

Abstract
We report a fast Staudinger reaction between perfluoroaryl azides (PFAAs) and aryl phosphines, occurring readily under ambient conditions. A rate constant as high as 18 M−1 s−1 was obtained between methyl 4-azido-2,3,5,6-tetrafluorobenzoate and methyl 2-(diphenylphosphanyl)benzoate in CD3CN/D2O. In addition, the iminophosphorane product was stable toward hydrolysis and aza-phosphonium ylide reactions. The PFAA-Staudinger reaction proved to be an excellent bioothorgonal reaction. PFAA-derivatized mannose and galactose were successfully transformed into cell surface glycans and efficiently labeled with phosphine-derivatized fluorescent bovine serum albumin.
Keywords: Staudinger reaction, perfluoroaryl azide, bioorthogonal reaction, metabolic labelling
COMMUNICATION
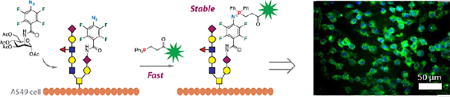
Perfluoroaryl azides (PFAAs) react with aryl phosphines in a fast reaction under ambient conditions, yielding kinetically stable iminophosphorane products. The PFAA-Staudinger reaction proved to be an excellent bioorthogonal reaction and was successfully applied to cell surface labeling.
The Staudinger reaction, the formation of an iminophosphorane from an azide and a phosphine,1 proceeds through nucleophilic attack of the phosphine on the distal nitrogen of the azide to form the phosphazide intermediate, which decomposes to the iminophosphorane upon release of N2.2,3 When exposed to water, the iminophosphorane is readily hydrolyzed to the corresponding amine and phosphine oxide. The reaction has recently gained renewed interest, owing to the search for highly efficient bioorthogonal reactions.4
The pioneering work applying the Staudinger reaction as a bioorthogonal reaction involves the introduction of an electrophilic ester trap on the phenyl phosphine (Fig. 1A).5 In the resulting Staudinger ligation reaction, the nucleophilic iminophosphorane attacks the ester intramolecularly to form a cyclic intermediate, which, upon hydrolysis, gives the conjugated amide product.6 The reaction has been successfully applied to metabolic engineering, where azide-derivatized N-acetylmannosamine was converted to sialic acid derivatives, allowing the expression of azido groups on the cell surface. Subsequent Staudinger ligation with biotin-derivatized phosphine followed by fluorescent avidin allowed the detection of cell surface glycans by flow cytometry. Although high background noise was often observed,7 the Staudinger ligation is considered superior for in vivo labelling compared to the strain-promoted alkyne-azide cycloaddition (SPAAC) reaction, where, for example, the alkynes could bind strongly to murine serum albumin and were sequestered from azide-labelled tissues.8
Figure 1.

A) Staudinger ligation. B) PFAA-Staudinger reaction leading to stable iminophosphorane.
Mechanistic studies on the Staudinger reaction between phenyl azides and triphenylphosphines revealed that the reaction rate can be enhanced by introducing electron-donating groups on the phosphine or electron-withdrawing groups on the phenyl azide.2,6 Since electron-rich phosphines are more susceptible to oxidation in the biological environment,8 the alternative of using electron-deficient azides would be a more logical solution. It was reported that iminophosphoranes formed from electron-deficient azides were more stable, and had lower hydrolysis rates.6,9–11 The P=N bond can be stabilized by steric hindrance.12,13 Encouraged by these findings, we envisaged efficient Staudinger reactions using perfluoroaryl azides (PFAAs). The F atoms lower the LUMO of the aryl azides, giving PFAAs unique reactivities towards, for example, enamines, thioacids and aldehydes.14–19 Here, we report that PFAAs undergo fast Staudinger reactions with aryl phosphines under ambient conditions to give stable iminophosphoranes (Fig. 1B). In addition, the reaction showed excellent bioothorgonality and was successfully applied for cell surface labeling.
When an equimolar amount of PFAA 1a and phosphine 2a (Fig. 2A) were mixed in acetonitrile (100 mM) at room temperature, the solution immediately turned yellow. The color remained for 4 min and was followed by evolution of nitrogen gas. The product, iminophosphorane 3aa, was obtained in quantitative yield (≥99%), and the structure was confirmed by single crystal x-ray crystallography (Fig. 2B).
Figure 2.

A) Reaction between 1a and 2a. B) X-ray single crystal structure of 3aa. C) Kinetic analysis; conditions: [1a]0 = 2.5 mM, [2a]0 = 2.5 mM, CD3CN, 25 °C; concentrations monitored by 1H NMR with spectra recorded every 60 s; kobs (2nd order): 3.68 ± 0.03 M−1 s−1
The Staudinger reaction can proceed through first- or second-order kinetics depending on whether the rate-limiting step is unimolecular decomposition or bimolecular formation of the intermediate phosphazide.20 In the present case, the reaction between 1a and 2a followed second order as can be seen from the kinetic analysis (Fig. 2C). The observed rate constant, 3.68 ± 0.03 M−1s−1, is six times higher than that between 1-azido-4-nitrobenzene and triphenylphosphine (0.611 M−1s−1),2 the fastest Staudinger reaction reported, and 1940 times higher than that of the classic Staudinger ligation reaction (kobs= 1.9 × 10−3 M−1s−1).6
The effect of solvent on the reaction rate was next investigated (Table 1). In general, the observed rate constant increased with the polarity of the solvent (Entries 1–4). Consistent with this observation is the rate acceleration with the addition of D2O (Entries 5–7), where an observed rate constant of 18.3 M−1s−1 was obtained with 50% D2O (v/v) (Entry 7). These results are in agreement with a polar transition state that is more stabilized in polar solvents.2
Table 1.
Effect of solvent on reaction rate between PFAA 1a and phosphine 2a.
| Entry | Solvent | kobs (M−1s−1)a | Yield (%)b |
|---|---|---|---|
| 1 | CDCl3 | 0.83 ± 0.03 | 79 |
| 2 | CD3CN | 3.68 ± 0.03 | 91 |
| 3 | (CD3)2CO | 2.37 ± 0.03 | 91 |
| 4 | CD3OD | 5.7 ± 0.1 | 95 |
| 5 | CD3CN/D2O (5/1) | 6.5 ± 0.1 | 97 |
| 6 | CD3OD/D2O (5/1) | 11.3 ± 0.3 | 96 |
| 7 | CD3CN/D2O (1/1) | 18.3 ± 0.7 | 97 |
2nd order; reactions followed with 1H NMR by monitoring the methyl protons in PFAA 1a over 30–45 min.
NMR yield at 30 min.
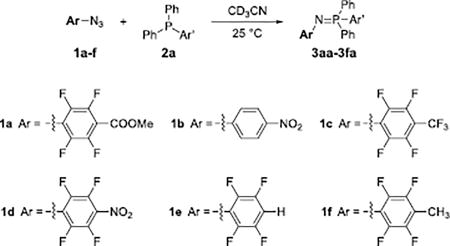
The scope of the PFAA structures was studied, and high reaction rates were observed for all PFAAs tested (Table 2). Furthermore, the more electron-withdrawing the substituent on the PFAA core, the higher the reaction rate is consistent with the kinetics of the Staudinger reaction.2 In addition, the reactions proved efficient, and high conversions (> 90%) were achieved under ambient conditions in all cases. Hammett analysis performed on the PFAAs (Graph S1) showed a linear correlation (R2 = 0.99) with a small, positive ρ value (0.43). These results suggest a buildup of negative charge and a small influence of the PFAA’s para-substituent on the reaction rate. The reaction was repeated with 1-azido-4-nitrobenzene (1b), the non-fluorinated analog of compound 1d. This reaction was not completed after 30 min (Entry 2, Table 2), and displayed a 32 times lower rate in comparison to the reaction with its perfluorinated analog 1d (Entry 4).
Table 2.
Scope of PFAAs.
| |||
|---|---|---|---|
|
| |||
| Entry | Azide | kobs (M−1s−1)a | Yield (%)b |
| 1 | 1a | 3.68 ± 0.03 | 91 |
| 2 | 1b | 0.153 ± 0.001 | 41 |
| 3 | 1c | 3.92 ± 0.07 | 95 |
| 4 | 1d | 4.97 ± 0.08 | 95 |
| 5 | 1e | 2.73 ± 0.06 | 93 |
| 6 | 1f | 0.91 ± 0.01 | 82 |
2nd order; reactions were followed with 1H NMR over the course of 30 min.
NMR yield at 30 min.
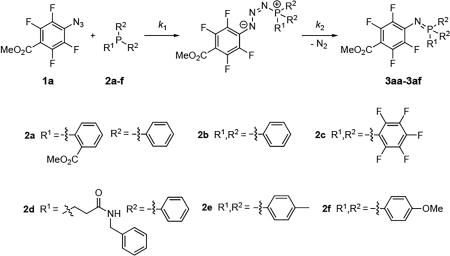
The scope of the phosphine was next investigated (Table 3). Introducing an electron-withdrawing methyl ester on the phenyl group increased the rate slightly (Entry 1 vs. Entry 2). Substituting the phenyl moieties with pentafluorophenyl groups (2c) strongly impeded the reaction as no product was observed after 30 min (Entry 3). This is consistent with the classic Staudinger reaction in which the rate decreases when using an electron-poor phosphine.2 When electron-donating groups were introduced (2d, 2e, 2f), the phosphine was immediately converted into the phosphazide (Fig. S16A–S18A) upon mixing with PFAA 1a. The decomposition of phosphazide was in this case the rate-limiting step, following first order kinetics (Fig. S16). The rate of phosphazide decomposition decreased with the electron donating ability of the aryl substituent (Entries 4–6). For the reaction with 2d, the isolated yield was lower in comparison to the triphenyl counterpart (63%) due to the production of oxidized phosphine.
Table 3.
Scope of phosphines
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Phosphine | k1,obs (M−1s−1)a | k2,obs × 103 (s−1)a | Yield (%)b |
| 1 | 2a | 3.68 ± 0.03 | - | 91 |
| 2 | 2b | 6.1 ± 0.7 | 4.1 ± 0.1 | 93 |
| 3 | 2c | - | - | NRc |
| 4 | 2d | - | 1.56 ± 0.02 | 96 |
| 5 | 2e | - | 1.31 ± 0.01 | 89 |
| 6 | 2f | - | 0.56 ± 0.01 | 62 |
Monitored by 1H NMR.
NMR yield at 30 min.
No reaction during the course of 30 min.
The iminophosphorane products proved stable over time. When solutions of 3aa or 3ab in CD3CN were stored under ambient conditions (Fig. S6–S7), or when 10% D2O was added (Fig. S8–S9), the 1H NMR spectra did not change during a course of 35 days. This demonstrates the high stability of the iminophosphorane towards hydrolysis. Compounds 3aa or 3ab were then mixed with an excess of CS2 or p-anisaldehyde in CD3CN. After 35 days, no new peaks were detected in the 1H NMR spectra (Fig. S10–S13), demonstrating the inability of these iminophosphoranes to undergo aza-Wittig reactions in consequence to the low nucleophilicity of the fluorinated iminophosphoranes.
The PFAA-Staudinger reaction was next tested for its bioorthogonality (Fig. 3), employing the widely-studied N-acetylneuraminic acid (Neu5Ac) metabolic pathway that also accommodates other monosaccharides.5,21 PFAA-derivatized acetylated Man (4a), Gal (4b), and Glc (4c) (Fig. 3) were synthesized by a straightforward protocol (cf. Supporting Information). The labelling reagents, P2g-BSA-FITC or P2h-BSA-FITC, were synthesized by treating BSA with fluorescein isothiocyanate (FITC) followed by phosphines 2g or 2h, and purified by dialysis. BSA was chosen for its ability to reduce non-specific protein adsorption. A549 cells were incubated with 4a, 4b, or 4c for 3 days,5 washed, and treated with P2g-BSA-FITC for 30 min. The extent of cell labelling was analysed using flow cytometry. Results showed that cells treated with 0.2 mM of 4a had ~4 times higher fluorescence intensity than the control (0 mM, Fig. 4A), indicating the successful expression of PFAA on the cell surface.
Figure 3.

Cell surface labelling experiment using PFAA-Staudinger reaction.
Figure 4.

Fluorescence intensity of labelled cells measured by flow cytometry. (A) A549 cells were incubated with 4a at 0, 0.04, 0.09, 0.2 mM for 3 days followed by P2g-BSA-FITC (1 µM) for 30 min. (B) A549 cells were incubated with 4a at 0, 0.09, 0.2 mM, 4b at 0.2 mM or 4c at 0.2 mM for 3 days followed by P2h-BSA-FITC (1 µM) for 30 min.
The experiment was then repeated using P2h-BSA-FITC following the same protocol as above, and the labeled cells were analyzed by flow cytometry. Similar to the results of P2g-BSA-FITC, cells treated with 0.2 mM of 4a displayed the highest fluorescence intensity (Fig. 4B). Furthermore, P2h-BSA-FITC showed higher labeling efficiency, displayed ~11 times higher intensity than the control (0 mM) compared to P2g-BSA-FITC (~4). P2h-BSA-FITC was thus chosen as the labeling agent to test PFAA 4b and 4c. As can be seen from Fig. 4B, 4b was as efficient as 4a, whereas 4c was much less efficient. Similar low labeling efficiency of GlcNAc derivatives was also observed by others, speculated as the restriction of GlcNAc into the sialic acid pathway, and the ability of GlcNAc analogs to produce multiple metabolic products.21–23
Fluorescence microscopy was furthermore used to image the labeled cells. For this, A549 cells were first incubated with 4a at 0.29 mM for 3 days and then treated with P2h-BSA-FITC for 1, 5, 10, 15, or 30 min. The green fluorescence that resulted from P2h-BSA-FITC was clearly seen on the cells (Fig. 5). Successful labeling was already visible at 5 min. High fluorescence intensities were observed at 15 min, and remained unchanged after 30 min.
Figure 5.

Fluorescence images of labelled cells. Cells were incubated with 0.3 mM 4a for 3 days, followed by P2h-BSA-FITC for 1, 5, 10, 15, or 30 min.
A549 cells were then incubated with 4a, 4b or 4c for 3 days, followed by P2h-BSA-FITC for 30 min, and the cell nuclei were stained with Hoechst 33342 which fluoresces blue. Green fluorescence was clearly seen on the cells (Fig. 6, I), where the signals were strong in the case of 4a and 4b (Fig. 6A, 6B), and much weaker in the case of 4c (Fig. 6C). This is consistent with the flow cytometry results (Fig. 4B) showing the lower labelling efficiency for 4c. Control samples prepared without treating the cells with 4 showed no visible green fluorescence (Fig. 6D).
Figure 6.

Fluorescence images of A549 cells treated for 3 days with 0.3 mM of (A) 4a, (B) 4b, (C) 4c, or (D) none, followed by 1 µM of P2h-BSA-FITC at 4 °C for 30 min. The cell nuclei were stained with Hoechst 33342 dye. I: Excitation at 488 nm showing the green FITC. II: Excitation at 358 nm showing the blue stained nuclei. III: Overlay of I and II.
In summary, we have developed a fast PFAA-Staudinger reaction, with rate constants of up to 18 M−1s−1 in CD3CN/D2O (1/1). The iminophosphorane product was found stable towards hydrolysis thus eliminating the need of an ester trap in the Staudinger ligation. The iminophosphorane was also inert towards aza-Wittig reagents. Furthermore, PFAA-derivatized ManNAc and GalNAc were taken up by A549 cells and successfully probed by the PFAA-Staudinger reaction. The labelling reaction was fast, carried out using low concentration of the labelling agent (1 µM) tagged with a conventional dye (FITC), affording clear images with low background noises.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (R01GM080295 and R21AI109896, to MY) for the financial support of this work. We acknowledge Dr. Andreas Fischer for his contribution in performing X-ray crystallography, and Dr. Nanjing Hao for his help in fluorescence microscopy.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Staudinger H, Meyer J. Helv. Chim. Acta. 1919;2:635. [Google Scholar]
- 2.Leffler JE, Temple RD. J. Am. Chem. Soc. 1967;89:5235. [Google Scholar]
- 3.Tian WQ, Wang YA. J. Chem. Theory Comput. 2005;1:353. doi: 10.1021/ct049918x. [DOI] [PubMed] [Google Scholar]
- 4.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Saxon E, Bertozzi CR. Science. 2000;287:2007. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 6.Lin FL, Hoyt HM, van Halbeek H, Bergman RG, Bertozzi CR. J. Am. Chem. Soc. 2005;127:2686. doi: 10.1021/ja044461m. [DOI] [PubMed] [Google Scholar]
- 7.Chang PV, Prescher JA, Hangauer MJ, Bertozzi CR. J. Am. Chem. Soc. 2007;129:8400. doi: 10.1021/ja070238o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sletten EM, Bertozzi CR. Acc. Chem. Res. 2011;44:666. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Berkel SS, van Eldijk MB, van Hest JCM. Angew. Chem. Int. Ed. 2011;50:8806. doi: 10.1002/anie.201008102. [DOI] [PubMed] [Google Scholar]
- 10.Banks RE, Sparkes GR. J. Chem. Soc., Perkin Trans. 1972;1:2964. [Google Scholar]
- 11.Banks RE, Prakash A. J. Chem. Soc., Perkin Trans. 1974;1:1365. [Google Scholar]
- 12.Palacios F, Alonso C, Aparicio D, Rubiales G, de los Santos JM. Tetrahedron. 2007;63:523. [Google Scholar]
- 13.Kennedy RD. Chem. Commun. 2010;46:4782. doi: 10.1039/c0cc00426j. [DOI] [PubMed] [Google Scholar]
- 14.Dommerholt J, van Rooijen O, Borrmann A, Guerra CF, Bickelhaupt FM, van Delft FL. Nat Commun. 2014;5 doi: 10.1038/ncomms6378. [DOI] [PubMed] [Google Scholar]
- 15.Xie S, Lopez SA, Ramström O, Yan M, Houk KN. J. Am. Chem. Soc. 2015;137:2958. doi: 10.1021/ja511457g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie S, Fukumoto R, Ramström O, Yan M. J. Org. Chem. 2015;80:4392. doi: 10.1021/acs.joc.5b00240. [DOI] [PubMed] [Google Scholar]
- 17.Xie S, Ramström O, Yan M. Org. Lett. 2015;17:636. doi: 10.1021/ol503655a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie S, Zhang Y, Ramstrom O, Yan M. Chem. Sci. 2016;7:713. doi: 10.1039/c5sc03510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie S, Manuguri S, Proietti G, Romson J, Fu Y, Inge AK, Wu B, Zhang Y, Häll D, Ramström O, Yan M. Proc. Natl. Acad. Sci. 2017 doi: 10.1073/pnas.1708556114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leffler JE, Tsuno Y. J. Org. Chem. 1963;28:902. [Google Scholar]
- 21.Wratil PR, Horstkorte R, Reutter W. Angew. Chem. Int. Ed. 2016;55:9482. doi: 10.1002/anie.201601123. [DOI] [PubMed] [Google Scholar]
- 22.Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, Yarema KJ. Glycobiology. 2009;19:1382. doi: 10.1093/glycob/cwp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, Bertozzi CR. J. Am. Chem. Soc. 2002;124:14893. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.