

Abstract
Reactive oxygen and nitrogen species damage cellular macromolecules including DNA. Cells have a robust base excision repair pathway to deal with this damage in both nuclear and mitochondrial genomes. However, mitochondria lack nucleotide excision repair. Evidence suggests that chronic oxidative stress can induce protective pathways lowering genotoxicity. Understanding oxidant injury to DNA and its repair is critical for our understanding the pathophysiology of a wide range of human disorders.
Graphical Abstract
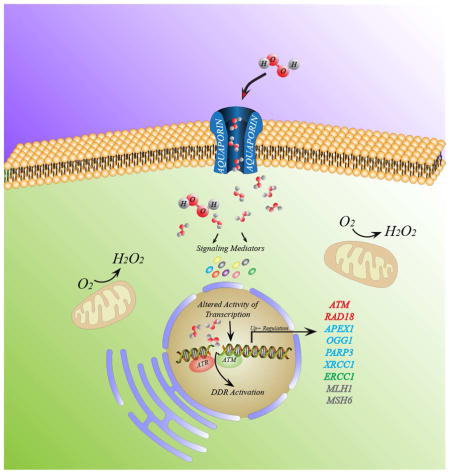
Model of how chronic ROS exposure provides a protective mechanism when cells are grown at intermittent low concentration (50μM) of H2O2 and during prolonged periods (seven days). H2O2 molecules diffuse into the cell mediated by the aquaporin cell membrane protein, triggering two main cell responses: i) activating redox-sensitive mediators (e. g. PI3K/AKT, ERK1, SRC), and ii) inducing DDR through ATM and ATR sensors (red). Both signaling events promote transactivation of transcription factors augmenting gene expression of DNA repair enzymes, mainly of the BER pathway (blue), NER (green) and mismatch repair (black) converging in the ROS protective phenotype.
1. Oxidant injury and macromolecule damage
The evolution of photosynthesis some 3.5 billion years ago provided oxygen to planet Earth and a subsequent explosion of life forms that used oxygen in their metabolism and energy production. During this process many organisms had to deal with a toxic brew of reactive oxygen and nitrogen species (RONS) that arose from both exogenous and endogenous sources. The primary endogenous sources are from mitochondria during oxidative phosphorylation, specific metabolic pathways often involved in xenobiotic detoxification, and NADPH oxidases. The later of enzymes was essential for allowing the evolution a billion years ago of ambeoid-like cells into macrophages in multi-cellular organisms, to help fight infections. The main exogenous sources that help shape evolution were ionizing and UV radiation and vast array of natural and man-made chemicals [1]. Some of these chemicals generate free radicals (molecules with unpaired electrons) during their metabolic conversion, such as carbon tetracholoride, whereas other chemicals, such as rotenone can cause free radicals by inhibiting key steps in oxidative phosphorylation, in this case at Complex I (Figure 1). The seminal discovery of enzymes (superoxide dismutases) that convert the superoxide radical anion to hydrogen peroxide by McCord and Fridovich in 1969 [2] triggered an enormous interest in how free radicals, and RONS might damage macromolecules, including protein, lipids and DNA, and how the accumulation of this damage can underlie the pathophysiology of a large number of human diseases. However critical thinkers in the free radical community, including Torren Finkel [3] and Barry Halliwell [4], suggest that RONS are important signaling molecules and that failure of anti-oxidant therapy may be in part due to our lack of sufficient knowledge of redox biology in compartments of living cells. This review discusses our current understanding of how oxidative stress induces DNA damage in both nuclear and mitochondrial compartments, the subsequent biochemical pathways that help to remove this damage, how chronic oxidative stress can upregulate DNA repair, and finally how faulty repair can cause pathological consequences. We have tried to highlight key studies and current challenges that are active areas of research; this review is therefore not comprehensive in nature.
Figure 1. Common reactive oxygen species generated in the mitochondria.

A. Superoxide radical anions are primarily made at Complexes I and III are converted to hydrogen peroxide, H2O2, by manganese superoxide dismutase, MnSOD (SOD2), and broken down to water by either glutathione peroxidase, GPx, or peroxiredoxin (Prx). Mitochondria make iron sulfur centers and in the presence of reduced iron, Fe2+, Fenton chemistry can generate the highly reactive hydroxyl radical that can attack all mitochondrial macromolecules including DNA causing loss of electron transport proteins encoded by the mtDNA and subsequent increases in superoxide production. At Complexes I, III, and IV, electron movement is coupled to proton movement into the inner membrane space which is harvested by Complex V the ATP synthase. B. Oxygen is consumed at Complex IV in a four electron reduction to form water. Adapted from [60].
2. Current views of oxidative DNA damage and repair in the mitochondrial and nuclear genomes
2.1 Repair of oxidative DNA damage by base excision repair (BER)
Over the last decade many excellent reviews on the topic of oxidative damage and repair to nuclear and mitochondria genomes have been published and the reader is encouraged to examine these excellent summaries, including: [5–11]. In fact, an entire recent issue of Free Radical Biology and Medicine (Volume 107, June 2017) has been dedicated to this topic [12]. One of the most common forms of oxidative DNA damage is 8-oxoguanine, 8-oxoG (Figure 2). The repair of this lesion is initiated by the same enzyme 8-oxoguanine glycosylase (OGG1) in the nucleus and mitochondria. OGG1 efficiently recognizes 8-oxoG removing the damaged base and through an intrinsic lyase activity cleaves on the 3′ side of remaining sugar to generate a 3′ ribose moiety and a 5′ phosphate. OGG1 shows slow turnover, due to avid binding to the abasic site product and is released by the action of apurinic endonuclease, APE1 that cleaves on the 5′ side of the sugar moiety to generate a 3′ hydroxyl and a one base gap. In the nucleus, and perhaps in the mitochondria (see section 2.1.4) this gap triggers activation of poly-ADP-polymerase (PARP1), which uses NAD to make chains of poly(ADP-ribose) on itself and other proteins. This parylation serves to help recruit DNA polymerase β, Polβ, the scaffold protein, XRCC1 and DNA ligase III [11]. Pol β serves to fill in the one base gap and DNA ligase I or III seals the nick. Many BER enzymes like OGG1, are either alternatively spliced or have a different start codon to encode a mitochondrial leader sequence for mitochondrial targeting. Mitochondrial repair of 8-oxoG, as in the nucleus, proceeds by the action of OGG1 and APE1. The resulting gap is filled in by DNA polymerase γ, Pol γ, but rather than stop at a one base insertion Pol γ likes to perform strand displacement. This 5′ flap needs to be processed by an endonuclease, which was believed to be DNA2 [13], but is apparently Exonuclease G (EXOG) (see 2.1.1) [14]. DNA ligase III, the only known ligase in the mitochondria is then required for sealing the repair patch [15].
Figure 2. Common forms of oxidative DNA damage and its repair.


A. Common oxidized purines, B. Common oxidized pyrimidines, C. Base excision repair pathway in the nucleus: i. 8-oxoguaine is 8oxoguanine glycosylase, OGG1, which removes the damaged base and produces a 3′ nick, ii. This intermediate is recognized by apurinic endonuclease, APE1 that facilitate OGG1 turnover, and nicks the 5′ of the sugar moiety to generate a one base gap, iii. DNA polymerase β, XRCC1 and PARP1 are recruited to the gap and pol β fills in gap (iv) which is ligated close (v) D. Base excision repair in mitochondria. DNA polymerase γ provides gap filling and strand displacement synthesis that is trimmed by Exonuclease G (ExoG) and ligated by DNA ligase III. Adapted from [13, 61]
2.1.1 Base excision repair and the role of EXOG in mitochondrial BER [14]
Sankar Mitra and coworkers discovered a mitochondrial protein, ExoG, with homology to the mitochondrial protein EndoG which is released during apoptosis. Through a comprehensive series of sophisticated experiments they found that EXOG, and not DNA2 or FEN1, was responsible for the removal of the 5′ flap generated during mtDNA repair. They found that depletion of EXOG caused mtDNA damage, increased ROS, loss of mitochondrial function and cell death [14].
2.1.2. DNA ligase III required for normal cell growth
Maria Jason’s laboratory used an elegant “pre-emptive complementation” approach were able to show that cells did not require DNA ligase III in the nucleus to proliferate, while cells lacking DNA ligase III in the mitochondria would not grow [15]. These data suggested that DNA ligase III activity in the mitochondria was critical for normal cell proliferation [15, 16].
2.1.3. Inhibiting mitochondria DNA ligase III
Building on this idea that loss of mitochondrial DNA ligase III activity my cause cytostatic or cytotoxic effects, Alan Tomkinson and coworkers developed a DNA ligase III inhibitor, L67 that specifically inhibited the growth of tumor cells through alterations in mitochondrial DNA transactions [17].
2.1.4. PARP1 mediator of BER in the nucleus and inhibitor of mitochondrial repair
Despite evidence for poly(ADP-ribose) glycohydrolase, PARG, in the mitochondria [18], an enzyme that breaks down poly(ADP-ribose) chains, the presence of PARP1 in the mitochondria has been controversial. Recently a strong case has been made that PARP1 has activity in the mitochondria, and unlike its positive role in the nucleus, it seems its activity in the mitochondria inhibits DNA repair and can exacerbate cell injury [19–21].
2.2 Are current genomic regions more susceptible to oxidant injury?
2.2.1 Nuclear regions including telomeres
Since DNA is negatively charged it has been speculated that divalent metal ions like Fe2+ might bind to specific DNA sequences and make DNA more prone to Fenton-chemistry attack of hydroxyl radical (Figure 1). Stuart Linn and coworkers mapped reactive sites on DNA and surprisingly, found the highest reactivity to be that with the consensus sequence, TTAGGG, the identical sequence found in telomeric DNA [22]. This has led to the concept that telomeric DNA, due to its G rich strand, may be more susceptible to ROS damage that the nuclear DNA overall.
2.2.2 Mitochondrial DNA is more susceptible to oxidant injury
In the late 1990’s we developed a quantitative PCR assay for the detection of DNA damage in specific sequences and we found that mtDNA is much more prone to oxidant injury from hydrogen peroxide than nuclear DNA [23, 24]. This oxidant injury leads to rapid mtDNA loss and subsequent decline in mitochondrial function [25]. Surprisingly, equal amounts of alkylation damage to mitochondrial DNA did not trigger the same cellular events. Part of the explanation could be the large amounts of Fe in the mitochondrial either involved in iron-sulfur (FeS) cluster synthesis or as FeS centers in key mitochondrial electron transport proteins are prone to attack by hydrogen peroxide.
3. Is DNA repair inducible after oxidative stress?
3.1 Current evidence
After the discovery of an inducible DNA repair adaptive response in E.coli [26], seven years later an analogous system was discovered in eukaryotes [27], followed by genetic and enzymatic support. Currently, in mammalian cells it is well documented that in vitro acute exposures to reactive oxygen species (ROS), activates metabolic antioxidant defenses and a sophisticated network of DNA damage-response (DDR) systems, among them, DNA repair mechanisms for genomic oxidative lesions, and cell-cycle checkpoint pathways [28, 29]. However, fewer studies have examined whether genomic damage responds in a redox homeostasis-related process and adaptation can occur during conditions of chronic low-dose exposures.
Recently we addressed this issue by reporting that chronic exposures to low concentrations of oxidative stimuli (50 μM hydrogen peroxide), over seven days, induces an adaptive response in C2C12 cells, with low genotoxicity [30]. These chronically exposed cells were found to become resistant to subsequent challenges of 10X-20X acute doses of hydrogen peroxide. More importantly, this adaptation was marked by the concurrent upregulation of mRNA levels for ATR, APE1, OGG1, PARP3, XRCC1, ERCC1, MLH1, MSH6 AND RAD18 genes associated with a G2 check point (important for cell survival) and DNA damage response pathways (see graphical abstract). Among the induced proteins, several are involved in base excision repair (BER) pathway (APE1, OGG1, PARP3 and XRCC1), which play a key role in the repair of oxidative DNA damages (Figure 2). MLH1 and MSH6 are also two key proteins in the mismatch repair (MMR) pathway, also implicated in the response to oxidative DNA damage, particularly of 8-oxoG residues [31].
Oxidative events at the promoter levels can generate transcriptional upregulation of DNA repair genes [32], resulting in an adaptive response only visible through a narrow window of oxidant dose, because DNA damage itself can interfere with transcription [29, 33]. This induction of repair systems after oxidative stress is reminescent of induction of base excision repair after alkylation damage [34]. Despite compensatory pathways, genome instability induced by chronic levels of ROS like those found in inflammation-related disorders, may be involved in spontaneous mutagenesis and the etiology of a wide variety of human diseases, including: aging, asthma, atherosclerosis, cancer, diabetes and neurodegeneration, Figure 3 [8, 35, 36].
Figure 3.

Several human diseases associated with chronic oxidative stress and subsequent genomic instability in nucleus or mitochondria.
3.2 Outlook
It is clear that some types of cells can adapt to moderate oxidative stress with protective mechanisms. As has been pointed out by several research teams, a low level of hydrogen peroxide acts as a signaling molecule, sometimes emanating from the mitochondria, and might help to explain the anti-oxidant paradox were both high levels of ROS or anti-oxidant scavengers become detrimental to human health [3, 37]. However, what specific cell types, how much stress and whether these stress responses are altered with age, are important questions that remain to be answered. Obtaining this knowledge will certainly have important implications in medicine. For example, perhaps small bouts of oxidant stress from routine moderate exercise helps muscles acquire the capacity to withstand subsequent higher doses of ROS, such as those that occur during strenuous exercise. A similar idea is pre-conditioning heart muscle to ischemia-reperfusion injury to prevent large myocardial infarctions. Clearly we do not have a sufficient battery of tests for assessing the status of redox balance in people and whether chronic oxidant stress or antioxidants may be beneficial. Mitochondrial DNA damage may be one such biomarker of oxidant injury [38, 39].
4. Are DNA repair proteins susceptible to oxidative injury: Iron-sulfur (Fe-S) centers in key nuclear DNA metabolizing enzymes
One intriguing finding is that many nuclear proteins that function during DNA transactions such as transcription, replication and DNA repair have Fe-S clusters Figure 4, [40]. It is important to note that Fe-S cluster assembly occurs in mitochondria and a key enzyme in nucleotide excision repair, XPD, a damage sensing helicase that requires a Fe-S center for function [41]. NER is known to remove some forms of oxidative lesions such as cyclopurine adducts in the nucleus, and one report suggests a role of XPD in reducing ROS stress in mitochondria, although this latter observation requires confirmation [41]. Thus in cells with mitochondrial dysfunction there may be a loss of mitochondria to form Fe-S clusters that could result in increased nuclear damage and lack of repair. Such a case may occur in the human disease Friedreich ataxia patients where both an increase in mitochondrial and nuclear damage was found. This DNA damage in peripheral white blood cells was associated with a pattern of gene expression consistent with DNA damage [38].
Figure 4. Iron-sulfur containing nuclear proteins involved in important DNA transactions.

Mitochondria are required for the synthesis of iron sulfur clusters (FeS), many important nuclear enzymes that work on DNA require FeS centers for normal function. Adapted from [40]
5. Mitochondrial-nuclear cross talk
It is clear that mitochondrially-generated ROS can induce protective as well as deleterious effects. As mentioned previously, mitochondrially-generated ROS may be important for triggering key nuclear events [3, 37, 42, 43]. Oxidant stress can induce a robust Nrf2 response which is protective to cells [44]. While too much ROS can cause cellular senescence [45, 46].
5.1. hTERT in the mitochondria
Another interesting feature of cross-talk between the mitochondria and the nucleus is the observation that the protein subunit of telomerase, hTERT, has a mitochondrial leader sequence and can be both deleterious and helpful during oxidant injury [47–50]
5.2. Does mitochondrial dysfunction cause nuclear problems?
While many reviews suggest that mitochondrially-generated ROS is sufficient to cause nuclear problems, direct evidence for such a mechanism is not clear and there are data both for and against.
5.2.1. Cases against
Early studies failed to show any nuclear damage following mitochondrial dysfunction induced by treatment with electron transport inhibitors that are known to increase mitochondrially-derived ROS [51, 52].
5.2.2. Cases for
However nuclear genome instability has been noted in human patients with pulmonary arterial hypertension that is associated with an increase in mitochondrial ROS production [53]. Also quite recently it has been shown that mitochondria with defects in a specialized repair system to remove topoisomerase-DNA protein cross-links lead to an increase in ROS generated by the mitochondria and subsequent nuclear damage [54].
6. Scientific Outlook
We live in an exciting time where new chemical biology advances are allowing manipulation of RNOS species in specific compartments of cells in culture and living tissue [42, 55, 56]. Low to moderate levels of oxidative stress are involved in a great diversity of physiological process. In evolutionary terms, it is reasonable to think that adaptive mechanisms have evolved to maintain oxidative homeostasis. However, it is captivating to note that the abrupt extension of average human life span in the past two centuries, overlaps with the steep rise in ROS-related diseases such as neurodegeneration, diabetes, cancer and age-related pathologies (Figure 3). Several questions arise: 1) Could this be due in part to pro-inflammatory immune responses that allow us to fight infections in our youth, which however, cause age-related deterioration of the base-line adaptive mechanisms later in life? 2) Can we replicate an adaptive responses in an aging model linked with these pathologies? 3) Can we use chemotherapeutics and/or nutraceuticals, stress reduction and exercise to help modulate the adaptive response to oxidative stress? One interesting starting point to these questions is to better understand what influences the highly conserved (BER) pathway, which plays a central role in the repair of oxidative DNA damages. Among the different enzymes involved in BER, one of the most interesting and critical ones is the multifunctional APE1/Ref-1 that clearly responds to oxidant stress as described above. The protein has been the target of biochemical manipulation with small molecules such as, E3330, to regulate its reversible nuclear redox activity [57]. While this compound has been targeted for chemotherapy of cancers, it may be extended to other age-related pathologies involving REDOX biology [58, 59].
Highlights.
Oxidative stress causes a wide spectrum of DNA lesions
These lesions are repaired in both nuclear and mitochondrial genomes
Chronic oxidative stress protects from genotoxic damage
Many humans diseases are associated with oxidant injury
Acknowledgments
BVH is supported by a NIH grant R33ES025606. Thanks to A. Castro for graphical assistance. We apologize in advance for lack of citations of critical papers due to limited space.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meyer JN, Leung MC, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, Bess AS. Mitochondria as a target of environmental toxicants. Toxicological sciences : an official journal of the Society of Toxicology. 2013;134:1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 3.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halliwell B. Free radicals and antioxidants: updating a personal view. Nutrition Reviews. 2012;70:257–265. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- 5.Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb Perspect Biol. 2013;5:a012641. doi: 10.1101/cshperspect.a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol. 2012;13:659–671. doi: 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- 7.Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. 2017;107:216–227. doi: 10.1016/j.freeradbiomed.2016.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Houten B, Hunter SE, Meyer JN. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front Biosci-Landmrk. 2016;21:42–54. doi: 10.2741/4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goellner EM, Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. Antioxidants & redox signaling. 2011;14:2491–2507. doi: 10.1089/ars.2010.3466. doi:2410.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadet J, Davies KJA, Medeiros MHG, Di Mascio P, Wagner JR. Formation and repair of oxidatively generated damage in cellular DNA. Free Radical Biology and Medicine. 2017;107:13–34. doi: 10.1016/j.freeradbiomed.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasad R, Dyrkheeva N, Williams J, Wilson SH. Mammalian Base Excision Repair: Functional Partnership between PARP-1 and APE1 in AP-Site Repair. PLoS One. 2015;10:e0124269. doi: 10.1371/journal.pone.0124269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cadet J, Davies KJA. Oxidative DNA damage & repair: An introduction. Free Radic Biol Med. 2017;107:2–12. doi: 10.1016/j.freeradbiomed.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copeland WC, Longley MJ. DNA2 resolves expanding flap in mitochondrial base excision repair. Mol Cell. 2008;32:457–458. doi: 10.1016/j.molcel.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tann AW, Boldogh I, Meiss G, Qian W, Van Houten B, Mitra S, Szczesny B. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5′-EXO/endonuclease) in their repair. J Biol Chem. 2011;286:31975–31983. doi: 10.1074/jbc.M110.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis AK, Van Houten B, Shuman S, McKinnon PJ, Jasin M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011;471:245–248. doi: 10.1038/nature09794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shokolenko IN, Fayzulin RZ, Katyal S, McKinnon PJ, Wilson GL, Alexeyev MF. Mitochondrial DNA ligase is dispensable for the viability of cultured cells but essential for mtDNA maintenance. J Biol Chem. 2013;288:26594–26605. doi: 10.1074/jbc.M113.472977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sallmyr A, Matsumoto Y, Roginskaya V, Van Houten B, Tomkinson AE. Inhibiting Mitochondrial DNA Ligase IIIalpha Activates Caspase 1-Dependent Apoptosis in Cancer Cells. Cancer Res. 2016;76:5431–5441. doi: 10.1158/0008-5472.CAN-15-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whatcott CJ, Meyer-Ficca ML, Meyer RG, Jacobson MK. A specific isoform of poly(ADP-ribose) glycohydrolase is targeted to the mitochondrial matrix by a N-terminal mitochondrial targeting sequence. Exp Cell Res. 2009;315:3477–3485. doi: 10.1016/j.yexcr.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunyanszki A, Szczesny B, Virag L, Szabo C. Mitochondrial poly(ADP-ribose) polymerase: The Wizard of Oz at work. Free Radic Biol Med. 2016;100:257–270. doi: 10.1016/j.freeradbiomed.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, Staras K, Mancini GM, McKinnon PJ, Wang ZQ, Wagner JD, Yoon G, Caldecott KW. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. 2017;541:87–91. doi: 10.1038/nature20790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szczesny B, Brunyanszki A, Olah G, Mitra S, Szabo C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res. 2014;42:13161–13173. doi: 10.1093/nar/gku1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J Biol Chem. 1999;274:962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]
- 23.Salazar JJ, Van Houten B. Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator of hydrogen peroxide in human fibroblasts. Mutat Res. 1997;385:139–149. doi: 10.1016/s0921-8777(97)00047-5. [DOI] [PubMed] [Google Scholar]
- 24.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furda AM, Marrangoni AM, Lokshin A, Van Houten B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA repair. 2012;11:684–692. doi: 10.1016/j.dnarep.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samson L, Cairns J. A new pathway for DNA repair in Escherichia coli. Nature. 1977;267:281–283. doi: 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- 27.Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science. 1984;223:594–597. doi: 10.1126/science.6695170. [DOI] [PubMed] [Google Scholar]
- 28.Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011;3:a000745. doi: 10.1101/cshperspect.a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013;41:8403–8420. doi: 10.1093/nar/gkt635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santa-Gonzalez GA, Gomez-Molina A, Arcos-Burgos M, Meyer JN, Camargo M. Distinctive adaptive response to repeated exposure to hydrogen peroxide associated with upregulation of DNA repair genes and cell cycle arrest. Redox Biol. 2016;9:124–133. doi: 10.1016/j.redox.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bridge G, Rashid S, Martin SA. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers (Basel) 2014;6:1597–1614. doi: 10.3390/cancers6031597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tudek B, Zdżalik-Bielecka D, Tudek A, Kosicki K, Fabisiewicz A, Speina E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radical Biology and Medicine. 2017;107:77–89. doi: 10.1016/j.freeradbiomed.2016.11.043. [DOI] [PubMed] [Google Scholar]
- 33.Saintigny Y, Chevalier F, Bravard A, Dardillac E, Laurent D, Hem S, Depagne J, Radicella JP, Lopez BS. A threshold of endogenous stress is required to engage cellular response to protect against mutagenesis. Sci Rep. 2016;6:29412. doi: 10.1038/srep29412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen KH, Yakes FM, Srivastava DK, Singhal RK, Sobol RW, Horton JK, Van Houten B, Wilson SH. Up-regulation of base excision repair correlates with enhanced protection against a DNA damaging agent in mouse cell lines. Nucleic Acids Res. 1998;26:2001–2007. doi: 10.1093/nar/26.8.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graupner A, Eide DM, Instanes C, Andersen JM, Brede DA, Dertinger SD, Lind OC, Brandt-Kjelsen A, Bjerke H, Salbu B, Oughton D, Brunborg G, Olsen AK. Gamma radiation at a human relevant low dose rate is genotoxic in mice. Sci Rep. 2016;6:32977. doi: 10.1038/srep32977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA repair. 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, Halweg C, Collins JB, Durr A, Fischbeck K, Van Houten B. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLoS Genet. 2010;6:e1000812. doi: 10.1371/journal.pgen.1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol Dis. 2014;70:214–223. doi: 10.1016/j.nbd.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim Biophys Acta. 2015;1853:1253–1271. doi: 10.1016/j.bbamcr.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Houten B, Kuper J, Kisker C. Role of XPD in cellular functions: To TFIIH and beyond. DNA Repair (Amst) 2016;44:136–142. doi: 10.1016/j.dnarep.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochemical Society Transactions. 2016;44:1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy Michael P, Holmgren A, Larsson N-G, Halliwell B, Chang Christopher J, Kalyanaraman B, Rhee Sue G, Thornalley Paul J, Partridge L, Gems D, Nyström T, Belousov V, Schumacker Paul T, Winterbourn Christine C. Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metabolism. 2011;13:361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmstrom KM, Kostov RV, Dinkova-Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016;1:80–91. doi: 10.1016/j.cotox.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correia-Melo C, Birch J, Passos JF. Powering senescence: The ugly side of mitochondria. Cell Cycle. 2016;15:2541–2542. doi: 10.1080/15384101.2016.1204852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Correia-Melo C, Passos JF. Demystifying the role of mitochondria in senescence. Mol Cell Oncol. 2016;3:e1162896. doi: 10.1080/23723556.2016.1162896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 48.Santos JH, Meyer JN, Skorvaga M, Annab LA, Van Houten B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 49.Singhapol C, Pal D, Czapiewski R, Porika M, Nelson G, Saretzki GC. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS One. 2013;8:e52989. doi: 10.1371/journal.pone.0052989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Listerman I, Sun J, Gazzaniga FS, Lukas JL, Blackburn EH. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013;73:2817–2828. doi: 10.1158/0008-5472.CAN-12-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann S, Spitkovsky D, Radicella JP, Epe B, Wiesner RJ. Reactive oxygen species derived from the mitochondrial respiratory chain are not responsible for the basal levels of oxidative base modifications observed in nuclear DNA of Mammalian cells. Free Radic Biol Med. 2004;36:765–773. doi: 10.1016/j.freeradbiomed.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 52.Cleaver JE, Brennan-Minnella AM, Swanson RA, Fong KW, Chen J, Chou KM, Chen YW, Revet I, Bezrookove V. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci U S A. 2014;111:13487–13492. doi: 10.1073/pnas.1414135111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Houten B. Pulmonary Arterial Hypertension Is Associated with Oxidative Stress-induced Genome Instability. Am J Respir Crit Care Med. 2015;192:129–130. doi: 10.1164/rccm.201505-0904ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang SC, Meagher M, Kassouf N, Hafezparast M, McKinnon PJ, Haywood R, El-Khamisy SF. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci Adv. 2017;3:e1602506. doi: 10.1126/sciadv.1602506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends in Pharmacological Sciences. 2017 doi: 10.1016/j.tips.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 56.He J, Wang Y, Missinato MA, Onuoha E, Perkins LA, Watkins SC, St Croix CM, Tsang M, Bruchez MP. A genetically targetable near-infrared photosensitizer. Nat Methods. 2016;13:263–268. doi: 10.1038/nmeth.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cesaratto L, Codarin E, Vascotto C, Leonardi A, Kelley MR, Tiribelli C, Tell G. Specific inhibition of the redox activity of ape1/ref-1 by e3330 blocks tnf-alpha-induced activation of IL-8 production in liver cancer cell lines. PLoS One. 2013;8:e70909. doi: 10.1371/journal.pone.0070909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fishel ML, Colvin ES, Luo M, Kelley MR, Robertson KA. Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines results in a hypersensitive response to retinoic acid-induced differentiation and apoptosis. Exp Hematol. 2010;38:1178–1188. doi: 10.1016/j.exphem.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environmental and molecular mutagenesis. 2010;51:391–405. doi: 10.1002/em.20575. [DOI] [PubMed] [Google Scholar]
- 61.Lee AJ, Wallace SS. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radical Biology and Medicine. 2017;107:170–178. doi: 10.1016/j.freeradbiomed.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]