation, while anti-inflammatory mediators ensure resolution of the inflammatory response. This latter aspect has gained interest in recent years because several classes of peptidic or non-peptidic endogenous factors have been identified as pro-resolving mediators [7]. Many pro- and anti-inflammatory signals and pro-resolving circuits converge on a group of receptors that integrate contrasting cues to determine the course of inflammation. Among these receptors is the formyl peptide receptor 2 (FPR2) [8,9], a G-protein coupled receptor that belongs to the formyl peptide receptor (FPR) family, which includes also the subtypes FPR1 and FPR3. FPRs are expressed in several immune cells, including neutrophils, monocytes/macrophages and microglia and are considered to play relevant roles in innate immunity and host defense mechanisms and chemotaxis [9]. FPR2 is functionally expressed in glial cells and astrocytes [10–12]. FPRs have homologs in fish and rodents [13,14]. For example, Fpr2, a homolog of human FPR2, is localized in rat satellite glial cells and neurons of dorsal root ganglions [15]. Expression of Fpr1 and Fpr2 was also reported in rat neuronal stem cells [16]. Recently, expression of Fpr, which cross-reacts with anti-human FPR2 antibodies, was detected in rat brain cortex and hippocampus [17].
A prominent and unusual feature of FPR2 is that it can be activated by a variety of structurally diverse agonists. Several endogenous peptides can activate FPR2 and are able to induce very different biological effects. For example, FPR2 can mediate pro-inflammatory effects if activated by N-formyl peptides, produced by bacteria and mitochondria to induce defense mechanisms, as well as by prion protein PrP(106-126) and the amyloidogenic peptides serum amyloid A and β-amyloid, which are associated with chronic inflammation and amyloidosis [9]. On the other hand, FPR2 can mediate anti-inflammatory effects if activated by annexin A1, a glucocorticoid-regulated protein that is involved in the adhesion mechanisms of leukocytes, as well as by lipoxin A4 (LXA4), which is an anti-inflammatory lipid, the first endogenous FPR2 ligand to be identified [9,18]. The ability of FPR2 to mediate contrasting effects is mechanistically related to the receptor dimerization that can be induced in a ligand-specific manner [19].
Preclinical evidence generated with pharmacological tools or with knockout and transgenic mice has suggested that LXA4 contributes to the resolution of inflammation through the activation of FPR2, which in turn modulates chemokine and cytokine synthesis, inhibits neutrophils infiltration, and promotion of phagocytosis [20–22]. Recently, stimulation of the resolution phase has been proposed as a new therapeutic perspective for the treatment of chronic inflammatory diseases such as chronic obstructive pulmonary disease, cystic fibrosis, rheumatoid arthritis, and also for CNS diseases [23,24]. With this respect, recent data demonstrate that LXA4 inhibits microglial activation and diminishes neuroinflammation after spinal cord hemisection [25]. Moreover, in rat hemorrhage, the beneficial role of LXA4 in suppression of inflammation mediated by FPR2 and p38 MAPK signaling pathways has been demonstrated [26]. It is worth emphasizing that the administration of LXA4 in a transgenic mouse model of Alzheimer’s Disease is able to stimulate a pro-resolving activation of microglia by reducing NF-κB activation and levels of pro-inflammatory cytokines and chemokines and by increasing levels of anti-inflammatory IL-10. This alternative activation of microglia translated into an improved phagocytic function with increased clearance of β-amyloid plaques, reduced synaptotoxicity, and improvement of cognition [27].
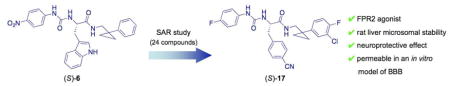
A wide range of chemically diverse non-peptidic FPR agonists have been identified so far [28], including benzimidazole derivatives exemplified by compound 1 [29], N-phenylurea derivatives (compound 2) [30], quinazolinones derivatives such as the highly specific non-peptide FPR2 agonist Quin-C1 (3) [31], and pyrazolone derivatives like the mixed FPR1/FPR2 agonist 4 (designated in most of publications as “Compound 43”) [32] (see Chart 1). Recently, we reported the identification of a group of 3-(1H-indol-3-yl)-2-[3-(4-substituted-phenyl)ureido]propanamide derivatives as agonists of human FPR2, exemplified by compound 5 (Table 1, Chart 1) [33,34]. The above agonists were characterized for their ability to induce intracellular Ca2+ release. Whereas agonists 3 and 4 exhibited anti-inflammatory properties in peripheral models of inflammation [35,36], there are no reports on the effects of FPR2 agonists in in vitro or in vivo models of neuroinflammation.
Chart 1.
Structural Formulas of Non-Peptidic FPR2 Agonists
Table 1.
Effect of the compounds on Ca2+ mobilization in FPR1- and FPR2-HL60 transfected cells and human neutrophils, and metabolic stability.
| |||||||
|---|---|---|---|---|---|---|---|
| EC50, μM (Efficacy %) | Metabolic stability (% remaining)a | ||||||
| Compd. | R1 | Ar | R2 | FPR2-HL60 | FPR1-HL60 | Neutrophils | |
| 5 |

|
0.19b | 0.26b | 0.086b | 4 | ||
| (R)-6 | NO2 |
|
H | N.A.b | N.A.b | N.A.b | 15 |
| (S)-6 | 7.6 ± 2.1 (60)b | N.A.b | N.A.b | 11 | |||
| (R)-7 | OCH3 |
|
H | 6.5 ± 1.8 (55) | N.A. | N.A. | 4 |
| (S)-7 | 0.11 ± 0.03 (125) | 0.95 ± 0.3 (105) | 1.4 ± 0.3 (145) | 3 | |||
| (R)-8 | F |
|
H | N.A. | N.A. | N.A. | 4 |
| (S)-8 | 2.0 ± 0.4 (70) | 18.8 ± 3.7 (60) | N.A. | 4 | |||
| (R)-9 | H |
|
H | N.A. | N.A. | N.A. | 4 |
| (S)-9 | 6.7 ± 1.9 (35) | 2.4 ± 0.7 (55) | 1.5 ± 0.4 (25) | 5 | |||
| (R)-10 | NO2 |
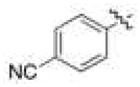
|
H | 1.5 ± 0.3 (90) | 4.7 ± 1.1 (75) | 4.1 ± 0.8 (150) | 38 |
| (S)-10 | 0.63 ± 0.2 (100) | 2.8 ± 0.6 (75) | 2.5 ± 0.7 (115) | 56 | |||
| (R)-11 | NO2 |
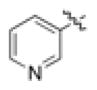
|
H | 2.9 ± 0.8 (60) | 1.5 ± 0.4 (145) | 0.12 ± 0.04 (90) | 45 |
| (S)-11 | 6.4 ± 1.5 (60) | 0.85 ± 0.3 (125) | 0.73 ± 0.2 (65) | 51 | |||
| (R)-12 | OCH3 |
|
OCF3 | N.A. | N.A. | N.A. | 43 |
| (S)-12 | N.A. | N.A. | N.A. | 51 | |||
| (R)-13 | F |
|
OCF3 | N.A. | N.A. | N.A. | 88 |
| (S)-13 | N.A. | N.A. | N.A. | 80 | |||
| (R)-14 | OCH3 |
|
4-CH3-3-F | N.A. | N.A. | N.A. | 10 |
| (S)-14 | 16.3 ± 3.6 (35) | 10.3 ± 2.2 (55) | 1.7 ± 0.4 (70) | 8 | |||
| (R)-15 | F |
|
4-CH3-3-F | N.A. | N.A. | N.A. | 37 |
| (S)-15 | 3.0 ± 0.8 (65) | 5.9 ± 1.6 (50) | 1.7 ± 0.5 (55) | 39 | |||
| (R)-16 | OCH3 |
|
3-Cl-4-F | 1.8 ± 0.3 (60) | 0.63 ± 0.2 (100) | 0.73 ± 0.2 (115) | 5 |
| (S)-16 | 3.4 ± 0.9 (60) | 0.45 ± 0.1 (115) | 0.33 ± 0.1 (155) | 10 | |||
| (R)-17 | F |
|
3-Cl-4-F | 0.3 ± 0.1 (90) | 5.4 ± 1.2 (55) | 1.3 ± 0.4 (95) | 20 |
| (S)-17 | 3.9 ± 1.1 (80) | 5.2 ± 1.4 (55) | 0.19 ± 0.05 (130) | 33 | |||
Percent of parent compound remaining after 30-min incubation.
Data taken from ref. 33.
Based on our previous studies, we aimed to identify new potent FPR2 agonists and to prove if the new ureidopropanamide derivatives display anti-inflammatory and pro-resolving effects in an in vitro model of neuroinflammation. Toward this aim, we have modified the structural framework of 5 taking into account that this agonist showed very low stability toward oxidative metabolism and was rapidly degraded after incubation with rat liver microsomes, thus implying that 5 has a pharmacokinetic profile not compatible with in vivo studies [34].
Results and Discussion
Study Design
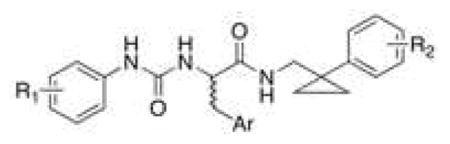
As indicated above, 5 is a potent mixed FPR1/FPR2 agonist in FPR2-transfected HL-60 cells and in human neutrophils [33] but displays very low stability toward oxidative metabolism (Table 1). Thus, in search of metabolically stable FPR2 agonists we selected from our chemical library various 3-(1H-indol-3-yl)-2-[3-(4-substituted-phenyl)ureido]propanamide derivatives and tested them for their susceptability to oxidative metabolism in vitro using rat liver microsomes. We assessed microsomal stability as the percentage of recovery of parent compound after a 30 minute incubation with liver microsomes preparation, as described previously [37]. Among the screened compounds, the enantiomeric pair (R)- and (S)-6, in which the cyclohexane ring was replaced with the cyclopropane ring, had a percentage of recovery (15% and 11%, respectively) higher than that of 5 (4%) (Table 1). In addition, the phenylcyclopropylmethyl moiety is more synthetically accessible as compared to the (5-methoxypyridin-2-yl)cyclohexylmethyl moiety of compound 5 [34]. Regarding interaction with FPR2, (R)-6 was previously characterized as a weak FPR2 antagonist, whereas (S)-6 was able to induce Ca2+ mobilization, albeit with low potency (EC50 = 7.6 μM) [33]. Therefore, considering the slightly more favorable metabolic stability profile and the synthetic accessibility, we selected (S)-6 as starting point for our study with the aim of improving both potency in FPR2 activation and metabolic stability in vitro. We systematically evaluated both enantiomers of the target compounds 7-17 (Table 1) because previous studies have shown that the interaction of these chiral ureidopropanamides with FPRs can be highly stereospecific [33]. We assessed the ability of the target compounds to induce Ca2+ mobilization in HL-60 cells transfected with h-FPR1 or h-FPR2 and in human neutrophils. Finally, considering that the ability to induce Ca2+ mobilization is a feature exhibited by both pro-inflammatory and pro-resolving FPR2 agonists, the compounds showing the best combination of potency and metabolic stability were further characterized in primary rat microglia cultures to assess their anti-inflammatory properties.
Chemistry
Synthesis of the target compounds is depicted in Scheme 1. The key amines 22 and 23 were prepared from the corresponding nitriles 18 [38] and 19 [39], respectively, after hydrogenation over Nickel-Raney. Amine 24 was prepared by reduction of nitrile 20 [40] with the borane-dimethyl sulfide complex in order to avoid reductive dehalogenation. The amines 21-24 were then condensed with the commercially available (R)-Boc- or (S)-Boc-amino acids 25-27 after activation with N-N′-carbonyldiimidazole to give the Boc-protected derivatives (R)- and (S)- 28-33. Subsequently, these latter compounds were deprotected with trifluoroacetic acid to give amines (R)- and (S)- 34-39. The target compounds were obtained by condensing amines (R)- and (S)- 34-39 with the appropriate phenylisocyanate, except for compounds (R)-8, (S)-14, (S)-15, and (R)-17, which were prepared by condensing the amines (R)-34, (S)-38, (R)-39 with the the appropriate aniline and N-N′-carbonyldiimidazole.
Scheme 1.

Synthesis of the target compounds (R)- and (S)-7-17.
aReagents and Conditions: (A) Raney-nickel, H2, 2 M ethanolic NH3; 5 atm; 50 °C, 15 h, 77–90% yield or borane-methyl sulfide complex 10 M, HCl, 81% yield; (B) N,N′-carbonyldiimidazole, r.t., overnight, 45–87% yield; (C) trifluoroacetic acid, r.t., 5 h, quantitative yield; (D) 4-substituted phenylisocianate, r.t., overnight, 10–80% yield; or 4-substitued aniline, N,N′-carbonyldiimidazole, r.t., overnight, 10–19% yield.
Intrinsic Activity and Metabolic Stability of the Target Compounds
The first structural modification on (R)- and (S)-6 was removal of the nitro substituent from the phenylureidic group or its replacement with –OCH3 or –F substituents. This structural modification was inspired by previous structure-activity relationship studies on ureidopropanamide derivatives, where the introduction of a methoxy group [33] or an halogen [41] in the same position led to an improvement in agonist potency. Indeed, (S)-7 and (S)-8 showed EC50 values lower than that of (S)-6. The effect was more pronounced for the methoxy derivative (S)-7 (Table 1). In contrast, removal of the nitro substituent of (S)-6 had little impact on its potency ((S)-9). Interestingly, replacement of the nitro group with a methoxy group in (R)-6 shifted the intrinsic activity from antagonism to agonism ((R)-7)) [33]. This effect was not observed for derivatives (R)-8 and (R)-9, which were not able to induce Ca2+ mobilization. Note that the replacement of the nitro group with other substituents led to a decrease in selectivity towards FPR1, as (S)-7, (S)-8, and (S)-9 were able to activate FPR1 as well. In particular, (S)-9 showed a slight preference for FPR1 over FPR2. Finally, similarly to (S)-6, compounds (S)-8 and (S)-9 behaved as partial agonists, whereas (S)-7 was characterized as a full agonist at both FPR1 and FPR2.
Considering metabolic stability, it can be noted that this structural modification had a negative influence, as the compounds (R)- and (S)-7-9, were less stable than (R)- and (S)-6 (Table 1). By comparing compounds 6 and 8, it can be speculated that the electronic properties of the substituent on the phenyl ureidic moiety can have limited effects on metabolic stability of this group of compounds.
We next evaluated the impact on both potency and metabolic stability of replacement of the tryptophan core in (R)- and (S)-6 with that of unnatural amino acids. This structural modification was inspired by the observation that different aminoacid cores were well tolerated in other FPR2 agonists with ureidopropanamide structure [28]. For this purpose, we selected the commercially available 4-cyanophenylalanine and 3-pyridylalanine. In general, this structural modification was beneficial. In fact, the introduction of 4-cyanophenylalanine led to a 10-fold increase in agonist potency, as shown by (S)-10 compared to (S)-6, whereas (S)-11, which bears the 3-pyridylalanine core, was only slighly more potent than (S)-6 (Table 1). Comparig the (R)- enantiomers with (R)-6, a shift from antagonism to agonism was observed, as (R)-10 and (R)-11 were able to induce Ca2+ mobilization although with lower potency than the (S)-enantiomers. It can be also noted that (R)- and (S)-10 behaved as full agonists, whereas (R)- and (S)-11 were characterized as partial agonists. Finally, this structural modification led to a substantial increase in FPR1 agonist activity; in particular, (S)-11 had an EC50 value for FPR1 in the low micromolar range. As for metabolic stability, both enantiomeric pairs showed a percentage of recovery higher than 40%, with (S)-10 being the most stable compound of the set (56% recovery) (Table 1). These data suggest that replacement of the tryptophan with an unnatural amino acid led to unfavorable interactions of the molecule with the metabolic enzymes.
On the basis of these data, we selected the 4-cyanophenylalanine as the amino acid core for further modifications to obtain new ureidopropanamide FPR2 agonists. First, we replaced the nitro substituent of (R)- and (S)-10 with –OCH3 or –F with the aim of increasing agonist potency. Additionally, we decorated the phenyl ring on the “right hand” of (R)- and (S)-10 with substituents that could prevent interaction of the molecule with metabolic enzymes in an effort to further increase metabolic stability (compounds 12-17). These modifications did not lead to improved FPR2 agonist potency. In particular, the presence of a substituent in the 4-position of the phenyl linked to the cyclopropyl ring negatively influenced the ability of these compounds to activate FPR2 (Table 1). This effect was more evident for compounds (R)- and (S)-12 and (R)- and (S)-13, which feature the bulky trifluoromethoxy group. Instead, (R)- and (S)-16 and (R)- and (S)-17, bearing the small F– substituent in 4-position, showed EC50 values comparable to those of (R)- and (S)-10 (Table 1). Moreover, decoration of the phenyl ring linked to the cyclopropyl moiety had different impact on the FPR1 interaction, with (R)- and (S)-16 being potent FPR1 full agonists. Introduction of bulky substituents on the phenyl linked to the cyclopropyl ring had a beneficial effect on metabolic stability. In fact, derivates (R)- and (S)-13 showed very high percentage recovery (> 80%) (Table 1). However, the methoxy-substituted derivatives showed lower stability than that of the fluoro-substituted counterparts. On the other hand, the presence of the unnatural amino acid core did not always translate into an improvement of metabolic stability. Derivatives (R)- and (S)-14, and (R)- and (S)-16 showed metabolic stabilities comparable to those of ureidopropanamides with the tryptophan core (Table 1).
Collectively, these data clearly indicate that the “right hand” of these ureidopropanamides plays an important role in the interaction with FPR2 because structural modifications in this part of the molecule greatly influence agonist potency. On the other hand, the steric hindrance of this part of the molecule seems to be an important requisite for metabolically stable compounds. Therefore, it is important to find the right balance in the dimension of the cyclopropyl phenyl moiety in order to combine high potency with good metabolic stability.
We next evaluated the ability of the new compounds to activate FPR2 in human neutrophils. The data show that neutrophils responded to all agonists that activated FPR-expressing HL-60 cells. The only exceptions were (R)-8 and (R)-9, which were able to induce Ca2+ mobilization in neutrophils but not in FPR-expressing HL-60 cells. Among the compounds studied, (R)-11 and (S)-17 exhibited EC50 values in the submicromolar range.
Half-life and Intrinsic Clearance of Selected Compounds
According to our paradigm for assessing metabolic stability of the new compounds, derivatives (R)- and (S)-10, (R)- and (S)-11, (R)-15, (R)- and (S)-17, which all showed recovery higher than 20%, were further characterized by evaluating half-life (t1/2) and intrinsic clearance (CLint) (Table 2). All selected compounds showed CLint much lower than that of 5, confirming that the new derivatives were more stable than 5 with respect to microsomal oxidative metabolism. Moreover, all compounds displayed t1/2 values exceeding 15 min, which is reported as the lower limit for predicted low clearance compounds [42]. Among the new compounds, (S)-10, (R)-11, and (S)-17 showed a good balance between potency and metabolic stability and were, thus, selected for further studies in order to assess their pharmacological properties in an in vitro model of neuroinflammation.
Table 2.
Half-life and Intrinsic Clearance of Selected Compounds.
| Compound | t1/2 (min) | CLint (μL/min/mg) |
|---|---|---|
|
| ||
| 5 | 1.1a | 1162.4a |
| (R)-10 | 95 | 7.29 |
| (S)-10 | 120 | 5.78 |
| (R)-11 | 99 | 7.0 |
| (S)-11 | 110 | 6.3 |
| (R)-17 | 47 | 14.7 |
| (S)-17 | 48 | 14.4 |
| (R)-15 | 59 | 10.04 |
Data taken from ref. 34.
Effect of Selected Compounds on Cell Viability and Metabolic Activity in Rat Primary Microglial Cells
Initially, we evaluated the effect of (S)-10, (R)-11, and (S)-17 on cell viability and metabolic activity in rat primary microglia cell cultures under basal conditions and after stimulation with lipopolysaccharide (LPS). It is well known that LPS is a primary component of endotoxin from Gram-negative bacteria cell walls [43,44], which binds mostly to Toll-like receptor 4 and induces intracellular signaling resulting in the activation of mitogen activated protein kinases (MAPKs) and NF-κB [45]. These proteins have been described as the key regulators of pro-inflammatory factors production such as cytokines and NO, which may have cytotoxic effects and may damage cells [46].
We evaluated the effect of (S)-10, (R)-11, and (S)-17 on cell viability by using the lactate dehydrogenase (LDH) and MTT tests, two biochemical assays that evaluate different aspects of cell death/viability processes. The LDH assay is a well-accepted paradigm to quantitatively assess cell death after damage of the plasma membrane, which results in increased LDH efflux from injured cells, and it is known that LPS induces impairements in cell membrane integrity. On the other hand, MTT test quantifies mitochondrial activity by measuring the formation of a dark blue formazan product formed by reduction of the tetrazolium ring of MTT in living cells. Thus, these tests provide an accurate and complete assessment of the impact of the selected compounds on the vital status of microglial cells in culture.
We first tested the effect of (S)-10, (R)-11, and (S)-17 at different doses under basal conditions. None of the compounds induced significant effects on LDH release or metabolic activity in the dose range of 0.5–10 μM. On the other hand, exposure of the microglial cells to a 50 μM dose of all compounds resulted in a significant increase in death processes. (S)-10 and (S)-17 also decreased the conversion of MTT dye at a 50 μM dose (Figure 1).
Figure 1.

Effect of the (S)-10 (A), (R)-11 (B) and (S)-17 (C) on cell integrity and mitochondrial activity in rat microglial cell cultures.
* vs control
# vs control + LPS
Next, we evaluated the effect of the selected compounds after stimulation of the microglial cells with LPS, which induces cell death processes by increasing LDH release as well as diminishing metabolic activity. Interestingly, pre-treatment with (S)-10, (R)-11, and (S)-17 effectively blocked LPS-induced cell death processes at the doses of 1 μM and 5 μM (Figure 1). On the other hand, no effect was observed on metabolic activity in LPS-stimulated microglial cell cultures (Figure 1). These data indicate that (S)-10, (R)-11, and (S)-17 did not induce either pro-inflammatory responses or cytotoxicity in microglial cells. Moreover, our FPR2 agonists showed protective properties against LPS treatment in the LDH assays.
Effect of Selected Compounds on Pro-Inflammatory Mediators Production in Rat Primary Microglial Cells
To further characterize the anti-inflammatory properties of our FPR2 agonists, we assessed their effect on the production of the pro-inflammatory mediators NO, IL-1β and TNF-α in rat primary microglial cell cultures. Under normal conditions, NO is involved in many physiological processes in the brain, such as regulation of proliferation, survival, differentiation of the neurons, synaptic activity, and neural plasticity [47]. However, excessive NO synthesis leads to the formation of reactive nitrogen species and neuronal cell death. Moreover, there is an intimate relation between microglial activation, NO production, and neuroinflammation in the brain [48]. Also, inflammatory cytokines, such as IL-1β and TNF-α, have physiological functions in the brain which include effects on neurite outgrowth, neurogenesis, neuronal survival, and synaptic pruning during brain development, synaptic transmission and synaptic plasticity [49]. However, overproduction and exaggerated release of cytokines is associated with neuronal dysfunction.
We evaluated effects of the selected FPR2 agonists on the production of NO, IL-1β, and TNF-α in rat primary microglial cultures under basal conditions and after stimulation with LPS. Furthermore, to check if the observed effect is mediated through the interaction with FPR2, the microglia cells were also pre-treated with the selective FPR2 antagonist WRW4.
(S)-10, (R)-11, and (S)-17 did not induce any change in NO level under basal conditions. The same effect was observed when the cells were treated with WRW4 alone or in combination with the selected agonists (Figure 2). Stimulation of the microglial cells with LPS increased the level of NO, which was significantly attenuated by (S)-10 (5 μM and 10 μM), whereas no effect was observed for (R)-11 and (S)-17. Moreover, pre-treatment of the microglial cells with WRW4 was not able to reverse the anti-inflammatory effect on NO secretion evoked by (S)-10, suggesting that this effect was mediated by the interaction with molecular targets different from FPR2 (Figure 2).
Figure 2.


Effect of the (S)-10 (A), (R)-11 (B) and (S)-17 (C) on NO production in rat microglial cell cultures.
* vs control
# vs control + LPS
(S)-10, (R)-11, and (S)-17 (0.5 μM-10 μM) did not induce any change in the intracellular levels of IL-1β and TNF-α production under basal conditions. As observed for NO production, the antagonist WRW4, alone or in combination with the selected agonists did not affect IL-1β and TNF-α production (Figures 3 and 4). Consistent with our previous studies, the stimulation with LPS induced a significant up-regulation of both IL-1β and TNF-α production in the microglial cells [3]. Interestingly, (S)-10, (R)-11, and (S)-17 showed anti-inflammatory effects, being able to effectively decrease LPS-evoked cytokine production (Figure 3 and 4). Pre-treatment with the antagonist WRW4 was able to block the effect of (S)-17 on both IL-1β and TNF-α secretion, and of (R)-11 only on IL-1β production. In the case of (S)-10, WRW4 did not abolish the observed effect on both pro-inflammatory cytokine release, suggesting that the anti-inflammatory effects of (S)-10 were mediated by other molecular targets (Figure 3 and 4). Considering that the inhibition of IL-1β and TNF-α production by (S)-17 did not exhibit a clear dose response in the range from 0.5 to 10 μM, we tested the effect of (S)-17 at lower doses. The data indicate that at 0.05 μM or 0.1 μM (S)-17 was not able to induce a significant effect on IL-1β and TNF-α production (Figure S4, Supplementary Material).
Figure 3.


Effect of the (S)-10 (A), (R)-11 (B) and (S)-17 (C) on IL-1β production in rat microglial cell cultures.
* vs control
# vs control + LPS
@@ vs agonist + LPS
Figure 4.


Effect of the (S)-10 (A), (R)-11 (B) and (S)-17 (C) on TNF-α production in rat microglial cell cultures.
* vs control
# vs control + LPS
@@ vs agonist + LPS
Collectively, the data indicated that the selected FPR2 agonists did not induce pro-inflammatory responses in resting microglial cells but exerted clear anti-inflammatory effects in the LPS-stimulated cells. In the case of (S)-17 the anti-inflammatory effect was mediated by FPR2, whereas (S)-10 and (R)-11 seem to exert these effects through other molecular targets.
Evaluation of Permeability in hCMEC/D3 Cells
A critical characterization for prospective CNS-acting drugs is the ability to cross the blood brain barrier (BBB), which acts as a highly lipophilic boundary. Compounds permeate BBB mainly by passive diffusion mechanism, and several efflux systems prevent the entrance of xenobiotics into CNS. To endorse the potential of compounds (S)-10, (R)-11, and (S)-17 as promising leads for the development of neuroprotective agents, in vitro transport studies were undertaken. To this end, we selected hCMEC/D3 cells, an immortalized human brain microvascular endothelial cell line, as an in vitro model of BBB. hCMEC/D3 cells stably maintain most of the unique structural and biochemical properties of brain endothelium in vivo, including tight junctions formation and the expression of multiple efflux transporters of the ATP-binding cassette superfamily [50].
The permeation rate of compounds (S)-10, (R)-11, and (S)-17 across the cell monolayer in both directions, i.e., apical-to-basolateral (AB) and basolateral-to-apical (BA), was assessed (Table 3). Moreover, we evaluated the efflux ratio (ER) between BA and AB fluxes because ER greater than 3 can be taken as a figure of undesirable interaction with the efflux transporters (Table 3) [51]. Compound (S)-17 showed good permeation rates in both directions and ER value below 3, thus envisaging good brain distribution and low interactions with the efflux transporters. On the other hand, (S)-10 and (R)-11 showed lower permeation rates, especially in AB direction, which is more strongly influenced by the interaction with the efflux transporters, suggesting that (S)-10 and (R)-11 might have low brain distribution.
Table 3.
Bidirectional Transport across hCMEC/D3 Cells of Compounds (S)-10, (R)-11, and (S)-17.
| Compound | PappBA (nm/sec)a | PappAB (nm/sec)b | ER(BA/AB) |
|---|---|---|---|
|
| |||
| (S)-10 | 253033 | 49785 | 5.06 |
| (R)-11 | 437544 | 119245 | 3.7 |
| (S)-17 | 375981 | 145521 | 2.6 |
Apparent permeability of the basolateral-to-apical transport;.
Apparent permeability of the apical-to-basolateral transport.
Conclusion
In summary, we have manipulated the structure of 5, a moderately potent ureidopropanamide FPRs agonist previously studied in our laboratories, with the aim of identifying new potent FPR2 agonists endowed with enhanced metabolic stability. Several of these new studied derivatives exhibited agonist potency comparable to that of 5 and significantly higher t1/2 and CLint, indicating that the aim of improving stability towards oxidative metabolism was achieved. Analysis of (S)-10, (R)-11, and (S)-17 in an in vitro model of neuroinflammation showed that they did not induce inflammatory responses in resting rat primary microglial cell cultures but they were able to significantly reduce the production of pro-inflammatory mediators when microglial cells were stimulated with LPS. In particular, compound (S)-17 was able to reduce IL-1β and TNF-α levels in LPS-stimulated microglial cells and this effect was mediated by FPR2 interaction because its effects were blocked by pre-treatment of the cells with the FPR2 antagonist WRW4. Moreover, bidirectional transport studies on hCMEC/D3 cells denoted good permeation rates of compound (S)-17 without suffering from likely efflux transporters interactions, thus suggesting good brain permeation and CNS distribution.
To the best of our knowledge, this is the first report on FPR2 agonists being studied in a model of neuroinflammation in order to evaluate their ability to induce anti-inflammatory responses in primary microglial cells. Among the studied agonists, (S)-17 emerges as a prospective pharmacological tool to study the effects of FPR2 activation in the CNS because it elicits FPR2-mediated anti-inflammatory effects in rat microglia and also displays suitable pharmacokinetic characteristics. We believe that these data are promising considering that very recent studies have indicated that stimulation of brain FPR2 with endogenous ligands such as LXA1 or annexin A1 [52] is able to inhibit microglial activation and diminish neuroinflammation in several pathological conditions, including Alzheimer’s Disease and neuropathic pain. This can open new therapeutic perspectives in the treatment of those CNS disorders characterized by neuroinflammation.
Experimental Section
1. Chemistry
Chemicals were purchased from Sigma-Aldrich, Alfa Aesar, and TCI Chemicals. Unless otherwise stated, all chemicals were used without further purification. Thin layer chromatography (TLC) was performed using plates from Merck (silica gel 60 F254). Column chromatography was performed with 1:30 Merck silica gel 60 Å (63–200 μm) as the stationary phase. Flash chromatographic separations were performed on a Biotage SP1 purification system using flash cartridges pre-packed with KP-Sil 32–63 μm, 60 Å silica. 1H and 13C NMR spectra were recorded on a Varian Mercury-VX spectrometer (300 MHz) or on a 500-vnmrs500 Agilent spectrometer (500 MHz). All chemical shift values are reported in ppm (δ). For enantiomeric pairs, NMR spectra for both enantiomers were recorded but the NMR spectrum of only the (R)-enatiomer is reported in the experimental section. Recording of mass spectra was done on an HP6890-5973 MSD gas chromatograph/mass spectrometer; only significant m/z peaks, with their percentage of relative intensity in parentheses, are reported. HRMS-ESI analyses were performed on a Bruker Daltonics MicrOTOF-Q II mass spectrometer, mass range 50–800 m/z, electrospray ion source in positive or negative ion mode. All spectra were in accordance with the assigned structures. The purity of the target compounds listed in Table 1 was assessed by RP-HPLC and combustion analysis. All compounds showed ≥ 98% purity. RP-HPLC analysis was performed on an Agilent 1260 Infinity Binary LC System equipped with a diode array detector using a Phenomenex Gemini C-18 column (250 × 4.6 mm, 5 μm particle size). All target compounds (Table 1) were eluted with CH3OH/H2O/Et3N, 8:2:0.01 at a flow rate of 1 mL/min. Elemental analyses (C,H,N) of the target compounds were performed on a Eurovector Euro EA 3000 analyzer. Analyses indicated by the symbols of the elements were within ± 0.4 % of the theoretical values. Enantiomeric purity of the target compounds (R)- and (S)-7-17 was assessed by chiral HPLC analysis on a Perkin-Elmer series 200 LC instrument using a Daicel ChiralCel OD column (250 mm × 4.6 mm, 5 μm particle size) and equipped with a Perkin-Elmer 785A UV/VIS detector setting λ = 230 nm. The compounds were eluted with n-hexane/EtOH, 4:1, v/v at a flow rate of 0.8 mL/min. All compounds showed enantiomeric excesses ≥ 98%.
The following compounds were prepared according to literature methods: 1-[(4-trifluoromethoxy)phenyl]cyclopropanecarbonitrile (18) [38]; 1-(3-chloro-4-fluorophenyl)-cyclopropanecarbonitrile (19) [39]; 1-(3-fluoro-4-methylphenyl)-cyclopropanecarbonitrile (20) [40]; 1-(phenylcyclopropyl)methylamine (21) [53]; (R)- and (S)-1-(1H-indol-3-ylmethyl)-2-[[(1-phenylcyclopropyl)methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((R)- and (S)- 28) [54]; (R)- and (S)-2-amino-3-(1H-indol-3-yl)-N-[(1-phenylcyclopropyl)methyl]propanamide ((R)- and (S)-34) [54].
General Procedure for the Synthesis of the Amines 22 and 23
Raney nickel was activated with 10 M KOH, then washed with H2O, then with EtOH to remove H2O. The catalyst was then taken up in 2 N ethanolic ammonia and the appropriate nitrile (0.87 mmol) was added to the mixture which was then hydrogenated under 5 bar pressure of hydrogen at 50 °C for 15 h. The reaction mixture was then filtered through Celite and concentrated in vacuo to give the desired amine as an oil.
[1-(4-Trifluoromethoxyphenyl)cyclopropyl]methanamine (22)
90% Yield. 1H NMR (CDCl3): δ 0.740.83 (m, 4H), 1.35 (br s, 2H), 2.76 (s, 2H), 7.13 (d, 2H, J= 8.3 Hz), 7.27 (d, 2H, J= 8.1 Hz). GC/MS: m/z 231 (M+, 7), 203 (100), 134 (67), 115 (35).
[1-(3-Fluoro-4-methylphenyl)cyclopropyl]methanamine (23)
77% Yield. 1H NMR (CDCl3): δ 0.72–0.81 (m, 4H), 1.35 (br s, 2H), 2.23 (s, 3H), 2.74 (s, 2H), 6.74–6.82 (m, 2H), 7.07 (t, 1H, JH-H= 7.6 Hz, JH-F= 8.2 Hz), GC/MS: m/z 179 (M+, 25), 151 (100), 147 (36), 133 (49), 109 (37).
[1-(3-Chloro-4-fluorophenyl)cyclopropyl]methanamine (24)
Borane-methyl sulfide complex as 10.0 M BH3 in excess methyl sulfide (4.6 mmol, 0.46 mL) was dropped into an ice-cooled solution of 1-(3-chloro-4-fluorophenyl)cyclopropylcarbonitrile (0.30 g, 1.5 mmol) in anhydrous THF (10 mL), under stirring. After being refluxed for 3–4 h, the reaction mixture was cooled at −10 °C and MeOH was added dropwise very carefully until gas evolution ceased. The mixture was treated with 3 N HCl (20 mL) and was refluxed for 1h. After cooling, the mixture was alkalized with 3 N NaOH and extracted with CH2Cl2 (5 × 20 mL). The collected organic layers were dried over Na2SO4 and the solvent was evaporated under pressure to give the pure amine as a colorless oil (0.25 g; 81% yield). 1H NMR (CDCl3): δ 0.73–0.82 (m, 4H), 1.37 (br s, 2H), 2.76 (s, 2H), 7.07 (t, 1H, JH-H= 8.2 Hz, JH-F= 8.8 Hz), 7.17–7.22 (m, 1H), 7.36 (dd, 1H, J= 7.0, 2.3 Hz). GC/MS: m/z 201 (M++2, 1), 199 (M+, 4), 171 (100), 147 (27), 133 (60), 109 (29).
General Procedure for the Synthesis of Boc-protected Derivatives (R)- and (S)-29–33
N,N′-Carbonyldiimidazole (1.1 mmol) was added to a solution of (R)- or (S)-Boc-protected amino acid (1.0 mmol), in anhydrous THF (10 mL) under N2. The reaction mixture was stirred at room temperature overnight, then a solution of the appropriate amine (1.0 mmol) in anhydrous THF was added. The reaction mixture was stirred at room temperature for 6 h. Then, the solvent was removed in vacuo and the residue was partitioned between EtOAc (20 mL) and H2O (2 × 20 mL). The aqueous layer was separated and extracted twice with EtOAc (20 mL). The collected organic layers were dried (Na2SO4) and evaporated in vacuo. The crude residue was purified through flash chromatography (gradient eluition from 30% to 70% ethyl acetate in n-hexane) to give pure target compound as a white solid.
(R)-1-(4-Cyanophenylmethyl)-2-[[(1-phenylcyclopropyl)methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((R)-29)
75% Yield. 1H NMR (CDCl3): δ 0.76–0.89 (m, 4H), 1.38 (s, 9H), 2.96 (dd, 1H, J= 13.5, 6.4 Hz), 3.07 (dd, 1H, J= 13.5, 7.6 Hz), 3.32 (dd, 1H, J= 14.1, 5.9 Hz), 3.4 (dd, 1H, J= 14.1, 5.9 Hz), 4.23–4.27 (m, 1H), 4.98 (br d, 1H), 5.8 (br t, 1H), 7.12–7.15 (m, 2H), 7.20–7.31 (m, 5H), 7.51 (d, 2H, J= 8.8 Hz). ESI+/MS m/z 442 (M+Na)+, ESI+/MS/MS m/z 342 (100).
(S)–1-(4-Cyanophenylmethyl)-2-[[(1-phenylcyclopropyl)methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((S)-29)
64% yield. ESI+/MS m/z 442 (M+Na)+, ESI+/MS/MS m/z 342 (100).
(R)-2-[[(1-Phenylcyclopropyl)methyl]amino]-2-oxoethyl]-1-(3-pyridinylmethyl)carbamic acid, 1,1-Dimethylethyl Ester ((R)-30)
58% Yield. 1H NMR (CDCl3): δ 0.80–0.85 (m, 4H), 1.37 (s, 9H), 2.93 (dd, 1H, J= 13.5, 7.0 Hz), 3.04 (dd, 1H, J= 14.1, 7.0 Hz), 3.38 (d, 2H, J= 5.3 Hz), 4.20–4.25 (m, 1H), 4.97 (br d, 1H), 5.91 (br t, 1H), 7.13–7.20 (m, 5H), 7.23–7.26 (m, 1H), 7.46 (d, 1H, J= 7.6 Hz), 8.39 (d, 1H, J= 1.8 Hz), 8.47 (dd, 1H, J= 4.7, 1.2 Hz). ESI+/MS m/z 418 (M+Na)+, ESI+/MS/MS m/z 318 (100).
(S)- 2-[[(1-Phenylcyclopropyl)methyl]amino]-2-oxoethyl]-1-(3-pyridinylmethyl)carbamic acid, 1,1-Dimethylethyl Ester ((S)-30)
45% Yield. ESI+/MS m/z 418 (M+Na)+, ESI+/MS/MS m/z 318 (100).
(R)-1-(4-Cyanophenylmethyl)-2-[[[(1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((R)-31)
84% Yield. 1H NMR (CDCl3): δ 0.79–0.89 (m, 4H), 1.38 (s, 9H), 2.99 (dd, 1H, J= 13.5, 6.5 Hz), 3.13 (dd, 1H, J= 13.5, 7.0 Hz), 3.35 (dd, 1H, J= 13.5, 5.5 Hz), 3.42 (dd, 1H, J= 13.5, 5.5 Hz); 4.26–4.27 (m, 1H), 4.87 (br s, 1H), 5.92 (br t, 1H), 7.13 (d, 2H, J= 8.5 Hz), 7.19–7.21 (m, 2H), 7.26–7.28 (m, 2H), 7.57 (d, 2H, J= 8.5 Hz). ESI+/MS: m/z 526 (M+Na)+, ESI+-MS/MS: m/z 426 (100).
(S)-1-(4-Cyanophenylmethyl)-2-[[[(1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((S)-31)
87% Yield. ESI+/MS: m/z 526 (M+Na)+. ESI+-MS/MS: m/z 426 (100).
(R)-1-(4-Cyanophenylmethyl)-2-[[[(1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]amino]-2-oxoethyl]carbamic acid, 1,1-Dimethylethyl Ester ((R)-32)
81% Yield. 1H NMR (CDCl3): δ 0.75–0.84 (m, 4H), 1.38 (s, 9H), 2.23 (s, 3H), 2.98 (dd, 1H, J= 13.5, 6.6 Hz); 3.10 (dd, 1H, J= 13.5, 6.5 Hz); 3.34 (d, 2H, J= 5.4 Hz); 4.20–4.25 (m, 1H), 4.94 (br d, 1H), 5.84 (br t, 1H), 6.74–6.82 (m, 2H), 7.07 (t, 1H, J= 8.0 Hz), 7.25 (d, 2H, J= 8.4 Hz), 7.55 (d, 2H, J= 8.1 Hz). ESI+/MS: m/z 474 (M+Na)+; ESI+-MS/MS: m/z 374 (100).
(S)-1-(4-Cyanophenylmethyl)-2-[[[(1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]amino]-2-oxoethyl]carbamic acid,1,1-dimethylethyl ester ((S)-32)
71% Yield. ESI+/MS: m/z 474 (M+Na)+, ESI+-MS/MS: m/z 374 (100).
(R)-2-[[[(1-(3-Chloro-4-fluorophenyl)cyclopropyl]methyl]amino]-2-oxoethyl]-1-(4-cyanophenylmethyl)-carbamic acid, 1,1-Dimethylethyl Ester ((R)-33)
77% Yield. 1H NMR (CDCl3): δ 0.78–0.89 (m, 4H), 1.39 (s, 9H), 2.98 (dd, 1H, J= 13.5, 7.0 Hz), 3.13 (dd, 1H, J= 13.5, 7.0 Hz), 3.32 (dd, 1H, J= 14.0, 5.5 Hz), 3.38 (dd, 1H, J= 14.0, 5.5 Hz), 4.24–4.28 (m, 1H), 4.94 (br s, 1H), 6.04 (br t, 1H), 7.02–7.06 (m, 2H), 7.20 (d, 1H, J=7.0 Hz), 7.28 (d, 2H, J= 8.5 Hz), 7.58 (d, 2H, J= 8.0 Hz). ESI+/MS: m/z 494 (M+Na)+, ESI+-MS/MS: m/z 394 (100).
(S)- 2-[[[(1-(3-Chloro-4-fluorophenyl)cyclopropyl]methyl]amino]-2-oxoethyl]-1-(4-cyanophenylmethyl)-carbamic acid, 1,1-Dimethylethyl Ester ((S)-33)
84% Yield. ESI+/MS: m/z 494 (M+Na)+, ESI+-MS/MS: m/z 394 (100).
General Procedure for the Synthesis of Amines (R)- and (S)-35–39
Trifluoroacetic acid (5 mL) was added to a solution of Boc-protected derivatives (R)- and (S)-29-33 (0.46 mmol) in CH2Cl2 (20 mL). The reaction mixture was stirred at room temperature for 5 h and basified with aqueous 1 M NaOH. The separated aqueous phase was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo to give the desired compounds as pale yellow semisolids that were used without further purification.
(R)-2-Amino-3-(4-cyanophenyl)-N-[(1-phenylcyclopropyl)methyl]propanamide ((R)-35)
43% Yield. 1H NMR (CDCl3): δ 0.87–0.97 (m, 4H), 1.72 (br s, 2H), 2.82 (dd, 1H, J= 14.0, 8.5 Hz), 3.21 (dd, 1H, J= 13.7, 4.4 Hz), 3.40 (dd, 1H, J= 13.9, 6.0 Hz), 3.45 (dd, 1H, J= 13.7, 6.0 Hz), 3.62 (dd, 1H, J= 8.0, 4.5 Hz), 7.19–7.24 (m, 4H), 7.27–7.31 (m, 3H), 7.58 (d, 2H, J= 8.2 Hz). ESI+/MS m/z 342 (M+Na)+, ESI+/MS/MS m/z 151(100).
(S)-2-Amino-3-(4-cyanophenyl)-N-[(1-phenylcyclopropyl)methyl]propanamide ((S)-35)
76% Yield. ESI+/MS m/z 342 (M+Na)+, ESI+/MS/MS m/z 151(100).
(R)-2-Amino-N-[(1-phenylcyclopropyl)methyl]-3-(3-pyridinyl)propanamide ((R)-36)
90% Yield. 1H NMR (CDCl3): δ 0.81–0.87 (m, 4H), 1.53 (br s, 2H), 2.71 (dd, 1H, J = 13.5, 8.2 Hz), 3.14 (dd, 1H, J= 14.1, 4.1 Hz), 3.36–3.49 (m, 2H), 3.55 (dd, 1H, J= 8.8, 4.1 Hz), 7.19–7.30 (m, 7H), 7.52 (d, 1H, J= 7.6 Hz), 8.43 (d, 1H, J= 1.7 Hz), 8.5 (dd, 1H, J= 5.3, 1.7 Hz). ESI+/MS m/z 318 (M+Na)+, ESI+/MS/MS m/z 226(100).
(S)-2-Amino-N-[(1-phenylcyclopropyl)methyl]-3-(3-pyridinyl)propanamide ((S)-36)
Quantitative yield. ESI+/MS m/z 318 (M+Na)+, ESI+/MS/MS m/z 226 (100).
(R)-2-Amino-3-(4-cyanophenyl)-N-[[1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl] propanamide ((R)-37)
92% Yield. 1H NMR (CDCl3): δ 0.82–0.93 (m, 4H), 1.47 (br s, 2H), 2.75 (dd, 1H, J= 13.5, 8.5 Hz), 3.23 (dd, 1H, J= 13.5, 4.5 Hz), 3.41 (dd, 1H, J= 14.0, 6.00 Hz), 3.46 (dd, 1H, J= 14.0, 6.00 Hz), 3.58 (dd, 1H, J= 8.8, 4.5 Hz), 7.12–7.15 (m, 2H), 7.25–7.28 (m, 3H), 7.29–7.31 (m, 2H), 7.59–7.62 (m, 2H). ESI−/MS: m/z 402 (M-H)−, ESI−-MS/MS: m/z 285 (100), 116 (12).
(S)-2-Amino-3-(4-cyanophenyl)-N-[[1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl] propanamide ((S)-37)
90% Yield. ESI−/MS: m/z 402 (M-H)−, ESI−-MS/MS: m/z 285 (100), 116 (13).
(R)-2-Amino-3-(4-cyanophenyl)-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl] propanamide ((R)-38)
71% Yield. 1H NMR (CDCl3): δ 0.76–0.82 (m, 2H), 0.83–0.88 (m, 2H), 1.47 (br s, 2H), 2.23 (s, 3H), 2.89–2.92 (m, 1H), 3.17–3.20 (m, 1H), 3.23–3.29 (m, 1H), 3.42–3.46 (m, 1H), 4.12–4.14 (m, 1H), 6.83–6.86 (m, 2H), 7.06–7.09 (m, 1H), 7.13 (br t, 1H), 7.22 (d, 2H, J= 8.5 Hz), 7.46 (d, 2H, J= 8.5 Hz). ESI−/MS: m/z 350 (M-H)−, ESI−-MS/MS: m/z 233 (100), 116 (67).
(S)-2-Amino-3-(4-cyanophenyl)-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl] propanamide ((S)-38)
94% yield. ESI−/MS: m/z 350 (M-H)−, ESI−-MS/MS: m/z 233 (100), 116 (61).
(R)-2-Amino-3-(4-cyanophenyl)-N-[[1-(3-chloro-4-fluorophenyl)cyclopropyl]methyl] propanamide ((R)-39)
47% Yield. 1H NMR (DMSO-d6): δ 0.73–0.80 (m, 2H), 0.83–0.88 (m, 2H), 2.30 (br s, 2H), 2.64 (dd, 1H, J= 13.0, 8.0 Hz), 2.88 (dd, 1H, J=13.0, 5.5 Hz), 3.20 (dd, 1H, J= 13.0, 5.5 Hz), 3.33–3.37 (m, 1H), 3.40–3.42 (m, 1H), 7.18–7.21 (m, 1H), 7.28 (d, 1H, J= 9.0), 7.34 (d, 2H, J= 8.5 Hz), 7.38 (dd, 1H, J= 6.5, 2.0 Hz), 7.70 (d, 2H, J= 8.4 Hz), 7.91 (br t, 1H). ESI−/MS: m/z 370 (M-H)−, ESI−-MS/MS: m/z 253 (100), 116 (25).
(S)-2-Amino-3-(4-cyanophenyl)-N-[[1-(3-chloro-4-fluorophenyl)cyclopropyl]methyl] propanamide ((S)-39)
95% Yield. ESI−/MS: m/z 370 (M-H)−, ESI−-MS/MS: 253 (100), 116 (21).
General Procedure for the Synthesis of the Final Compounds (Procedure A)
To a solution of the amine (R)- and (S)-34-39 (1.0 mmol) in anhydrous THF, a solution of the appropriate 4-substitued phenylisocyanate (1.2 mmol) in the same solvent (10 mL) was added and the reaction mixture was stirred at room temperature overnight. After removing the solvent in vacuo, the residue was taken up in CHCl3 (20 mL) and washed with H2O (2 × 20 mL). The separated organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude residue was chromatographed (CHCl3/AcOEt, 1:1 as the eluent). When necessary, the obtained solid was further purified by crystallization from MeOH to give the final compounds.
(R)-3-(1H-Indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]-N-[(1-phenylcyclopropyl)methyl] propanamide ((R)-7)
19% Yield. 1H NMR (CDCl3): δ 0.56–0.74 (m, 4H), 3.06 (dd, 1H, J= 14.6, 8.2 Hz), 3.19–3.25 (m, 3H), 3.76 (s, 3H), 4.63–4.65 (m, 1H), 5.91 (br d, 1H), 6.00 (br t, 1H), 6.73 (d, 2H, J= 8.8 Hz), 6.83 (d, 1H, J= 1.8 Hz), 6.91–6.94 (m, 3H), 7.04–7.22 (m, 7H), 7.32 (d, 1H, J= 8.2 Hz), 7.62 (d, 1H, J= 8.4 Hz), 7.97 (br s, 1H). 13C NMR (DMSO-d6): 171.9; 158.9; 154.8; 147.9; 136.5; 131.5; 128.3; 127.4; 125.6; 125.0; 123.0; 121.7; 119.8; 118.8; 114.5; 111.1; 109.7; 55.8; 53.6; 47.2; 36.4; 25.3; 12.7; 12.6. ESI−/MS: m/z 481 (M-H)−; ESI−-MS/MS: m/z 358 (100), 332 (44).
(S)-3-(1H-Indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]-N-[(1-phenylcyclopropyl)methyl] propanamide ((S)-7)
22% Yield. ESI−/MS: m/z 481 (M-H)−; ESI−-MS/MS: m/z 358 (100), 332 (43).
(S)-2-[3-(4-Fluorophenyl)ureido]-3-(1H-indol-3-yl)-N-[(1-phenylcyclopropyl) methyl]propanamide ((S)-8)
41% Yield. 1H NMR (DMSO-d6): δ 0.64–0.81 (m, 4H), 2.84 (dd, 1H, J= 14.6, 7.0 Hz), 2.99 (dd, 1H, J= 14.6, 5.3 Hz), 3.22 (dd, 1H, J= 13.5, 5.3 Hz), 3.38 (dd, 1H, J= 13.5, 5.8 Hz), 4.51–4.44 (m, 1H), 6.22 (br d, 1H), 6.92 (t, 2H, J= 7.6 Hz), 6.98–7.00 (m, 2H), 7.10–7.21 (m, 6H), 7.28–7.34 (m, 3H), 7.5 (d, 1H, J= 8.2 Hz), 8.02 (t, 1H, J= 5.3Hz), 8.66 (s,1H), 10.78 (s, 1H). ESI−/MS: m/z 469 (M-H)−, ESI−-MS/MS: m/z 229 (6), 332 (14), 358 (100). 13C NMR (DMSO-d6): 170.7; 158.3; 154.6; 147.9; 136.5; 135.0; 128.3; 127.4; 125.6; 125.0; 123.1; 119.3; 118.8; 115.7; 109.6; 53.7; 47.0; 36.4; 25.3; 12.7. ESI−/MS: m/z 469 (M-H)−; ESI−-MS/MS: m/z 358 (100), 332 (12).
(R)-3-(1H-Indol-3-yl)-N-[(1-phenylcyclopropyl)methyl]-2-[3-(phenyl)ureido]propanamide ((R)-9)
35% Yield. 1H NMR (CDCl3): δ 0.49–0.74 (m, 4H), 3.09 (dd, 1H, J= 14.6, 8.8 Hz), 3.19–3.25 (m, 2H), 3.72–3.76 (m, 1H), 4.65–4.73 (m, 1H), 6.09 (br s, 1H), 6.40 (br t, 1H), 6.83 (br s, 1H), 6.91–6.93 (m, 2H), 6.97–7.01 (m, 1H), 7.06–7.13 (m, 4H), 7.16–7.21 (m, 5H), 7.32 (d, 1H, J= 8.2 Hz), 7.52 (br t, 1H), 7.65 (d, 1H, J= 8.2 Hz), 7.97 (s, 1H). 13C NMR (DMSO-d6): 171.7; 154.5; 147.9; 139.4; 136.5; 128.9; 128.3; 128.0; 127.4; 123.3; 121.6; 119.8; 118.8; 110.1; 109.5; 58.7; 46.9; 25.7; 12.7; 12.6. ESI−/MS: m/z 451 (M-H)−; ESI−-MS/MS: m/z 358 (100), 332 (16).
(S)-3-(1H-Indol-3-yl)-N-[(1-phenylcyclopropyl)methyl]-2-[3-(phenyl)ureido]propanamide ((S)-9)
15% Yield. ESI−/MS: m/z 451 (M-H)−, ESI−-MS/MS: m/z 358 (100), 332 (10).
(R)-3-(4-Cyanophenyl)-2-[3-(4-nitrophenyl)ureido]-N-[(1-phenylcyclopropyl)methyl]propanamide ((R)-10)
69% Yield. 1H NMR (DMSO-d6): δ 0.69–0.84 (m, 4H), 2.79 (dd, 1H, J= 13.5, 7.6 Hz), 2.96 (dd, 1H, J= 13.5, 5.2 Hz), 3.18 (dd, 1H, J= 13.5, 5.3 Hz), 3.48 (dd, 1H, J= 14.1, 6.4 Hz), 4.51–4.58 (m,1H), 6.61 (br d, 1H), 7.11–7.17 (m, 2H), 7.19–7.27 (m, 5H), 7.53 (d, 2H, J= 9.1 Hz), 7.66 (d, 2H, J= 8.2 Hz), 8.01 (d, 2H, J= 8.8 Hz), 8.22 (br t, 1H), 9.41 (br s, 1H). 13C NMR (DMSO-d6): 170.9; 154.1; 147.2; 143.7; 141.0; 132.3; 130.8; 128.5; 128.4; 126.5; 125.6; 119.3; 117.3; 109.7; 53.7; 47.0; 25.3; 12.7. ESI−/MS: m/z 482 (M-H)−, ESI−-MS/MS: m/z 344 (100).
(S)-3-(4-Cyanophenyl)-2-[3-(4-nitrophenyl)ureido]-N-[(1-phenylcyclopropyl)methyl]propanamide ((S)-10)
39% Yield. ESI−/MS: m/z 482 (M-H)−; ESI−-MS/MS: m/z 344 (100).
(R)-2-[3-(4-Nitrophenyl)ureido]-N-[(1-phenylcyclopropyl)methyl]-3-(3-pyridinyl)-propanamide ((R)-11)
35% Yield. 1H NMR (DMSO-d6): δ 0.73–0.85 (m, 4H), 2.73 (dd, 1H, J= 13.5, 7.6 Hz), 2.90 (dd, 1H, J= 14.1, 4.7 Hz), 3.22 (dd, 1H, J= 14.1, 4.7 Hz), 3.45 (dd, 1H, J= 13.5, 5.9 Hz), 4,51–4.53 (m, 1H), 6.65 (d, 1H, J= 7.6 Hz), 7.13–7.26 (m, 5H), 7.37 (m, 1H), 7.53 (d, 2H, J= 8.9 Hz), 8.1 (d, 2H, J= 9.3 Hz), 8.22–8.30 (m, 3H), 8.36 (d, 1H, J= 4.7 Hz), 9.45 (s, 1H). 13C NMR (DMSO-d6): 171.1; 154.1; 150.7; 148.0; 147.2; 143.8; 141.0; 137.1; 133.2; 128.6; 128.5; 126.5; 125.6; 123.6; 117.2; 53.7; 47.1; 36.4; 25.3; 12.7; 12.6. ESI−/MS: m/z 458 (M-H)−, ESI−-MS/MS: m/z 320 (100).
(S)- 2-[3-(4-Nitrophenyl)ureido]-N-[(1-phenylcyclopropyl)methyl]-3-(3-pyridinyl)-propanamide ((S)-11)
28% Yield. ESI−/MS: m/z 458 (M-H)−; ESI−-MS/MS: m/z 320 (100).
(R)-3-(4-Cyanophenyl)-2-[3-(4-methoxyphenyl)ureido]-N-[[1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl]propanamide ((R)-12)
72% Yield. 1H NMR (DMSO-d6): δ 0.70–0.87 (m, 4H), 2.75 (dd, 1H, J= 13.5, 8.1 Hz), 2.92 (dd, 1H, J= 14.1, 5.1 Hz), 3.19 (dd, 1H, J= 14.1, 5.1 Hz), 3.42 (dd, 1H, J= 14.1, 6.0 Hz), 3.66 (s, 3H), 4.46–4.53 (m, 1H), 6.20 (br d, 1H), 6.78 (d, 2H, J= 8.7 Hz), 7.17–7.21 (m, 4H), 7.26 (d, 2H, J= 8.4 Hz), 7.36 (d, 2H, J= 8.7 Hz), 7.68 (d, 2H, J= 8.1 Hz), 8.16 (br t, 1H), 8.39 (s, 1H). 13C NMR (DMSO-d6): 171.2; 158.9; 154.8; 144.8; 140.8; 140.2; 132.1; 131.7; 130.9; 126.0; 119.8; 118.6; 114.5; 113.9; 97.2; 55.8; 53.7; 47.0; 25.3; 12.7; 12.6. ESI−-MS: m/z 551 (M-H)−, ESI−-MS/MS: m/z 428 (46), 402 (23), 385 (100).
(S)-3-(4-Cyanophenyl)-2-[3-(4-methoxyphenyl)ureido]-N-[[1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl]propanamide ((S)-12)
76% Yield. ESI−-MS: m/z 551 (M-H)−, ESI−-MS/MS: m/z 428 (50), 385 (100).
(R)-3-(4-Cyanophenyl)-2-[3-(4-fluorophenyl)ureido]-N-[[1-(4-trifluoromethoxyphenyl)cyclopropyl]methyl]propanamide ((R)-13)
64% Yield. 1H NMR (DMSO-d6): δ 0.72–0.74 (m, 2H), 0.80–0.82 (m, 2H), 2.76 (dd, 1H J= 13.5, 8.5 Hz), 2.92 (dd, 1H, J= 13.5, 5.5 Hz), 3.20 (dd, 1H, J= 13.5, 5.5 Hz), 3.44 (dd, 1H, J= 14.0, 6.5 Hz), 4.49–4.53 (m, 1H), 6.32 (br d, 1H), 7.01–7.05 (m, 2H), 7.20 (d, 2H, J= 8.5 Hz), 7.25 (d, 2H, J= 8.5 Hz) 7.29–7.34 (m, 2H), 7.35–7.38 (m, 2H), 7.69 (d, 2H, J= 8.5 Hz), 8.24 (br t, 1H), 8.65 (s, 1H). 13C NMR (DMSO-d6): 171.2; 162.9; 154.8; 144.8; 140.8; 140.2; 135.0; 132.1; 130.9; 129.7; 126.0; 119.3; 115.7; 113.9; 53.5; 47.1; 36.4; 25.3; 12.7; 12.6. ESI−/MS: m/z 539 (M-H)−, ESI−-MS/MS: m/z 428 (94), 385 (100).
(S)-3-(4-Cyanophenyl)-2-[3-(4-fluorophenyl)ureido]-N-[[1-(4-trifluoromethoxyphenyl) cyclopropyl]methyl]propanamide ((S)-13)
80% Yield. ESI−/MS: m/z 539 (M-H)−, ESI−-MS/MS: m/z 428 (86), 385 (100).
(R)-3-(4-Cyanophenyl)-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]-2-[3-(4-methoxyphenyl)ureido]propanamide ((R)-14)
13% Yield. 1H NMR (DMSO-d6): δ 0.74–0.78 (m, 2H), 0.81–0.86 (m, 2H), 2.14 (s, 3H), 2.78 (dd, 1H, J= 14.0, 8.0 Hz), 2.94 (dd, 1H, J= 14.0, 5.5 Hz), 3.18 (dd, 1H, J= 14.0, 5.5 Hz), 3.44 (dd, 1H, J= 14.0, 6.5 Hz), 3.67 (s, 3H), 4.48–4.52 (m, 1H), 6.26 (br d, 1H), 6.78 (d, 2H, J= 8.8 Hz), 6.95–6.98 (m, 1H), 7.02 (d, 1H, J= 1.5 Hz), 7.12 (t, 1H, J= 8.0 Hz), 7.21 (d, 2H, J= 8.8 Hz), 7.26 (d, 2H, J= 8.3 Hz), 7.67 (d, 2H, J= 8.5 Hz), 8.18 (br t, 1H), 8.46 (s, 1H). 13C NMR (DMSO-d6): 170.8; 161.1; 158.9; 154.8; 148.6; 140.8; 132.1; 130.9; 130.2; 120.5; 119.8; 114.5; 111.9; 55.8; 53.7; 47.0; 25.2; 14.5; 12.6. ESI–/MS: m/z 499 (M-H)−, ESI−-MS/MS: m/z 376 (42), 333 (100).
(R)-3-(4-Cyanophenyl)-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]-2-[3-(4-fluorophenyl)ureido]propanamide ((R)-15)
10% Yield. 1H NMR (DMSO-d6): δ 0.71–0.86 (m, 4H), 2.14 (s, 3H), 2.79 (dd, 1H, J= 13.7, 7.8 Hz), 2.94 (dd, 1H, J= 13.7, 5.4 Hz), 3.19 (dd, 1H, J= 14.2, 5.0 Hz), 3.44 (dd, 1H, J= 14.2, 6.4 Hz), 4.51–4.52 (m, 1H), 6.36 (br d, 1H), 6.96 (td, 2H, J= J= 8.5, 1.5 Hz), 7.01–7.05 (m, 2H), 7.12 (t, 1H, J= 8.0 Hz), 7.25 (d, 2H, J= 7.8 Hz), 7.30–7.35 (m, 2H), 7.67 (d, 2H, J= 8.5 Hz), 8.19 (br t, 1H), 8.70 (s, 1H). 13C NMR (DMSO-d6): 171.7; 162.9; 154.8; 148.6; 140.9; 132.1; 130.9; 130.2; 120.8; 120.5; 119.3; 118.6; 115.7; 111.9; 109.8; 53.7; 47.0; 36.4; 25.2; 14.5; 12.6. ESI−/MS: m/z 487 (M-H)−, ESI−-MS/MS: m/z 376 (75), 333 (100).
(R)-N-[[1-(3-Chloro-4-fluorophenyl)cyclopropyl]methyl]-3-(4-cyanophenyl)-2-[3-(4-methoxyphenyl)ureido]propanamide ((R)-16)
19% Yield. 1H NMR (DMSO-d6): δ 0.72–0.87 (m, 4H), 2.76 (dd, 1H, J= 13.5, 7.5 Hz), 2.92 (dd, 1H, J= 13.5, 5.5 Hz), 3.14 (dd, 1H, J= 14.0, 5.0 Hz), 3.44 (dd, 1H, J= 14.0, 6.5 Hz), 3.67 (s, 3H), 4.50 (td, 1H, J= 8.5, 6.0 Hz), 6.25 (br d, 1H), 6.78 (d, 2H, J= 9.0 Hz), 7.19–7.22 (m, 2H), 7.24–7.27 (m, 4H), 7.45 (d, 1H, J= 7.5 Hz), 7.69 (d, 2H, J= 8.5 Hz), 8.22 (br t, 1H), 8.42 (s, 1H). 13C NMR (DMSO-d6): 170.8; 158.9; 155.7; 154.8; 147.0; 140.8; 132.1; 131.7; 132.1;130.9; 126.4; 120.3; 118.6; 119.8; 116.5; 114.5; 55.8; 53.7; 47.2; 36.4; 25.3; 12.7; 12.6. ESI−-MS: m/z 519 (M-H)−, ESI−-MS/MS: m/z 396 (50), 370 (30), 353 (100).
(S)-N-[[1-(3-Chloro-4-fluorophenyl)cyclopropyl]methyl]-3-(4-cyanophenyl)-2-[3-(4-methoxyphenyl)ureido]propanamide ((S)-16)
50% Yield. ESI−-MS: m/z 519 (M-H)−, ESI−-MS/MS: m/z 396 (50), 370 (32), 353 (100).
(S)- 3-(4-Cyanophenyl)-N-[[1-(3-chloro-4-fluorophenyl)cyclopropyl]methyl]-2-[3-(4-fluorophenyl)ureido]propanamide ((S)-17)
34% Yield. 1H NMR (DMSO-d6): δ 0.73–0.89 (m, 4H), 2.77 (dd, 1H, J= 13.7, 7.8 Hz), 2.93 (dd, 1H, J= 13.7, 5.3 Hz), 3.15 (dd, 1H, J= 14.2, 5.4 Hz), 3.44 (dd, 1H, J= 14.2, 7 Hz), 4.48–4.53 (m, 1H), 6.30 (br d, 1H), 7.01–7.06 (m, 2H), 7.25 (t, 2H, J= 7.8 Hz), 7.29–7.33 (m, 4H), 7.45 (dt, 1H, J= 6.0, 2.0 Hz), 7.69 (d, 2H, J= 6.4 Hz), 8.24 (br t, 1H), 8.63 (s, 1H). 13C NMR (DMSO-d6): 171.5; 158.3; 157,1; 156.4; 155.2; 154.9; 144.0; 141.6; 136.9; 132.3; 131.0; 130.7; 129.5; 119.5; 119.3; 116.9; 116.7; 115.7; 115.5; 109.6; 53.7; 46.9; 25.13; 12.7; 12.6. ESI−/MS: m/z 507 (M-H)−, ESI−-MS/MS: m/z 396 (100), 353 (89).
General Procedure for the Synthesis of the Final Compounds (Procedure B)
N,N′-Carbonyldiimidazole (1.1 mmol) was added to a solution of aniline (1.0 mmol), in anhydrous THF (10 mL), under N2. The reaction mixture was stirred at room temperature overnight, then a solution of the amine (R)-34, (S)-38, or (S)-39, (1.0 mmol) in anhydrous THF was added. The reaction mixture was stirred until the reagents disappeared monitoring by TLC. Then, the solvent was removed in vacuo and the residue was partitioned between EtOAc (20 mL) and H2O (20 mL). The separated aqueous layer was extracted twice with EtOAc (20 mL), then the collected organic layers were dried (Na2SO4) and evaporated in vacuo. The crude residue was chromatographed to give pure target compound as a white solid. When necessary, the obtained solid was further purified by crystallization from MeOH to give the final compound.
(R)-2-[3-(4-Fluorophenyl)ureido]-3-(1H-indol-3-yl)-N-[(1-phenylcyclopropyl) methyl]propanamide ((R)-8)
19% Yield. 1H NMR (DMSO-d6): δ 0.65–0.80 (m, 4H), 2.84 (dd, 1H, J= 14.6, 7.0 Hz), 2.99 (dd, 1H, J= 14.6, 5.3 Hz), 3.22 (dd, 1H, J= 13.5, 5.3 Hz), 3.38 (dd, 1H, J= 13.5, 5.8 Hz), 4.51–4.44 (m, 1H), 6.22 (br d, 1H), 6.92 (t, 2H, J= 7.6 Hz), 6.98–7.00 (m, 2H), 7.10–7.21 (m, 6H), 7.28–7.34 (m, 3H), 7.5 (d, 1H, J= 8.2 Hz), 8.02 (t, 1H, J= 5.3Hz), 8.66 (s,1H), 10.78 (s, 1H). 13C NMR (DMSO-d6): 170.7; 158.3; 154.6; 147.9; 136.5; 135.0; 128.3; 127.4; 125.6; 125.0; 123.1; 119.3; 118.8; 115.7; 109.6; 53.7; 47.0; 36.4; 25.3; 12.7. ESI−/MS: m/z 469 (M-H)−, ESI−-MS/MS: m/z 229 (6), 332 (14), 358 (100).
(S)-3-(4-Cyanophenyl)-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]-2-[3-(4-methoxyphenyl)ureido]propanamide ((S)-14)
13% Yield. 1H NMR (DMSO-d6): δ 0.74–0.78 (m, 2H), 0.81–0.86 (m, 2H), 2.14 (s, 3H), 2.78 (dd, 1H, J= 14.0, 8.0 Hz), 2.94 (dd, 1H, J= 14.0, 5.5 Hz), 3.18 (dd, 1H, J= 14.0, 5.5 Hz), 3.44 (dd, 1H, J= 14.0, 6.5 Hz), 3.67 (s, 3H), 4.48–4.52 (m, 1H), 6.26 (br d, 1H), 6.78 (d, 2H, J= 8.8 Hz), 6.95–6.98 (m, 1H), 7.02 (d, 1H, J= 1.5 Hz), 7.12 (t, 1H, J= 8.0 Hz), 7.21 (d, 2H, J= 8.8 Hz), 7.26 (d, 2H, J= 8.3 Hz), 7.67 (d, 2H, J= 8.5 Hz), 8.18 (br t, 1H), 8.46 (s, 1H). 13C NMR (DMSO-d6): 170.8; 161.1; 158.9; 154.8; 148.6; 140.8; 132.1; 130.9; 130.2; 120.5; 119.8; 114.5; 111.9; 55.8; 53.7; 47.0; 25.2; 14.5; 12.6. ESI−/MS: m/z 499 (M-H)−, ESI−-MS/MS: m/z 376 (39), 333 (100).
(S)-3-(4-Cyanophenyl)-2-[3-(4-fluorophenyl)ureido]-N-[[1-(3-fluoro-4-methylphenyl)cyclopropyl]methyl]propanamide ((S)-15)
10% Yield. 1H NMR (DMSO-d6): δ 0.71–0.86 (m, 4H), 2.14 (s, 3H), 2.79 (dd, 1H, J= 13.7, 7.8 Hz), 2.94 (dd, 1H, J= 13.7, 5.4 Hz), 3.19 (dd, 1H, J= 14.2, 5.0 Hz), 3.44 (dd, 1H, J= 14.2, 6.4 Hz), 4.51–4.52 (m, 1H), 6.36 (br d, 1H), 6.96 (td, 2H, J= 8.5, 1.5 Hz), 7.01–7.05 (m, 2H), 7.12 (t, 1H, J= 8.0 Hz), 7.25 (d, 2H, J= 7.8 Hz), 7.30–7.35 (m, 2H), 7.67 (d, 2H, J= 8.5 Hz), 8.19 (br t, 1H), 8.70 (s, 1H). 13C NMR (DMSO-d6): 171.7; 162.9; 154.8; 148.6; 140.9; 132.1; 130.9; 130.2; 120.8; 120.5; 119.3; 118.6; 115.7; 111.9; 109.8; 53.7; 47.0; 36.4; 25.2; 14.5; 12.6. ESI−/MS: m/z 487 (M-H)−, ESI−-MS/MS: m/z 376 (77), 333 (100).
(R)-3-(4-Cyanophenyl)-N-[[1-(3-chloro-4-fluorophenyl)cyclopropyl]methyl]-2-[3-(4-fluorophenyl)ureido]propanamide ((R)-17)
15% Yield. 1H NMR (DMSO-d6): δ 0.73–0.89 (m, 4H), 2.77 (dd, 1H, J= 13.7, 7.8 Hz), 2.93 (dd, 1H, J= 13.7, 5.3 Hz), 3.15 (dd, 1H, J= 14.2, 5.4 Hz), 3.44 (dd, 1H, J= 14.2, 7 Hz), 4.48–4.53 (m, 1H), 6.30 (br d, 1H), 7.01–7.06 (m, 2H), 7.25 (t, 2H, J= 7.8 Hz), 7.29–7.33 (m, 4H), 7.45 (dt, 1H, J= 6.0, 2.0 Hz), 7.69 (d, 2H, J= 6.4 Hz), 8.24 (br t, 1H), 8.63 (s, 1H). 13C NMR (DMSO-d6): 171.5; 158.3; 157,1; 156.4; 155.2; 154.9; 144.0; 141.6; 136.9; 132.3; 131.0; 130.7; 129.5; 119.5; 119.3; 116.9; 116.7; 115.7; 115.5; 109.6; 53.7; 46.9; 25.13; 12.7; 12.6. ESI−/MS: m/z 507 (M-H)−, ESI−-MS/MS: m/z 396 (92), 353 (100).
2. Stability Assays in Rat Liver Microsomes
Test compounds were pre-incubated at 37 °C with rat liver microsomes (Tebu-Bio, Milan, Italy) (1.0 mg/mL microsomal protein) at 10 μM final concentration in 100 mM potassium phosphate buffer (pH 7.4) for 10 min. Metabolic reactions were initiated by the addition of the NADPH regenerating system (containing 10 mM NADP, 50 mM glucose-6-phosphate, and 10 unit/mL glucose-6-phosphate dehydrogenase, final glucose-6-phosphate dehydrogenase concentration, 1 unit/mL). Aliquots were removed at specific time endpoints (0, 5, 15, 30 and 60 min) and immediately mixed with an equal volume of cold acetonitrile containing the internal standard. Test compound incubated with microsomes without NADPH regenerating system was included. Quenched samples were centrifuged at 4500 rpm for 15 min and the supernatants were injected for quantification analysis. Samples (100 μL) were analyzed by using an Agilent 1260 Infinity Binary LC System equipped with a diode array detector (Open Lab software was used to analyze the chromatographic data) and a Phenomenex Gemini C-18 column (250 × 4.6 mm, 5 μm particle size). The samples were eluted using CH3CN/20 mM ammonium formate pH 5.5 (70:30, v/v) as eluent (1 mL/min). Concentrations were quantified by measuring the area under the peak.
The percentage of the parent compound remaining after a 30-min incubation has been calculated according to the equation:
where Cparent is ligand concentration after incubation with microsome fraction and NADPH regenerating system and Ccontrol is ligand concentration after incubation with microsome fraction only.
The in vitro half life (t1/2) was calculated using the expression t1/2=0.693/b, where b is the slope found in the linear fit of the natural logarithm of the fraction remaining of the parent compound vs incubation time [55]. In vitro half-life was then used to calculate the intrinsic plasma clearance (CLint) according to the following equation:
3. Ca2+ Mobilization Assay in Transfected HL-60 Cells and Human Neutrophils
Cell Culture
Human promyelocytic leukemia HL-60 cells stably transfected with FPR1 (FPR1-HL-60 cells) or FPR2 (FPR2-HL-60 cells) (kind gifts from Dr. Marie-Josephe Rabiet, INSERM, Grenoble, France) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg/ml streptomycin, 100 U/ml penicillin, and G418 (1 mg/mL), as described previously [33]. Wild-type HL-60 cells were cultured under the same conditions, but without G418.
Isolation of Human Neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as previously described [56]. Isolated neutrophils were washed twice and resuspended in HBSS without Ca2+ and Mg2+ (HBSS−). Neutrophil preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion.
Ca2+ Mobilization Assay
Changes in intracellular Ca2+ were measured with a FlexStation II scanning fluorometer (Molecular Devices, Sunnyvale, CA) for human neutrophils and HL-60 cells, as described previously [33]. All active compounds were evaluated in parent (wild-type) HL-60 cells for supporting that the agonists are inactive in non-transfected cells. Human neutrophils or HL-60 cells, suspended in HBSS− containing 10 mM HEPES, were loaded with Fluo-4 AM dye (Invitrogen, Carlsbad, CA) (1.25 μg/mL final concentration) and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS− containing 10 mM HEPES, resuspended in HBSS containing 10 mM HEPES and Ca2+ and Mg2+ (HBSS+), and aliquotted into the wells of a flat-bottomed, half-area-well black microtiter plates (2 × 105 cells/well). The compound of interest was added from a source plate containing dilutions of test compounds in HBSS+, and changes in fluorescence were monitored (λex= 485 nm, λem= 538 nm) every 5 s for 240 s at room temperature after automated addition of compounds. Maximum change in fluorescence, expressed in arbitrary units over baseline, was used to determine agonist response. Responses were normalized to the response induced by 5 nM fMLF for FPR1-HL-60 cells and neutrophils, or 5 nM WKYMVM for FPR2-HL-60 cells, which were assigned a value of 100%. Curve fitting (5–6 points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose–response curves generated using Prism 5 (GraphPad Software Inc., San Diego, CA).
4. Evaluation of Anti-inflammatory Properties in Rat Primary Microglial Cell Cultures
Cell culture
Primary cultures of microglial cells were prepared from cortices of 1–2-day-old Sprague-Dawley rat pups as previously described [3]. Briefly, after decapitation, brains were removed immediately, and cerebral cortices were cut into small pieces. The minced tissue was incubated in dissecting medium HBSS (Gibco, USA) containing glucose, BSA and HEPES with 0.025% trypsin at 37 °C for 20 min. The trypsinization process was stopped by adding trypsin inhibitor from Glycine max (soybean) (Sigma-Aldrich, USA). A completely dissociated suspension of the tissue was prepared by mild trituration. Next, cells were plated at the density of 3x105 cells/cm2 in culture medium consisting of DMEM with GlutaMax and high glucose (4.5 g/L) supplemented with heat-inactivated 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin on poly-L-lysine-coated 75-cm2 culture flasks. After 3 days, culture medium was removed and replaced with fresh medium. On the 9th day in vitro (37 °C, 5% CO2), flasks were agitated on a horizontal shaker. After centrifugation, cells were resuspended in culture medium, and cell viability was determined by trypan blue exclusion. The cells were plated at a final density of 2x105 cells/well in 24-well plates or 4x104 cells/well in 96-well plates. The purity of microglial cell cultures was assessed using an anti-Iba-1 antibody and anti-CD11b antibody. More than 95% of cells were stained positively. Two days after plating, the cells were used for experiments.
Cell treatment
In all experiments, cells were pre-treated for 1 h with various concentrations of FPR2 agonists (S)-10, (R)-11 and (S)-17 and then stimulated for 24 hours with the lipopolysaccharide (LPS; 100 ng/ml) (Escherichia coli 0111:B4, Sigma-Aldrich, USA). Control (un-stimulated) cells were treated with vehicle. Additionally, in experiments where the secretion of NO or cytokines were measured, the FPR2 antagonist WRW4 (Alomone Labs, Israel) was added 30 min before agonists (Figure 5).
Figure 5.

Schematic Diagram Representing the Schedule of the Experiments on Rat Primary Microglial Cell Cultures.
Cell viability test
Cell viability was determined by the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Sigma Aldrich, Germany) assay. Microglial cells were seeded into 96-well plates at a density of 4 × 104 per well, with 100 μL of culture medium, and incubated for 48 h to allow cell adherence. At 24 h after treatment with different concentrations of tested compounds, MTT (at 0.15 mg/mL) was added to each well and incubated for 2 h at 37 °C. Next, culture medium was discarded, and 0.1 M HCl in isopropanol was added to dissolve the formazan dye. The absorbance value was measured using a multiwell spectrophotometer Infinite® 200 PRO Detector (TECAN, Switzerland) at 570 nm. The data were normalized to the absorbance in the vehicle-treated cells (100%) and expressed as a percentage of the control ± SEM.
Lactate dehydrogenase (LDH) test
To quantify the cell death, the level of lactate dehydrogenase release from the damaged cells into the culture media was measured 24 h after treatment. Cell culture supernatants were collected from each well of the 96-well plates and were incubated with the appropriate reagent mixture according to the supplier’s instructions (Cytotoxicity Detection Kit, Roche, Germany) at room temperature for 20 min. In this test, the amount of formazan salt, formed after the conversion of lactate to pyruvate and then by reduction of tetrazolium salt, is proportional to the LDH activity in the sample. The intensity of the red color formed in the assay, measured at a wavelength of 490 nm (Infinite® 200 PRO Detector, TECAN, Switzerland) is proportional to LDH activity and also to the number of damaged cells. The data were normalized to the activity of LDH released from vehicle-treated cells (100%) and expressed as a percentage of the control ±SEM.
NO release assay
Nitric oxide (NO) secreted in microglial culture medium was measured by a Griess reaction. After 24 h of treatment of microglia, 50 μL of supernatant was collected and mixed with an equal volume of Griess reagent (0.1% N-1-naphthylethylenediamine dihydrochloride and 1% sulfanilamide in 5% phosphoric acid) in a 96-well plate and incubated for 10 min at room temperature. Absorbance was measured at 540 nm in a microplate reader (Infinite® 200 PRO Detector, TECAN, Switzerland). The data were normalized to the NO released from vehicle-treated cells (100%) and expressed as a percentage of the control ±SEM.
Enzyme-linked immunosorbent assay (ELISA)
The medium of microglial cells for TNF-α and IL-1β was collected at 24 h after treatment. The protein levels of the cytokines TNF-α, IL-1β, (R&D Systems, USA) in the culture medium were measured using commercially available enzyme-linked immunosorbent assay kits according to the manufacturers’ instructions. The detection limits were as follows: TNF-α, 5 pg/ml; IL-1β, 5 pg/ml. Inter-assay precision were as follows: TNF-α: <8.8%; IL-1β <4.4%; intra-assay precision: TNF-α: <2.1%; IL-1β: <3.9%.
5. Evaluation of permeability in hCMEC/D3 cells
Cell cultures
hCMEC/D3 cells, a primary human brain microvascular endothelial stabilized cell line, were a kind gift from Prof. Pierre-Olivier Couraud (Institut Cochin, Centre National de la Recherche Scientifique UMR 8104, INSERM U567, Paris, France) and were cultured according to Weksler et al [50]. Cells were seeded at 50,000/cm2 density, and grown for 7 days up to confluence in Transwell devices (0.4 μm diameter pores-size, Corning Life Sciences, Chorges, France), to allow the formation of a competent BBB. Before each experiment, the transendothelial electrochemical resistance (TEER) and the permeability coefficients of dextran- fluorescein isothiocyanate (FITC), [14C]-sucrose, and [14C]-inulin were measured and taken as parameters of paracellular transport across hCMEC/D3 monolayer [55]. The TEER value was between 29 and 40 Ω cm2, the dextran-FITC permeability coefficient was 0.017 ± 0.005 × 10−3 cm min−1, the [14C]-sucrose permeability coefficient was 1.17 ± 0.08 × 10−3 cm min−1, the [14C]-inulin permeability coefficient was 0.37 ± 0.08 × 10−3 cm min−1. These values supported the functional integrity of the BBB monolayer [57].
Permeability of compounds through hCMEC/D3 cell monolayer
hCMEC/D3 cells, seeded as reported above in Transwell devices, were incubated at day 7 with free medium, then washed and rinsed with sterile PBS for 2 h at 37°C. 100 μM compounds were added in the upper or lower chamber for 2 h. After this incubation time, the medium in each chamber was collected and the amount of compound recovered was measured spectrophotometrically (λ = 230 nm) using a Synergy HTX Multi-Mode Reader (Bio-Tek Instruments, Winooski, VT).
Standard calibration curves were prepared at maximum absorption wavelength of each compound using PBS as solvent and were linear (r2 = 0.999) over the range of tested concentration (from 5 to 10 μM). Each compound was tested in triplicate, and the experiments were repeated three times. Data are reported as the apparent permeability (Papp), in units of nm/s, determined as indicated in the following equation:
where Va is the volume in the acceptor well, Area is the surface area of the membrane and time is the total transport time, [drug]acceptor is the concentration of the drug measured by UV-spectroscopy, and [drug]initial is the initial drug concentration in the AP or BL chamber.
Efflux ratio (ER) was calculated using the following equation: ER =Papp, BA/Papp, AB, where Papp, BA is the apparent permeability of basal-to-apical transport, and Papp, BA is the apparent permeability of apical-to-basal transport.
Supplementary Material
HIGHLIGHTS.
Novel FPR2 agonists were synthesized and in vitro metabolic stability was evaluated
The most interesting compounds showed in vitro neuroprotective properties
(S)-17 reduced IL-1β and TNFα levels in LPS-stimulated primary rat microglia cells
(S)-17 showed good permeation rate in an in vitro model of blood brain barrier
FPR2 agonists have potential for resolving neuroinflammation in CNS disorders
Acknowledgments
FUNDING SOURCES
This research was supported in part by National Institutes of Health IDeA Program COBRE Grant GM110732; USDA National Institute of Food and Agriculture Hatch project 1009546; Montana University System Research Initiative: 51040-MUSRI2015-03; and the Montana State University Agricultural Experiment Station. This research was supported in part by the the statutory funds of Institute of Pharmacology, Polish Academy of Sciences.
The authors wish to thank Dr. Iris Chiara Salaroglio for technical assistance in permeability studies with hCMEC/D3 cells.
ABBREVIATIONS
- FPR2
Formyl peptide receptor 2
- NO
nitric oxide
- IL-1β
interleukin 1β
- LPS
lipopolysaccharide
- TNF-α
tumor necrosis factor α
Footnotes
Elemental Analysis of Target Compounds; 1H and 13C spectra of compounds (S)-10, (R)-11 and (S)-17. Effect of low doses of (S)-17 on IL-1β (A) and TNF-α (B) production in rat microglial cell cultures.
Authors contributions
E.L. and M.L. conceived the project and wrote the manuscript. MLS synthesized the compounds and EL performed metabolic stability studies. LNK and IAS performed Ca2+ mobilization assays. ABK designed the experiments on rat primary microglia cultures and analyzed the data. JS and KC performed the experiments on rat primary microglia cultures. CR performed the permeability studies. MTQ and R.P. edited and revised the manuscript. All authors read and approved the final draft of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Graeber MB, Streit WJ. Microglia: biology and phatology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- 2.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Œlusarczyk J, Trojan E, Głombik K, Budziszewska B, Kubera M, Lasoñ WW, Popiołek-Barczyk K, Mika J, Wêdzony K, Basta-Kaim A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neurosci. 2015;9:82. doi: 10.3389/fncel.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136–153. doi: 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang N, Serhan CN, Dahlén SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 9.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacy M, Jones J, Whittemore SR, Haviland DL, Wetsel RA, Barnum SR. Expression of the receptors for the C5a anaphylatoxin, interleukin-8 and FMLP by human astrocytes and microglia. J Neuroimmunol. 1995;61:71–78. doi: 10.1016/0165-5728(95)00075-d. [DOI] [PubMed] [Google Scholar]
- 11.Cui Y, Le Y, Yazawa H, Gong W, Wang JM. Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer’s disease. J Leukocyte Biol. 2002;72:628–635. [PubMed] [Google Scholar]
- 12.Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med. 2007;204:245–252. doi: 10.1084/jem.20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu G, Tang W, Wang L, Wang C, Wang X. Identification of a uniquely expanded V1R (ORA) gene family in the Japanese grenadier anchovy (Coilia nasus) Mar Biol. 2016;163:126. doi: 10.1007/s00227-016-2896-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bufe B, Schumann T, Zufall F. Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J Biol Chem. 2012;287:33644–33655. doi: 10.1074/jbc.M112.375774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei L, Zhang J, Zhao F, Su T, Wei H, Tian J, Li M, Shi J. Annexin 1 exerts anti-nociceptive effects after peripheral inflammatory pain through formyl-peptide-receptor-like 1 in rat dorsal root ganglion. Br J Anaesth. 2011;107:948–958. doi: 10.1093/bja/aer299. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Zhang L, Chen X, Xue X, Guo Q, Liu M, Zhao J. Formylpeptide receptors promote the migration and differentiation of rat neural stem cells. Sci Rep. 2016;6:25946. doi: 10.1038/srep25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abd-Elghafour BA, El-Sayed NM, Ahmed AA, Zaitone SA, Moustafa YM. Aspirin and (or) omega-3 polyunsaturated fatty acids protect against corticohippocampal neurodegeneration and downregulate lipoxin A4 production and formyl peptide receptor-like 1 expression in pentylenetetrazole-kindled rats. Can J Physiol Pharmacol. 2017;95:340–348. doi: 10.1139/cjpp-2016-0060. [DOI] [PubMed] [Google Scholar]
- 18.Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180:253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooray SN, Thomas Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJL, Flower RJ, Perretti M. Ligand-specific conformational change of the G-protein–coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci. 2013;110:18232–18237. doi: 10.1073/pnas.1308253110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devchand PR, Arita M, Hong S, Bannenberg G, Moussignac RL, Gronert K, Serhan CN. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17:652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- 21.Sordi R, Menezes-de-Lima O, Horewicz V, Scheschowitsch K, Santos LF, Assreuy J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int Immunopharmacol. 2013;2:283–292. doi: 10.1016/j.intimp.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D’Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perretti M, Leroy X, Bland EJT. Montero-Melendez Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci. 2015;36:737–755. doi: 10.1016/j.tips.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. 2015;760:49–63. doi: 10.1016/j.ejphar.2015.03.083. [DOI] [PubMed] [Google Scholar]
- 25.Martini AC, Berta T, Forner S, Chen G, Bento AF, Ji RR, Rae GA. Lipoxin A4 inhibits microglial activation and reduces neuroinflammation and neuropathic pain after spinal cord hemisection. J Neuroinflammation. 2016;13:75. doi: 10.1186/s12974-016-0540-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Z, Hu Q, Xu L, Guo ZN, Ou Y, He Y, Yin C, Sun X, Tang J, Zhang JH. Lipoxin A4 reduces inflammation through formyl peptide receptor 2/p38 MAPK signaling pathway in subarachnoid hemorrhage rats. Stroke. 2016;47:490–497. doi: 10.1161/STROKEAHA.115.011223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medeiros R, Kitazawa M, Passos GF, Baglietto-Vargas D, Cheng D, Cribbs DH, LaFerla FM. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182:1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corminboeuf O, Leroy X. FPR/ALXR agonists and the resolution of inflammation. J Med Chem. 2015;58:537–559. doi: 10.1021/jm501051x. [DOI] [PubMed] [Google Scholar]
- 29.Frohn M, Xu H, Zou X, Chang C, McElvaine M, Plant MH, Wong M, Tagari P, Hungate R, Burli RW. New ‘chemical probes’ to examine the role of the hFPRL1 (or ALXR) receptor in inflammation. Bioorg Med Chem Lett. 2007;17:6633–6637. doi: 10.1016/j.bmcl.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 30.Forsman H, Kalderen C, Nordin A, Nordling E, Jensen AJ, Dahlgren C. Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem Pharmacol. 2011;81:402–411. doi: 10.1016/j.bcp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 31.He M, Cheng N, Gao WW, Zhang M, Zhang YY, Ye RD, Wang MW. Characterization of Quin-C1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol Sin. 2011;32:601–610. doi: 10.1038/aps.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burli RW, Xu H, Zou X, Muller K, Golden J, Frohn M, Adlam M, Plant MH, Wong M, McElvain M, Regal K, Viswanadhan VN, Tagari P, Hungate R. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett. 2006;16:3713–3718. doi: 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 33.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Leopoldo M, Lucente E, Lacivita E, De Giorgio P, Quinn MT. 3-(1H-Indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: molecular modeling of chiral recognition by FPR2. Biochem Pharmacol. 2013;85:404–416. doi: 10.1016/j.bcp.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacivita E, Schepetkin IA, Stama ML, Kirpotina LN, Colabufo NA, Perrone R, Khlebnikov AI, Quinn MT, Leopoldo M. Novel 3-(1H-indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]propanamides as selective agonists of human formyl-peptide receptor 2. Bioorg Med Chem. 2015;23:3913–3924. doi: 10.1016/j.bmc.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He HQ, Liao D, Wang ZG, Wang ZL, Zhou HC, Wang MW, Ye RD. Functional characterization of three mouse formyl peptide receptors. Mol Pharmacol. 2013;83:389–398. doi: 10.1124/mol.112.081315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kao W, Gu R, Jia Y, Wei X, Fan H, Harris J, Zhang Z, Quinn J, Morand EF, Yang YH. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. Br J Pharmacol. 2014;171:4087–4096. doi: 10.1111/bph.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lacivita E, Podlewska S, Speranza L, Niso M, Satała G, Perrone P, Perrone-Capano C, Bojarski AJ, Leopoldo M. Structural modifications of the serotonin 5-HT7 receptor agonist N-(4-cyanophenylmethyl)-4-(2-diphenyl)-1-piperazinehexanamide (LP-211) to improve in vitro microsomal stability: a case study. Eur J Med Chem. 2016;120:363–379. doi: 10.1016/j.ejmech.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Huang A, Moretto A, Janz K, Lowe M, Bedard PW, Tam S, Di L, Clerin V, Sushkova N, Tchernychev B, Tsao DHH, Keith JC, Jr, Shaw GD, Schaub RG, Wang Q, Kaila N. Dicovery of 2-[1-(4-chlorophenyl)cyclopropyl]-3-hydroxy-8-(trifluoromethyl)quinoline-4-carboxylic acid (PSI-421), a P-selectin inhibitor with improved pharmacokinetic properties and oral efficacy in models of vascular injury. J Med Chem. 2010;53:6003–6017. doi: 10.1021/jm9013696. [DOI] [PubMed] [Google Scholar]
- 39.Galley G, Goergler A, Groebke Zbinden K, Norcross R. 4,5-Dihydro-oxazol-2yl derivatives. US2010/29589. Patent. 2010
- 40.Pivetti F, Fornaretto MG, Re M. Process for the preparation of derivatives of 1-(2- halobiphenyl-4-yl)-cyclopropanecarboxylic acid. US2011039934. Patent. 2011
- 41.Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Identification of novel small-molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol Pharmacol. 2010;77:159–170. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di L, Kerns EH, Ma XJ, Huang Y, Carter GT. Applications of high throughput microsomal stability assay in drug discovery. Comb Chem High Throughput Screening. 2008;11:469–476. doi: 10.2174/138620708784911429. [DOI] [PubMed] [Google Scholar]
- 43.Molteni M, Gemma S, Rossetti C. The role of Toll-Like receptor 4 in infectious and noninfectious inflammation. Mediators Inflammation. 2016;2016:6978936. doi: 10.1155/2016/6978936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 45.Rousseau MC, Hsu RY, Spicer JD, McDonald B, Chan CH, Perera RM, Giannias B, Chow SC, Rousseau S, Law S, Ferri LE. Lipopolysaccharide-induced toll-like receptor 4 signaling enhances the migratory ability of human esophageal cancer cells in a selectin-dependent manner. Surgery. 2013;154:69–77. doi: 10.1016/j.surg.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 46.Slusarczyk J, Trojan E, Glombik K, Piotrowska A, Budziszewska B, Kubera M, Popiolek-Barczyk K, Lason W, Mika J, Basta-Kaim A. Anti-inflammatory properties of tianeptine on lipopolysaccharide-induced changes in microglial cells involve toll-like receptor-related pathways. J Neurochem. 2016;136:958–970. doi: 10.1111/jnc.13452. [DOI] [PubMed] [Google Scholar]
- 47.Džoljić E, Grbatinić I, Kostić V. Why is nitric oxide important for our brain? Funct Neurol. 2015;30:159–163. doi: 10.11138/FNeur/2015.30.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuste JE, Tarragon E, Campuzano CM, Ros-Bernal FF. Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci. 2015;9:322. doi: 10.3389/fncel.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marin I, Kipnis J. Learning and memory … and the immune system. Learn Mem. 2013;20:601–606. doi: 10.1101/lm.028357.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M, Tricoire-Leignel H, Nicotra A, Bourdoulous S, Turowski P, Male DK, Roux F, Greenwood J, Romero IA, Couraud PO. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 51.Hitchcock SA. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J Med Chem. 2012;55:4877. doi: 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]
- 52.Ries M, Loiola R, Shah UN, Gentleman SM, Solito E, Sastre MM. The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-β peptide. J Neuroinflammation. 2016;13:234. doi: 10.1186/s12974-016-0692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marinier A, Quesnelle CA, Dodier M, Roy S, Gill P, Wittman MD, Langley DR. Thiazolyl compounds useful as kinase inhibitors. US2010/048581. Patent. 2010
- 54.Lacivita E, Lucente E, Kwizera C, Antunes IF, Niso M, De Giorgio P, Perrone R, Colabufo NA, Elsinga PH, Leopoldo M. Structure-activity relationship study towards non-peptidic positron emission tomography (PET) radiotracer for gastrin releasing peptide receptors: Development of [18F] (S)-3-(1H-indol-3-yl)-N-[1-[5-(2-fluoroethoxy)pyridin-2-yl]cyclohexylmethyl]-2-methyl-2-[3-(4-nitrophenyl)ureido]propionamide. Bioorg Med Chem. 2017;25:277–292. doi: 10.1016/j.bmc.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 55.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 56.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Quinn MT. High-throughput screening for small-molecule activators of neutrophils: Identification of novel N-formyl peptide receptor agonists. Mol Pharmacol. 2007;71:1061–1074. doi: 10.1124/mol.106.033100. [DOI] [PubMed] [Google Scholar]
- 57.Riganti C, Salaroglio IC, Pinzòn-Daza ML, Caldera V, Campia I, Kopecka J, Mellai M, Annovazzi L, Couraud PO, Bosia A, Ghigo D, Schiffer D. Temozolomide down-regulates P-glycoprotein in human blood-brain barrier cells by disrupting Wnt3 signaling. Cell Mol Life Sci. 2014;71:499–516. doi: 10.1007/s00018-013-1397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.