

Abstract
Mycobacteria, including the bacterial pathogen that causes human tuberculosis, possess distinctive pathways for synthesizing and utilizing the non-mammalian disaccharide trehalose. Trehalose metabolism is essential for mycobacterial viability and has been linked to in vitro biofilm formation, which may bear relevance to in vivo drug tolerance. Previous research has shown that some trehalose analogues bearing modifications at the 6-position inhibit growth of various mycobacterial species. In this work, 2-, 5-, and 6-position-modified trehalose analogues were synthesized using our previously reported one-step chemoenzymatic method and shown to inhibit growth and biofilm formation in the two- to three-digit micromolar range in Mycobacterium smegmatis. The trehalose-specific ABC transporter LpqY-SugABC was essential for antimicrobial and anti-biofilm activity, suggesting that inhibition by monosubstituted trehalose analogues requires cellular uptake and does not proceed via direct action on extracellular targets such as antigen 85 acyltransferases or trehalose dimycolate hydrolase. Although the potency of the described compounds in in vitro growth and biofilm assays is moderate, this study reports the first trehalose-based mycobacterial biofilm inhibitors and reinforces the concept of exploiting unique sugar uptake pathways to deliver inhibitors and other chemical cargo to mycobacteria.
TOC image
Introduction
Tuberculosis (TB), which is caused by the bacterial pathogen Mycobacterium tuberculosis, remains a major global health problem. Every year, 10 million incident cases of TB occur, of which half a million are multi- or extensively-drug-resistant forms (MDR- and XDR-TB).[1] Although most cases of TB are curable, the disease still claims an estimated 1.5 million lives annually.[1] A contributing factor to these problems is the complex and lengthy treatment used to clear M. tuberculosis infection, which for drug-susceptible TB requires a combination of 3–4 drugs delivered over the course of 6–9 months.[2] Such challenging treatment regimens, coupled with increasing antibiotic resistance, underscore the need for new anti-tubercular compounds. As discussed recently, trehalose metabolism has received increasing attention as a target for the development of novel anti-tubercular agents.[3,4] Trehalose (1), a non-mammalian disaccharide, is essential for mycobacterial viability and virulence. Trehalose is responsible for the transport of mycolic acids—long-chain (C60–C90), α-branched, β-hydroxy fatty acids—to the exterior of the cell, where they are used to construct the thick, hydrophobic mycobacterial outer membrane, or “mycomembrane.”[5] As shown in Figure 1, cytoplasmic trehalose is converted to trehalose monomycolate (TMM),[6] which is then transported across the plasma membrane by MmpL3.[7,8] TMM is then processed by antigen 85 (Ag85) mycoloyltransferases, producing the major glycoconjugates of the mycomembrane: trehalose dimycolate (TDM) and arabinogalactan mycolate (AGM).[9–12] Free trehalose released through Ag85 catalysis is recycled back into the cell by the trehalose-specific ABC transporter LpqY-SugABC.[13] In addition, trehalose was recently implicated in mycobacterial biofilm formation. As revealed by Ojha and co-workers in 2010, TDM can be broken down by TDM hydrolase (TDMH) to release free mycolic acid,[14] which serves as a pivotal component of the biofilm extracellular matrix in mycobacteria.[14,15] Deletion of TDMH impairs biofilm formation in M. smegmatis, suggesting that trehalose-mediated mycolic acid export makes an important contribution to the biofilm extracellular matrix in this organism.[14]
Figure 1.
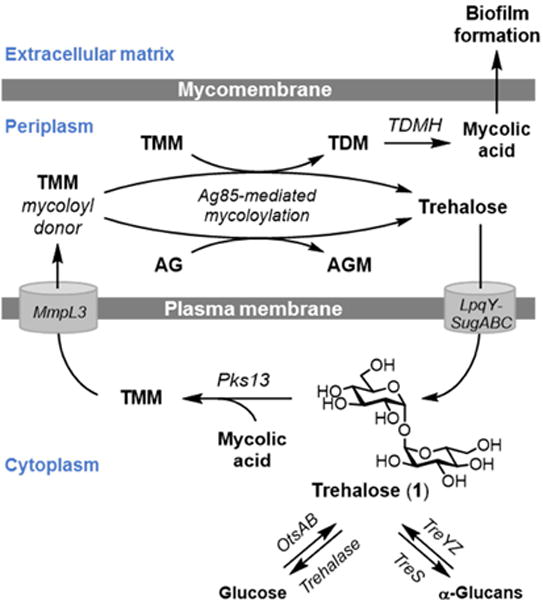
Trehalose (1) acts as a carrier of mycolic acids from the cytoplasm to the cell’s exterior, where mycolic acids are covalently attached to sugars to build the mycomembrane, or released in free form and incorporated into the biofilm extracellular matrix. Free extracellular trehalose generated through these processes is recycled back into the cell by the trehalose-specific transporter LpqY-SugABC. AG, arabinogalactan; AGM, arabinogalactan mycolate; TDM, trehalose dimycolate; TMM, trehalose monomycolate.
Given the involvement of trehalose in the production of the mycomembrane and biofilms—both of which contribute to the intrinsic drug tolerance that is highly characteristic of mycobacteria—inhibitors of trehalose metabolism are of high interest for TB drug development. Various strategies have been used to develop inhibitors targeting trehalose metabolism, including the rational design of trehalose-based inhibitors.[3] The first example of a trehalose-based inhibitor was reported by Belisle and co-workers, who observed that 6-azido-6-deoxy-α,α′-trehalose (6-TreAz, 8) inhibited growth of M. aurum on solid medium (MIC 200 μg/mL).[10] Treatment of M. aurum with a sub-inhibitory concentration of 6-TreAz led to marked reductions in TMM, TDM, and AGM, suggesting that the compound indeed acted on trehalose-mediated mycolic acid export.[10] Following on this discovery, the Barry and Davis groups chemically synthesized and evaluated a panel of trehalose analogues for antimicrobial activity against M. tuberculosis, also observing that 6-position-modified analogues exhibited growth inhibition (MIC 100–200 μg/mL).[16] TDM analogues have also been evaluated for growth inhibition in mycobacteria.[17–19] The promising activity of trehalose analogues as mycobacterial growth inhibitors motivated us to further investigate the activity of this class of compounds. While progress in this area is typically impeded by the difficulty of synthesizing trehalose derivatives, our recently developed chemoenzymatic method allowed easy access to a variety of structures for analysis. Here, we report the evaluation of a panel of monosubstituted trehalose analogues for inhibition of growth and biofilm formation in the model organism M. smegmatis. Our study identifies several novel trehalose-based inhibitors, demonstrates for the first time that trehalose analogues can selectively inhibit biofilm formation, and reveals that trehalose analogues’ antimicrobial and anti-biofilm activities are dependent upon cellular internalization by the trehalose-specific transporter LpqY-SugABC.
Results and Discussion
As noted above, prior research showed that trehalose analogues bearing azido and halogen modifications at the 6-position were moderate inhibitors of mycobacterial growth.[10,16] Furthermore, in our earlier research developing azido trehaloses for metabolic labeling applications,[20] we observed that 2-azido-2-deoxy-α,α′-trehalose (2-TreAz, 4) inhibited growth of M. smegmatis with potency similar to 6-TreAz, whereas analogues containing azido groups at the 3- and 4-positions did not. Therefore, in the present work we decided to focus mainly on 2- and 6-position-modified trehalose analogues with azido, deoxy, fluoro, and stereochemical modifications; we also evaluated 5-deoxy-5-thio-α,α′-D-trehalose (5-ThioTre, 9), which has one of its ring oxygens replaced with a sulfur atom (2–9, Scheme 1).
Scheme 1.
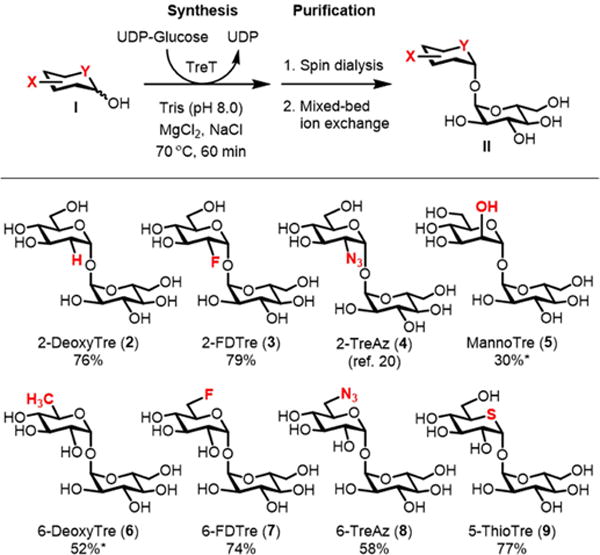
TreT-catalyzed synthesis of trehalose analogues. An asterisk (*) indicates that size exclusion chromatography was used to remove residual unreacted monosaccharide after the spin dialysis and ion exchange steps. 2-TreAz (4) was chemically synthesized as described in ref. 20.
Most of the compound panel was accessible using a one-step chemoenzymatic approach developed by our lab,[21] which circumvents the lengthy multi-step chemical syntheses typically used to access unsymmetrical trehalose analogues.[22] Since our initial report on this method, which described the semi-preparative synthesis of several panel members (2, 3, 8, and 9), we developed optimized reaction conditions and a convenient purification procedure that were applied to the preparation of the target compounds (see Experimental section for detailed procedures and Supplementary Data for NMR spectra). Briefly, trehalose synthase (TreT) from Thermoproteus tenax was used to convert commercially available monosaccharides (I) and UDP-glucose to the corresponding trehalose analogues (II) (Scheme 1). After incubating for 60 min at 70 °C, the product was purified from the reaction mixture simply by spin dialysis and mixed-bed ion exchange, which rapidly removed enzyme and all ionic species, respectively. Using this procedure, trehalose analogues 2, 3, 7, 8, and 9 were generated in 58–79% yield. For analogues 5 and 6, the monosaccharide acceptor was not completely consumed, so product purification required a final size-exclusion chromatography step to separate the disaccharide product from the unreacted monosaccharide, which resulted in lower yields. 2-TreAz (4) could not be synthesized chemoenzymatically because 2-azido-2-deoxy-D-glucose was not accepted as a substrate by TreT. Instead, we chemically synthesized 2-TreAz (4) using a reported 9-step synthesis starting from trehalose.[20] The example of 2-TreAz illustrates the efficiency advantage that chemoenzymatic synthesis offers versus chemical synthesis, while simultaneously underscoring the need for continued method development to improve the tolerance of TreT for poor substrates such as 2-azido-2-deoxy-D-glucose.
In addition to evaluating the compound panel for growth inhibition, we decided to perform the first investigation of trehalose analogues’ anti-biofilm activity for two reasons. First, as noted above, recent data from Ojha’s group demonstrated the important role of trehalose-mediated mycolic acid export in M. smegmatis biofilm formation.[14] Second, during preliminary planktonic growth inhibition experiments in our lab, we observed that treatment with some trehalose analogues induced a clumping phenotype that was not present in untreated or trehalose-treated controls, potentially indicating a change in cell envelope structure that modulated bacterial adhesion (Figure 2). Based on these data, we hypothesized that trehalose analogues might selectively impair biofilm formation.
Figure 2.

Appearance of M. smegmatis cultured under planktonic growth conditions in the presence of exogenous trehalose (1), trehalose analogues (2, 8, or 9), or left untreated (−).
We assayed compounds 1–9 for growth and biofilm inhibition in M. smegmatis using methods adapted from Teng and Dick.[23] Starter cultures of each bacterial strain were grown to logarithmic phase in M63 medium containing 0.05% Tween 80 to prevent cellular aggregation. From these cultures, the planktonic growth and biofilm experiments were initiated in parallel. For planktonic growth, cells were diluted to an OD600 of 0.05 with M63 medium containing 0.05% Tween 80 in polystyrene 96-well plates and treated with 0–500 μM of trehalose analogues. Growth of planktonic culture at 37 °C was assessed by monitoring OD600 after 48 h. For biofilm formation, cells from starter cultures were first washed in M63 medium lacking Tween 80, then diluted to an OD600 of 0.05 with M63 medium (again lacking Tween 80) in polyvinylchloride (PVC) 96-well plates and treated with 0–500 μM of trehalose analogues. After incubation at 37 °C for 48 h, biofilm content adhered to the PVC surface was quantified using the established crystal violet assay,[24,25] which has been successfully used to measure M. smegmatis biofilms.[26–28] To provide insight into the mechanism of trehalose analogue activity, these experiments were performed in three strains of M. smegmatis: (i) the mc2155 wild type strain; (ii) the ΔsugC mutant, which lacks the trehalose-specific transporter LpqY-SugABC; and (iii) the ΔsugC::sugC complement, which has the transporter restored.[13,21]
Dose-response curves for selected compounds are shown in Figure 3 (see Figure S1 in the Supplementary Data for dose-response curves for the remaining compounds). Minimum inhibitory concentration (MIC) and minimum biofilm inhibitory concentration (MBIC) values for all compounds are shown in Table 1. As expected, unmodified trehalose (1) had no discernable effect on M. smegmatis growth or biofilm formation. However, almost all of the trehalose analogues exhibited activity. 6-TreAz (8), which as noted above is a known growth inhibitor of M. aurum and M. tuberculosis, showed partial inhibition of planktonic growth in M. smegmatis at a similarly high concentration (500 μM). Interestingly, the anti-biofilm activity of 6-TreAz was significantly more pronounced (MBIC 50 μM) than its antimicrobial activity. 6-DeoxyTre (6) and 6-FluoroTre (7) both showed inhibition profiles similar to 6-TreAz. Most of the analogues bearing modifications at the 2-position also showed activity. 2-DeoxyTre (2) and 2-FDTre (3) both exhibited anti-biofilm activity (MBICs 50–100 μM) and, consistent with the 6-position-modified analogues, they affected biofilm formation to a greater extent than they affected planktonic growth. 2-TreAz (4) was active at higher concentrations but did not appear to exhibit a major difference in potency between the two assays. The sole trehalose stereoisomer tested, MannoTre (5), had no effect on planktonic growth or biofilm formation, perhaps indicating that eliminating the hydrogen bonding ability of the 2-position was responsible for the inhibitory activity of the other 2-position-modified analogues. Finally, 5-ThioTre (9) showed only slight growth inhibition at 500 μM, but exhibited clear anti-biofilm activity (MBIC 100 μM). In general, compounds 2–4 and 6–9 partially inhibited planktonic growth at the highest concentration tested (500 μM) but did not completely prevent growth as compared to the positive control compound isoniazid. On the other hand, in the biofilm assays, lower concentrations of the same trehalose analogues could inhibit biofilm formation to the same level that isoniazid did.
Figure 3.
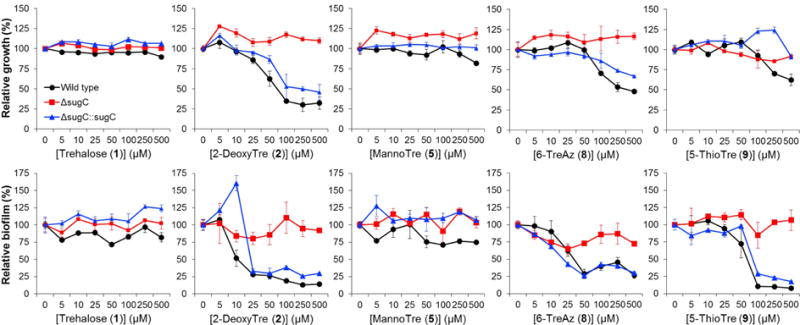
Dose- and transport-dependent inhibition of M. smegmatis planktonic growth (top row) and biofilm formation (bottom row) for selected analogues (dose-response curves for the remaining analogues are shown in Figure S1 of the Supplementary Data). M. smegmatis wild type (black circles), ΔsugC mutant (red squares), and ΔsugC::sugC complement (blue triangles) were cultured either in planktonic growth- or biofilm-promoting conditions in the presence of trehalose (1) or trehalose analogues (2–9) for 48 h. Planktonic growth and biofilm content were assessed by OD600 and the crystal violet assay, respectively. Data were normalized to untreated controls. Error bars represent the standard deviation from three replicate experiments. Data are representative of at least two independent experiments.
Table 1.
Minimum inhibitory concentration of compounds 1–9 for planktonic growth (MIC) and biofilm formation (MBIC) in wild-type M. smegmatis.
| Compound | MIC (μM) | MBIC (μM) |
|---|---|---|
| Trehalose (1) | > 500 | > 500 |
| 2 - DeoxyTre (2) | > 500a | 50 |
| 2 - FDTre (3) | > 500a | 100 |
| 2 - TreAz (4) | > 500a | 500 |
| MannoTre (5) | > 500 | > 500 |
| 6 - DeoxyTre (6) | > 500a | 100 |
| 6 - FDTre (7) | > 500a | 100 |
| 6 - TreAz (8) | > 500a | 50 |
| 5 - ThioTre (9) | > 500a | 100 |
Partial inhibition (25–75%) of planktonic growth was observed.
As shown in the dose-response curves (Figures 3 and S1), all of the analogues’ inhibitory activity was dependent on the trehalose transporter LpqY-SugABC. This result confirms that the compounds act on trehalose metabolism and provides some hints as to their mechanism of action. Rather than inhibiting extracellular Ag85 and/or TDMH directly, the trehalose analogues tested in this study first require transport into the cell to have an effect on growth or biofilm formation. What happens after analogue uptake is currently unknown, although we can speculate by reinterpreting earlier findings in light of our data. Belisle’s observation that 6-TreAz treatment reduced TMM, TDM, and AGM levels in M. aurum[10] suggests that this compound disrupted step(s) involved in the synthesis or transport of these species, presumably following internalization by LpqY-SugABC. Belisle also showed that 6-TreAz treatment led to the accumulation of an unknown intermediate. Later, Bertozzi’s group observed that when M. smegmatis was fed TreAz analogues—including 6-TreAz—the corresponding azide-modified TMM (AzTMM) was generated in a LpqY-SugABC-dependent manner,[20] which may account for the accumulated intermediate detected by Belisle. It is possible that accumulated unnatural TMM species could impair normal MmpL3-mediated transport of TMM and/or Ag85-catalyzed elaboration of TMM to TDM. In the case of 6-TreAz and other 6-position-modified analogues, their corresponding modified TMM products would be incapable of being converted to TDM, which would reduce the substrate pool for TDMH. Since the mycolic acid produced by TDMH-catalyzed TDM hydrolysis is required for biofilm formation in M. smegmatis,[14] this mechanism could account for the selective retardation of biofilm formation. As with 6-TreAz, 2-TreAz treatment of M. smegmatis also generated an unnatural AzTMM intermediate(s),[20] which likewise could disrupt normal TMM transport and subsequent mycoloylation processes. Thus, given the available data and the similar behaviors and potencies of the compounds assayed herein, we speculate that the trehalose analogues evaluated in this study share a similar mechanism of action that generally may involve LpqY-SugABC-dependent impairment of trehalose-mediated shuttling of mycolic acids to their intended destination in the mycomembrane or biofilm extracellular matrix. It is also possible that trehalose analogues could act on other metabolic pathways that utilize trehalose, such as the production of extracellular glucans or trehalose polyphleates, which are not required for growth but could be constituents of the biofilm extracellular matrix.[5, 39, 40]
The dependence of trehalose analogue inhibition on LpqY-SugABC-mediated uptake demonstrated in this study has broader implications for targeting mycobacteria. Mammalian cells do not possess dedicated trehalose uptake machinery, so it is possible that LpqY-SugABC could be targeted for mycobacteria-specific delivery of trehalose-based inhibitors or probes within an infected host. In this vein, Bertozzi previously showed that LpqY-SugABC could be exploited to deliver TreAz analogues to mycobacteria, enabling click chemistry-mediated fluorescence detection in vitro.[20] Our group recently demonstrated that [19F]fluorine-modified trehalose (FDTre) analogues can accumulate in the cytoplasm of M. smegmatis via LpqY-SugABC-mediated uptake, which motivates the investigation of [18F]-FDTre radiotracers as M. tuberculosis-selective positron emission tomography (PET) imaging probes.[29] As shown in the present work, it is conceivable that LpqY-SugABC could be similarly exploited to deliver antimicrobial or anti-biofilm cargo to mycobacteria.
Conclusion
In this report, we described the first investigation of trehalose analogues as inhibitors of mycobacterial biofilm formation. We capitalized on our previously reported chemoenzymatic method to obtain a panel of unsymmetrical, monofunctionalized trehalose analogues, which were then evaluated for antimicrobial and anti-biofilm formation in non-pathogenic M. smegmatis. Most of the compounds exhibited weak inhibition of planktonic growth, which was consistent with earlier work in M. aurum and M. tuberculosis. Interestingly, several compounds had more potent anti-biofilm activity, including trehalose analogues with modifications at the 6-position. These results suggest that it may be possible to target trehalose metabolism to selectively block biofilm formation in M. smegmatis. Whether this strategy can be extended to M. tuberculosis or other mycobacterial pathogens is presently unclear. Like M. smegmatis, M. tuberculosis forms biofilms that are rich in mycolic acids[15] and it possesses a TDMH enzyme which is known to hydrolyze TDM to form free mycolic acids.[30] However, TDMH deletion in an attenuated strain of M. tuberculosis did not impair biofilm formation, suggesting that other mechanisms of extracellular mycolic acid accumulation may be operating to build the biofilm extracellular matrix in M. tuberculosis.[30] In any case, because trehalose is responsible for carrying the large majority of mycolic acids to cell envelope components (i.e., not only to TDM, but also to TMM and AGM), its associated metabolic pathways remain of interest for targeting biofilm formation in M. tuberculosis. Given the increasing attention on understanding mycobacterial biofilms and identifying compounds that inhibit or disperse them,[31–35] further studies on the connections between trehalose metabolism and biofilms are needed. Finally, our data showed that the trehalose-specific transporter LpqY-SugABC is essential for the antimicrobial and anti-biofilm activity of the tested trehalose analogues, which provides insight into the compounds’ mechanism of action and suggests that hijacking trehalose import is an attractive strategy for delivering chemical payload to mycobacterial cells.
Experimental
General experimental
Materials and reagents were obtained from commercial sources without further purification unless otherwise noted. Monosaccharides were obtained from CarboSynth or Sigma Aldrich. Trehalose dihydrate was obtained from Acros. UDP-Glucose was obtained from Abcam. 2-TreAz (4) was obtained using a reported chemical synthesis.[20] Analytical TLC was performed on glass-backed silica 60 Å plates (thickness 250 μm) from Dynamic Adsorbents and detected by charring with 5% H2SO4 in ethanol. NMR spectra were obtained at room temperature on Varian INOVA 500 or Varian Mercury 300 instruments, with 1H and 13C NMR spectra recorded at 500 and 125 MHz, respectively.
General method for TreT-catalyzed trehalose analogue synthesis
To a 15 mL conical tube was added 20 mM monosaccharide, 40 mM UDP-glucose, and 20 mM MgCl2. TreT in Tris buffer (50 mM Tris, 300 mM NaCl, pH 8.0), plus additional Tris buffer if needed, were added to achieve a final volume of 4 mL and a final protein concentration of 10 μM. The reaction was incubated at 70 °C with shaking at 300 rpm for 1 h, then the tube was cooled by placing it on ice. An Amicon Ultra-15 centrifugal filter unit (nominal molecular weight limit 10 kDa) was pre-rinsed with 3 mL DI water three times by centrifugation at 3900 × g for 20 min to remove trace glycerol in the membrane. After transferring the cooled enzymatic reaction mixture to the pre-rinsed centrifugal filter unit, it was spun at 3900 × g for 20 min. The upper chamber of the centrifugal filter unit was rinsed two times with 3 mL of DI water and centrifuged again using the same speed and time. After discarding the upper chamber of the centrifugal filter unit, mixed-bed ion-exchange resin (3 g of Bio-Rad Bio-Rex RG 501-X8) was added to the tube and stirred for 1 h at room temperature. Next, the supernatant was filtered. The remaining resin was rinsed two times with 5 mL of DI water and the supernatant was filtered and combined with the rest of the product. TLC was performed using n-butanol/EtOH/DI water 5:3:2 to assess conversion of the glucose analogue starting material to the trehalose analogue product. The purified product was concentrated by rotary evaporation. For reactions that did not go to completion, the desired trehalose analogues were separated from unreacted monosaccharide using size-exclusion chromatography. In this case, the crude material following rotary evaporation was re-dissolved in DI water and loaded onto a 100 × 1 cm column of Bio-Gel P2 extra-fine polyacrylamide gel media, then eluted with DI water.
2-Deoxy-α,α-D-trehalose (2-DeoxyTre, compound 2)
From 12.6 mg of 2-deoxy-D-glucose, obtained 19.1 mg 2-DeoxyTre (76%). 1H and 13C NMR spectra of the product matched the literature[36] (see Supplementary Data).
2-Deoxy-2-fluoro-α,α-D-trehalose (2-FDTre, compound 3)
From 14.2 mg of 2-deoxy-2-fluoro-D-glucose, obtained 21.1 mg 2-FDTre (79%). 1H and 13C NMR spectra of the product matched the literature[29] (see Supplementary Data).
α-D-glucopyranosyl-(1➔1)-α-D-mannopyranoside (MannoTre, compound 5)
From 13.6 mg of D-mannose, obtained 7.8 mg MannoTre (30%). Size-exclusion chromatography was required to separate the product from residual unreacted D-mannose. 1H and 13C NMR spectra of the product matched the literature[37] (see Supplementary Data).
6-Deoxy-α,α-D-trehalose (6-DeoxyTre, compound 6)
From 15.3 mg of 6-deoxy-D-glucose, obtained 15.8 mg 6-DeoxyTre (52%). Size-exclusion chromatography was required to separate the product from residual unreacted 6-deoxy-D-glucose. 1H and 13C NMR spectra of the product matched the literature[36] (see Supplementary Data).
6-Deoxy-6-fluoro-α,α-D-trehalose (6-FDTre, compound 7)
From 15.6 mg of 6-deoxy-6-fluoro-D-glucose, obtained 21.9 mg 6-FDTre (74%). 1H and 13C NMR spectra of the product matched the literature[29] (see Supplementary Data).
6-Azido-6-deoxy-α,α-D-trehalose (6-TreAz, compound 8)
From 16.6 mg of 6-azido-6-deoxy-D-glucose, obtained 17.3 mg 6-TreAz (58%). 1H and 13C NMR spectra of the product matched the literature[38] (see Supplementary Data).
5-Deoxy-5-Thio-α,α-D-trehalose (5-ThioTre, compound 9)
From 15.4 mg of 5-thio-D-glucose, obtained 21.8 mg 5-ThioTre (77%). 1H and 13C NMR spectra of the product matched the literature[21] (see Supplementary Data).
Growth and biofilm inhibition experiments
Starter cultures of M. smegmatis wild type, ΔsugC mutant, or ΔsugC::sugC complement were generated by inoculating a single colony from a freshly streaked agar plate into 3 mL M63 medium (M63 salts minimal medium supplemented with 2% glucose, 0.5% casamino acids, 1 mM MgSO4, and 0.7 mM CaCl2) containing 0.05% Tween 80 in a culture tube. Starter cultures were incubated at 37 °C with shaking until reaching log phase, 24–36 h.
To initiate planktonic growth experiments, an aliquot of the starter culture was diluted with M63 medium containing 0.05% Tween 80 to an OD600 value of 0.1. 50 μL aliquots of this cell suspension were added to the wells of a sterile polystyrene 96-well plate, followed by addition of the appropriate volumes of (i) trehalose analogue stock solution and (ii) M63 medium containing 0.05% Tween 80 to obtain the desired final volume (100 μL), OD600 value (0.05), and concentration of trehalose analogue (0–500 μM). Isoniazid was used as the positive control drug at a concentration of 100 μg/mL because its minimal biofilm inhibitory concentration (MBIC) for M. smegmatis was previously reported to be approximately 80 μg/mL.[23] M63 medium containing 0.05% Tween 80 was used as the blank. Empty wells were filled with 100 μL DI water to minimize evaporation. The contents of each well were mixed thoroughly by pipetting up and down, then a lid was placed on the 96-well plate and it was incubated in a humidity chamber at 37 °C with shaking for 48 h. The contents of each well were mixed by pipetting up and down, then the final OD600 reading was taken in a Tecan F200 Pro multimodal microplate reader. Minimal inhibitory concentration (MIC) was defined as the concentration of compound at which no growth above the positive control (100 μg/mL isoniazid) was observed.
Biofilm experiments were run in parallel with planktonic growth experiments. To initiate biofilm formation experiments, 1 mL of the starter culture was transferred to microcentrifuge tubes and centrifuged at 3900 × g for 5 min. The supernatant was discarded and the cell pellet was re-suspended with 1 mL M63 medium without Tween 80. This was repeated once more, then the OD600 value of the cells was measured. An aliquot of this culture was diluted with M63 medium without Tween 80 to an OD600 value of 0.1. 50 μL aliquots of this cell suspension were added to the wells of a sterile U-bottom polyvinylchloride (PVC) 96-well plate, followed by incubation in the presence or absence of trehalose analogues as described above for the growth inhibition experiments. After incubating for 48 h, the biofilm content adhered to the PVC surface was quantified using the biofilm crystal violet assay adapted from the literature.[23,25,26] Briefly, the wells of the PVC 96-well plate were rinsed three times with DI water, then 125 μL of 0.1% aqueous solution of crystal violet (CV) was added. The plate was incubated at room temperature for 4 h, then rinsed three times with DI water. Biofilm-associated CV was extracted from each well using 200 μL of 95% ethanol at room temperature for 1 h, then the extracts were transferred to a flat-bottom 96-well plate and optical density at 570 nm was measured. Minimal biofilm inhibitory concentration (MBIC) was defined as the concentration of compound at which no biofilm formation above the positive control (100 μg/mL isoniazid) was observed.
Supplementary Material
Highlights.
A panel of trehalose analogues was prepared using a one-step chemoenzymatic synthesis method.
Trehalose analogues were shown to inhibit growth and biofilm formation in Mycobacterium smegmatis.
Several trehalose analogues were shown to be selective inhibitors of biofilm formation.
Trehalose analogue activity was dependent upon cellular uptake via the trehalose-specific transporter LpqY-SugABC.
Acknowledgments
This work was funded by a grant from the National Institutes of Health (R15 AI117670) to B.M.S. and P.J.W., as well as Cottrell College Scholar Awards from the Research Corporation to B.M.S. (22525) and P.J.W. (20185). L.M.M. was supported by a Provost’s Fellowship from CMU. K.M.Z. was supported by the American Chemical Society SEED Summer Research Internship Program. R. Kalscheuer is thanked for providing bacterial strains.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. Global Tuberculosis Report. 2016 [Google Scholar]
- 2.Mitchison DA. The diagnosis and therapy of tuberculosis during the past 100 years. Am J Respir Crit Care Med. 2005;171:699–706. doi: 10.1164/rccm.200411-1603OE. [DOI] [PubMed] [Google Scholar]
- 3.Thanna S, Sucheck SJ. Targeting the trehalose utilization pathways of Mycobacterium tuberculosis. Med Chem Commun. 2015;7:69–85. doi: 10.1039/C5MD00376H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Neill MK, Piligian BF, Olson CD, Woodruff PJ, Swarts BM. Tailoring trehalose for biomedical and biotechnological applications. Pure Appl Chem. 2017 doi: 10.1515/pac-2016-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalscheuer R, Koliwer-Brandl H. Genetics of mycobacterial trehalose metabolism. Microbiol Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0002-2013. [DOI] [PubMed] [Google Scholar]
- 6.Gavalda S, Bardou F, Laval F, Bon C, Malaga W, Chalut C, Guilhot C, Mourey L, Daffé M, Quémard A. The polyketide synthase Pks13 catalyzes a novel mechanism of lipid transfer in mycobacteria. Chem Biol. 2014;21:1660–1669. doi: 10.1016/j.chembiol.2014.10.011. doi: http://dx.doi.org/10.1016/j.chembiol.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Grzegorzewicz AE, Pham H, Gundi VAKB, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SEM, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M, Korduláková J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol. 2012;8:334–341. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z, Meshcheryakov VA, Poce G, Chng SS. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sathyamoorthy N, Takayama K. Purification and characterization of a novel mycolic acid exchange enzyme from Mycobacterium smegmatis. J Biol Chem. 1987;262:13417–13423. [PubMed] [Google Scholar]
- 10.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 11.Backus KM, Dolan MA, Barry CS, Joe M, McPhie P, Boshoff HI, Lowary TL, Davis BG, Barry CE., 3rd The three Mycobacterium tuberculosis antigen 85 isoforms have unique substrates and activities determined by non-active site regions. J Biol Chem. 2014;289:25041–25053. doi: 10.1074/jbc.M114.581579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quémard A. New insights into the mycolate-containing compound biosynthesis and transport in mycobacteria. Trends Microbiol. 2016;24:725–738. doi: 10.1016/j.tim.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 13.Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2010;107:21761–21766. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ojha AK, Trivelli X, Guerardel Y, Kremer L, Hatfull GF. Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J Biol Chem. 2010;285:17380–17389. doi: 10.1074/jbc.M110.112813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR, Hatfull GF. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol. 2008;69:164–174. doi: 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backus KM, Boshoff HI, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE, 3rd, Davis BG. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat Chem Biol. 2011;7:228–235. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rose JD, Maddry JA, Comber RN, Suling WJ, Wilson LN, Reynolds RC. Synthesis and biological evaluation of trehalose analogs as potential inhibitors of mycobacterial cell wall biosynthesis. Carbohydr Res. 2002;337:105–120. doi: 10.1016/s0008-6215(01)00288-9. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Elchert B, Hui Y, Takemoto JY, Bensaci M, Wennergren J, Chang H, Rai R, Chang CW. Synthesis of trehalose-based compounds and their inhibitory activities against Mycobacterium smegmatis. Bioorg Med Chem. 2004;12:6397–6413. doi: 10.1016/j.bmc.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 19.Sanki AK, Boucau J, Umesiri FE, Ronning DR, Sucheck SJ. Design, synthesis and biological evaluation of sugar-derived esters, a-ketoesters and a-ketoamides as inhibitors for Mycobacterium tuberculosis antigen 85C. Mol BioSyst. 2009;5:945–956. doi: 10.1039/b902284h. [DOI] [PubMed] [Google Scholar]
- 20.Swarts BM, Holsclaw CM, Jewett JC, Alber M, Fox DM, Siegrist MS, Leary JA, Kalscheuer R, Bertozzi CR. Probing the mycobacterial trehalome with bioorthogonal chemistry. J Am Chem Soc. 2012;134:16123–16126. doi: 10.1021/ja3062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbanek BL, Wing DC, Haislop KS, Hamel CJ, Kalscheuer R, Woodruff PJ, Swarts BM. Chemoenzymatic synthesis of trehalose analogues: rapid access to chemical probes for investigating mycobacteria. ChemBioChem. 2014;15:2066–2070. doi: 10.1002/cbic.201402288. [DOI] [PubMed] [Google Scholar]
- 22.Sarpe VA, Kulkarni SS. Regioselective protection and functionalization of trehalose. Trends Carbohydr Res. 2013;5:8–33. [Google Scholar]
- 23.Teng R, Dick T. Isoniazid resistance of exponentially growing Mycobacterium smegmatis biofilm culture. FEMS Microbiol Lett. 2003;227:171–174. doi: 10.1016/S0378-1097(03)00584-6. [DOI] [PubMed] [Google Scholar]
- 24.O’Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol. 1998;28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 25.O’Toole GA. Microtiter dish biofilm formation assay. J Vis Exp. 2011:2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Recht J, Martínez A, Torello S, Kolter R, Martinez A, Torello S, Kolter R. Genetic analysis of sliding motility in Mycobacterium smegmatis. J Bacteriol. 2000;182:4348–4351. doi: 10.1128/jb.182.15.4348-4351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Recht J, Kolter R. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J Bacteriol. 2001;183:5718–5724. doi: 10.1128/JB.183.19.5718-5724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SPS, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, Ng SH, Nanjundappa M, Ravindran S, Seah PG, Thayalan P, Lim SH, Lee BH, Goh A, Barnes WS, Chen Z, Gagaring K, Chatterjee AK, Pethe K, Kuhen K, Walker J, Feng G, Babu S, Zhang L, Blasco F, Beer D, Weaver M, Dartois V, Glynne R, Dick T, Smith PW, Diagana TT, Manjunatha UH. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med. 2013;5:214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 29.Rundell SR, Wagar ZL, Meints LM, Olson CD, O’Neill MK, Piligian BF, Poston AW, Hood RJ, Woodruff PJ, Swarts BM. Deoxyfluoro-D-trehalose (FDTre) analogues as potential PET probes for imaging mycobacterial infection. Org Biomol Chem. 2016;14:8598–8609. doi: 10.1039/c6ob01734g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Kulka K, Montelaro RC, Reinhart TA, Sissons J, Aderem A, Ojha AK. A hydrolase of trehalose dimycolate induces nutrient influx and stress sensitivity to balance intracellular growth of Mycobacterium tuberculosis. Cell Host Microbe. 2014;15:153–163. doi: 10.1016/j.chom.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Islam MS, Richards JP, Ojha AK. Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Rev Anti Infect Ther. 2012;10:1055–1066. doi: 10.1586/eri.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishida S, Arai M, Niikawa H, Kobayashi M. Inhibitory effect of cyclic trihydroxamate siderophore, desferrioxamine E, on the biofilm formation of Mycobacterium species. Biol Pharm Bull. 2011;34:917–920. doi: 10.1248/bpb.34.917. [DOI] [PubMed] [Google Scholar]
- 33.Arai M, Niikawa H, Kobayashi M. Marine-derived fungal sesterterpenes, ophiobolins, inhibit biofilm formation of Mycobacterium species. J Nat Med. 2013;67:271–275. doi: 10.1007/s11418-012-0676-5. [DOI] [PubMed] [Google Scholar]
- 34.Ackart DF, Lindsey EA, Podell BK, Melander RJ, Basaraba RJ, Melander C. Reversal of Mycobacterium tuberculosis phenotypic drug resistance by 2-aminoimidazole-based small molecules. Pathog Dis. 2014;70:370–378. doi: 10.1111/2049-632X.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, Hassani M, Huszar S, Trefzer C, Ma Z, Kaneko T, Mdluli KE, Franzblau S, Chatterjee AK, Johnsson K, Mikusova K, Besra GS, Futterer K, Robbins SH, Barnes SW, Walker JR, Jacobs WR, Jr, Schultz PG. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci U S A. 2013;110:E2510–2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin FL, van Halbeek H, Bertozzi CR. Synthesis of mono- and dideoxygenated α,α-trehalose analogs. Carbohydr Res. 2007;342:2014–2030. doi: 10.1016/j.carres.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bar-Guilloux É, Defaye J, Driguez H, Robic D. Synthèse, conformation et affinité tréhalasique des α-d-glucopyranosyl-α-d-xylopyranoside, α-d-glucopyranosyl-α-d-mannopyranoside et α-d-allopyranosyl-α-d-glucopyranoside. Carbohydr Res. 1975;45:217–236. [Google Scholar]
- 38.Hanessian S, Lavallee P. Synthesis of 6-amino-6-deoxy-α,α-trehalose. Positional isomer of trehalosamine. J Antibiot. 1972;25:683–684. doi: 10.7164/antibiotics.25.683. [DOI] [PubMed] [Google Scholar]
- 39.Burbaud S, Laval F, Lemassu A, Daffé M, Guilhot C, Chalut C. Cell Chem Biol. 2016;23:278–289. doi: 10.1016/j.chembiol.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Llorens-Fons M, Perez-Trujillo M, Julian E, Brambilla C, Alcaide F, Byrd TF, Luquin M. Front Microbiol. 2017;8:1402. doi: 10.3389/fmicb.2017.01402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.