

Abstract
The interplay between the primary and secondary coordination spheres is crucial to determining the properties of transition metal complexes. To examine these effects, a series of trigonal bipyramidal Co–OH complexes have been prepared with tripodal ligands that control both coordination spheres. The ligands contain combination of either urea or sulfonamide groups that control the primary coordination sphere through anionic donors in the trigonal plane and the secondary coordination sphere through intramolecular hydrogen bonds. Variations in the anion donor strengths were evaluated using electronic absorbance spectroscopy and a qualitative ligand field analysis to find that deprotonated urea donors are stronger field ligands than deprotonated sulfonamides. Structural variations were found in the CoII–O bond lengths that range from 1.953(4) to 2.051(3) Å; this range in bond lengths were attributed to the differences in the intramolecular hydrogen bonds that surround the hydroxido ligand. A similar trend was observed between the hydrogen bonding networks and the vibrations of the O–H bonds. Attempts to isolate the corresponding CoIII–OH complexes were hampered by their instability at room temperature.
Graphical Abstract
A series of trigonal bipyramidal Co–OH complexes have been prepared with tripodal ligands to examine the effects of both primary and secondary coordination spheres. The ligands contain a combination of either urea or sulfonamide groups that control the primary coordination sphere through anionic donors in the trigonal plane and the secondary coordination sphere through intramolecular hydrogen bonds. Structural and spectroscopic trends were found that correlate with the structures within the secondary coordination spheres.
Introduction
Regulating the properties and function of metal complexes is often achieved though control of their primary and secondary coordination spheres.1–6 Many systems been developed to correlate the changes in the primary coordination sphere with experimental observations. Less is known, however, about how the secondary coordination sphere effects the properties of metal ions. In an attempt to determine the influence of the secondary coordination sphere, ligands have been prepared that contain either H-bond donors or acceptors in order to understand the effects of intramolecular hydrogen bonding (H-bonding) networks on metal ion chemistry.1–14 For example, Fout has reported systems that regulate the secondary coordination sphere by using ligands that can tautomerize to function as either H-bond acceptors or donors.15–17 In addition, we have developed a family of symmetrical and hybrid tripodal ligands to probe how systematic changes in the H-bonding networks affect function.1–5 In one study we examined the effects of intramolecular H-bonds on the activation of dioxygen by a series of 4-coordinate CoII complexes with urea/amidate tripodal ligands.18 This series of ligands allowed us to vary the number of H-bond donors proximal to the CoII centers from three to zero, while maintaining similar primary coordination spheres, resulting in a relatively small variation of −0.25 V in their one-electron oxidation potentials. We thus attributed the differences in reactivity of the CoII complexes with O2 to the presence of varying numbers of H-bond donors within the secondary coordination sphere.
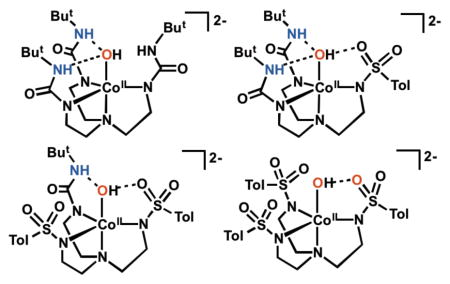
We now report a new series of tripodal ligand that contain urea/sulfonamido groups to further examine the effects of intramolecular H-bonding networks (Figure 1). Aspects of the coordination chemistry with the symmetrical tripods (tris[(N′-tert-butylureaylato)-N-ethylene] aminato) ([H3buea]3−) and N, N′, N″-(nitrilotris(ethane-2,1-diyl))tris(4-methylbenzene-sulfonamido) ([TST]3−) have been described previously,3,4,19–21 and we introduce here two new hybrid ligands N-(2-(bis(2-(3-(tert-butyl)ureido)ethyl)amino)ethyl)-4-methylbenzene-sulfonamido ([H22tol]3−) and N, N′-(((2-(3-(tert-butyl)ureido)ethyl)azanediyl)bis(ethane-2,1-diyl))bis(4-methylbenzenesulfonamido) ([H1tol]3−). Ligand [H22tol]3− contains two H-bond donors from the urea groups and a single H-bond acceptor from a lone sulfonamide group. In [H1tol]3− this pattern is reversed with two sulfonamide groups and one urea unit. This series of ligands allowed us to probe H-bond donor/acceptor relationships and examine their effects on structural and physical properties within a set of Co–OH complexes. In addition, we have qualitatively assessed how changes in the number of sulfonamide donors to the Co centers affects the electronic structure of the complexes, in particular the changes in the energies of the d-orbitals.
Figure 1.
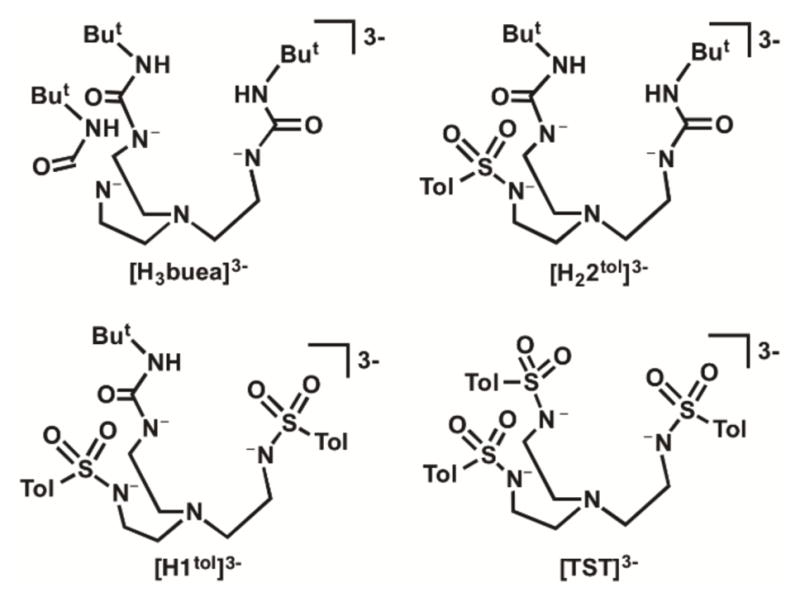
Tripodal ligands used in this study.
Experimental Section
Materials
N, N′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(4-methylbenzene-sulfonamide) and the ligand precursors 1,1′,1″-(nitrilotris(ethane-2,1-diyl))tris(3-(tert-butyl)urea) (H6buea), N, N′, N″-(nitrilotris(ethane-2,1-diyl)tris(4-methylbenzenesulfonamide) (H3TST), and the metal complex K2[CoII(OH)(H3buea)] were synthesized using literature procedures.22–24 For K2[CoII(OH)(H3buea)] a DMA/Et2O diffusion was used to crystallize the salt instead of DMA-MeCN/Et2O diffusion. The metal precursor Co(OAc)2 was purchased from Sigma Aldrich in ≥ 99% purity and used as received. Potassium hydride (KH) as a 30% dispersion in mineral oil was filtered with a glass frit and washed with pentane and Et2O and dried by vacuum and stored under an argon atmosphere. The syntheses of the metal complexes were conducted in an argon atmosphere.
Physical Methods
Room temperature electronic absorbance spectra were collected on a Cary 50 spectrometer and low-temperature electronic absorbance spectra were collected on a 8453 Agilent UV-vis spectrophotometer equipped with an Unisoku Unispeks cryostat with a 1.00 cm quartz cuvette. Electronic paramagnetic resonance (EPR) spectra were recorded using a Bruker EMX spectrometer equipped with an ER041XG microwave bridge, an Oxford Instrument liquid-helium quartz cryostat, and a dual mode cavity (ER4116DM). Fourier-transform infrared (FTIR) spectra were collected on a Varian 800 Scimitar series FTIR spectrometer in Nugol. 1H and 13C nuclear magnetic resonance (NMR) spectroscopies were conducted using a Bruker DRX500 spectrometer. Solution magnetic moments were determined by the Evan’s method using a Bruker DRX500 spectrometer.25 Cyclic voltammetric experiments were conducted using a CHI600C electrochemical analyzer. A 2.0 mm glassy carbon electrode was used as the working electrode at scan velocities of 100 mV s−1. A cobaltocenium/cobaltocene couple (CoCp2 +/CoCp2) (ΔEp = 0.136 V) was used to monitor the Ag wire reference electrode and all potentials are reference to the Fc+/Fc couple.
Preparative Methods
N, N′-(((2-(3-(tert-butyl)ureido)ethyl)azanediyl)bis(ethane-2,1-diyl))bis(4-methylbenzenesulfonamide) (H41tol)
N, N′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(4-methylbenzenesulfonamide) (2.507 g, 4.510 mmol) was dissolved in 120 mL of dichloromethane (CH2Cl2) and tert-butyl isocyanate (0.507 g, 5.11 mmol) dissolved in 60 mL of CH2Cl2 was added dropwise and stirred overnight. Volatiles were removed under reduced pressure to give a light yellow powder that was further dried under vacuum. The product was dissolved in ethyl acetate (EtOAc) and eluted through a silica plug with EtOAc to give 2.50 g (82%) of the desired product as a white solid. FTIR (KBr disc, cm−1): 3406, 3289, 2965, 2925, 2870, 1648, 1598, 1550, 1496, 1452, 1324, 1214, 1158, 1093, 950, 815, 707, 660, 551. 1H NMR (500 MHz, CDCl3, ppm): 1.32 (s, 9H), 2.42 (s, 6H), 2.14 (s, 6H), 2.46 (t, 2H), 2.51 (t, 4H), 2.96 (m, 4H), 3.15 (q, 2H), 4.96 (s, 1H), 5.22 (t, 1H), 5.89 (br s, 2H), 7.31 (d, 4H), 7.79 (d, 4H). 13C NMR (500 MHz, CDCl3, ppm): 158.41, 143.53, 136.86, 129.87, 127.15, 53.39, 50.28, 41.12, 37.59, 29.53, 21.60. HRMS (ES+) Exact mass calcd for C25H39N5O5S2Na [H41tol + Na]+, 576.2290. Found 576.2270.
tert-butyl (2-(bis(2-(3-(tert-butyl)ureido)ethyl)amino)ethyl)carbamate (6)
tert-butyl (2- (bis(2-aminoethyl)amino)ethyl)carbamate (1.292 g, 5.234 mmol) was dissolved in 60 mL of CH2Cl2 and a 60 mL solution of tert-butyl isocyanate (1.036 g, 10.45 mmol) was added dropwise over 30 min. The reaction was allowed to stir overnight under N2, after which the solvent was removed under vacuum to afford a white powder. This solid was dissolved in 5:100 solution of methanol: CH2Cl2 and was eluted through a silica plug with a 5:100 solution of methanol: CH2Cl2 to give 1.254 g (54%) of white solid. 1H NMR (500 MHz, CDCl3, ppm): 1.34 (s, 18H), 1.44 (s, 9H), 2.48 (m, 6H), 3.12 (m, 6H), 5.02 (m, 3H), 5.65 (s, 2H). 13C NMR (500 MHz, CDCl3, ppm): 158.71, 156.78, 79.44, 56.16, 55.43, 50.08, 39.10, 38.09, 29.67, 28.49. HRMS (ES+) [C21H44N6O4 + H]+, 445.3502. Found 445.3511.
1,1′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3-(tert-butyl)urea) (7)
tert-butyl (2-(bis(2-(3-(tert-butyl)ureido)ethyl)amino)ethyl)carbamate (6.415 g, 14.43 mmol) was dissolved in 40 mL of CH2Cl2, cooled to 0 ºC and 40 mL of trifluoroacetic acid was added dropwise under N2. The solution was allowed to stir for an additional 2 h and the volatiles were removed under reduced pressure to yield a colorless oil. This oil was stirred with 4 M NaOH until a pH of 11 was achieved. This solution was then extracted with 10×20 mL of CH2Cl2 and dried over Na2SO4. The solvent was removed by vacuum to afford 3.73 g (75%) of the desired product as a white solid. 1H NMR (500 MHz, CDCl3, ppm): 1.31 (s, 18H), 2.47 (m, 6H), 2.59 (t, 2H), 3.19 (q, 4H), 5.08 (s, 2H), 5.62 (m, 2H). 13C NMR (500 MHz, CDCl3, ppm): 158.70, 56.02, 50.07, 39.63, 38.10, 29.65 HRMS (ES+) [C16H36N6O2 + Na], 367.2798. Found 367.2808.
N-(2-(bis(2-(3-(tert-butyl)ureido)ethyl)amino)ethyl)-4-methylbenzenesulfonamide (H52tol)
1,1′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3-(tert-butyl)urea) (1.210 g, 3.517 mmol) and Et3N (0.613 g, 6.06 mmol) were dissolved in EtOAc (60 mL) and treated dropwise with 4-toluenesulfonyl chloride(0.674 g, 3.54 mmol) in EtOAc (60 mL) over 1 h. The mixture was allowed to stir overnight under N2, after which the resultant solution was washed with 2×20 mL of H2O and 15 mL of brine followed by drying over Na2SO4. The colorless solution was dried under reduced pressure to yield a white solid that was washed with diethyl ether (Et2O) and vacuumed dried overnight to yield 1.236 g (70.5%) of the desired white solid. 1H NMR (500 MHz, CDCl3, ppm): 1.32 (s, 18H), 2.40 (s, 3H), 2.47 (s, 6H), 2.99 (s, 2H), 3.14 (s, 4H), 5.22 (s, 2H), 5.60 (s, 2H), 6.22 (s, 1H), 7.31 (d, 2H), 7.76 (d, 2H). 13C NMR (500 MHz, CDCl3, ppm): 158.85, 143.31, 137.37, 129.80, 126.93, 55.53, 50.05, 46.30, 41.52, 37.87, 29.60, 21.55. HRMS (ES+) [C23H42SN6O4 + Na]+, 521.2886. Found 521.2872.
K2[CoII(OH)(H1tol)]
Solid KH (0.0289 g, 0.723 mmol) was treated with a 4 mL DMA solution of H41Tol (0.1003 g, 0.1811 mmol) and stirred until bubbling ceased. Co(OAc)2 (0.0322 g, 0.182 mmol) was then added as one portion and the resultant solution stirred for 20 min to produce a dark purple heterogeneous mixture. Water (3.5 μL, 0.19 mmol) was added via syringe that produced an immediate color change to pink. The mixture was stirred for 1.5 h followed by filtration through a medium porous-glass filter and the filtrate was concentrated to 1 mL under vacuum. To the pink solution 25 mL of Et2O was added that caused the precipitation of a pink solid which was collected on a medium porous-glass filter, washed with copious amounts of Et2O, and dried under vacuum. The pink solid was dissolved in 2 mL of DMA and 10 mL of Et2O was allowed to diffuse resulting in pink precipitate that was collected and dried on a glass fritted filter. 0.0586 g (45%) of the desired pink solid was obtained. FTIR (Nugol, cm−1): 3602, 3581, 3214, 3143, 1651, 1600, 1516, 1463, 1376, 1356, 1323, 1220, 1136, 1079, 1045, 1020, 976, 832, 809, 662. 604. UV/vis (DMA, nm (M−1 cm−1)): λmax (ε) = 470 (103), 493 (120), 528 (129), 610 (81), 637 (75), 690 (54), 820 (30). μeff (DMSO, 298K): 4.10 μB. Anal. Calcd (found) for C25H37CoK2N5O6S2: C, 42.60 (42.58); H, 5.29 (5.47); N, 9.94 (9.95). EPR (X-band, Perpendicular, DMA, 77K): g = 3.9
K2[CoII(OH)(H22tol)]
This salt was prepared using a similar procedure described above for K2[CoII(OH)(H1tol)] with H52tol (0.1974 g, 0.3958 mmol), KH (0.0649 g, 1.62 mmol), Co(OAc)2 (0.0710 g, 0.401 mmol), and H2O (7 μL, 0.4 mmol). The salt was isolated as a fine pink/purple powder after diffusion of Et2O into the DMA solution. The pink/purple solid was collected on a medium porous-glass filter, and washed with 20 mL MeCN, 20 mL of THF, and 20 mL of Et2O and dried by vacuum to give 0.1422 g (55 %) of the desired product. FTIR (Nugol mull, cm−1) 3625, 3218, 3142, 1646, 1593, 1514, 1464, 1400, 1378, 1357, 1330, 1268, 1228, 1221, 1132, 1125, 1105, 1074, 1048, 1019, 977, 891, 839, 817, 795, 720, 660. UV/Vis (DMA, nm (M−1 cm−1)): λ (ε) = 472 (80), 488 (90), 528 (120), 767 (30). μeff (DMSO, 298 K, Evan’s Method): 4.26 μB. Anal. Calcd (found) for C23H40CoK2N6O5S.DMA: C, 44.01 (44.28); H, 6.70 (6.74); N, 13.31 (13.17). EPR (X-band, DMA, 77K): g = 3.8.
K2[CoII(OH)TST]
H3TST (0.3254 g, 0.5345 mmol) was dissolved in 8 mL of DMA and added to 3 equiv of neat KH (0.0653 g, 1.63 mmol) and stirred until bubbling ceased. Co(OAc)2 (0.0951 g, 0.537 mmol) was added and stirred for 1 h to produce a dark blue mixture. The blue mixture was filtered through a glass fritted filter and H2O (12 μL, 0.67 mmol) was added to the filtrate to give a pink solution that was stirred for 15 min. KH (0.0222 g, 0.555 mmol) was added to the solution and it was stirred until bubbling ceased. The solution was filtered through a medium porous-glass filter and the filtrate was split into two vials and 10 mL of Et2O was allowed to diffuse which resulted in dark purple crystals that were washed with 20 mL of CH2Cl2 and 30 mL of Et2O and dried by vacuum to give 0.2407 g (59%) of the desired purple solid. FTIR (Nugol mull, cm−1) 3569, 1645, 1597, 1493, 1464, 1376, 1246, 1142, 1112, 1081, 1044, 1017, 974, 932, 872, 815, 735, 710, 663, 599, 556. UV/Vis (DMA, nm (M−1 cm−1)): λ (ε) = 522 (170), 660 (65), 890 (35). μeff (DMSO, 298K, Evan’s Method): 4.11 μB. Anal. Calcd (found) for C27H36CoK2N4O7S3.0.5CH2Cl2: C, 41.06 (41.16); H, 4.41 (4.52); N, 6.97 (6.79). EPR (X-band, 77K, perpendicular): g = 3.7.
Oxidation of CoII–OH Complexes
A stock solution of the CoII–OH complex was prepared in DMA (20 mL) at room temperature. A 3 mL aliquot of the solution of CoII–OH was transferred to a 1 cm quartz cuvette and was sealed with a rubber septum before being transferred to a precooled 8453 Agilent UV-vis spectrophotometer equipped with an Unisoku Unispeks cryostat and allowed to equilibrate to the desired temperature for at least 15 min. A stock solution of [FeCp2]BF4 was prepared in DMA. To the CoII–OH solution, 1 equiv of [FeCp2]BF4 was added via gas-tight syringe. The high energy band corresponding to the CoIII species between 400 – 500 nm was monitored to determine the half-lives of the CoIII–OH compounds (Table S1) and treated as 2nd order reaction: half-lives were determined from 1/k[CoIII complex].
Molecular Structure Determination
A Bruker SMART APEX II diffractometer was used molecular structure determination. The APEX226 program package was used to determine the unit-cell parameters and for data collection. The raw data was processed using SAINT27 and SADABS28 to yield the reflection data file. Subsequent calculations were carried out using the SHELXTL29 program. The structures were solved by direct methods and refined to convergence. Crystallographic data of the structure determination are given in Table 1. Selected bond lengths and angles are given in Table 2 and the molecular structures of the anions for each salt are shown in Figure 2. The structure of K2[CoII(OH)(TST)] contained three anions within the asymmetric that were chemically the same. Two of the anions have disorder within the [TST]3− ligand: the anion containing Co2 has two tolyl groups that are disordered and anion containing Co3 has one disordered tolyl group. For the anion containing Co2 the disorder was modeled using multiple components using partial site-occupancy factors (50:50 for both rings); for the anion with Co3 the disorder was modeled with 50:50 site occupancy. The anion containing Co3 also had partial disorder within the tren backbone that was modeled as 50:50 disorder. The anion of [CoIITST(OH)]2− that was not disordered is shown in Figure 2 and its metrical parameters were used in Table 2.
Table 1.
Crystallographic Data for K2[CoIITST(OH)], K2[CoIIH22tol(OH))].2DMA.
| Salt | K2[CoIITST(OH)] | K2[CoIIH22tol(OH)]•2DMA |
|---|---|---|
| Empirical Formula | C81H102Co3K6N12O21S9 | C31H58CoK2N8O7S |
| fw | 2277.65 | 824.04 |
| T (K) | 88(2) | 88(2) |
| space group | P21/c | P1̄ |
| a (Å) | 19.4569(14) | 11.9841(7) |
| b (Å) | 32.796(2) | 12.9122(7) |
| c (Å) | 18.3848(13) | 14.8689(8) |
| α (deg) | 90 | 68.6652(7) |
| β (deg) | 111.2036(9) | 87.4545(8) |
| γ (deg) | 90 | 69.8767(7) |
| Z | 4 | 2 |
| V (Å3) | 10943.1(13) | 2003.73(19) |
| δcalcd (Mg/m3) | 1.384 | 1.366 |
| R1 | 0.0804 | 0.0378 |
| wR2 | 0.2098 | 0.0942 |
| GOF | 1.058 | 1.025 |
Table 2.
Selected bond distances and angles for the three structurally characterized CoII—OH complexes
| Distances (Å) or angles (deg) | [CoIITST(OH)]2− | [CoIIH22tol(OH)]2− | [CoIIH3buea(OH)]2−a |
|---|---|---|---|
| Co1—N1 | 2.224(5) | 2.213(2) | 2.216(5) |
| Co1—N2 | 2.081(5) | 2.039(2) | 2.079(4) |
| Co1—N3 | 2.147(4) | 2.175(2) | 2.075(4) |
| Co1—N4 | 2.128(6) | 2.049(2) | 2.046(4) |
| Co1—O1 | 1.953(4) | 2.015(2) | 2.051(4) |
| d(O1…O2) | 2.808(6) | 2.950(2) | – |
| d(O1…N5) | – | 2.741(2) | 2.829(5) |
| d(O1…N6) | – | 2.851(3) | 2.781(5) |
| d(O1…N7) | – | – | 2.802(5) |
| N1—Co1—N2 | 78.7(2) | 80.74(6) | 80.2(1) |
| N1—Co1—N3 | 78.43(18) | 79.12(6) | 80.7(1) |
| N1—Co1—N4 | 78.5(2) | 81.04(6) | 80.2(2) |
| N2—Co1—N3 | 126.7(2) | 108.69(6) | 108.1(1) |
| N2—Co1—N4 | 110.6(3) | 117.79(7) | 125.0(2) |
| N3—Co1—N4 | 111.6(2) | 125.09(6) | 108.1(1) |
| O1—Co1—N1 | 172.1(2) | 176.40(6) | 179.3(1) |
ref. 40
Figure 2.

Thermal ellipsoid plots of [CoIIH22tol(OH)]2− (A), and [CoIITST(OH)]2− (B). Thermal ellipsoids are drawn at the 50% probability level and only urea and hydroxido hydrogen atoms are shown for clarity.
Results and Discussion
Design Considerations and Synthesis
We have previously utilized symmetrical tripodal ligand based on amides,18,30–33 ureas,3,4,24 and sulfonamides4,34 to study the effects of the secondary coordination sphere on the chemistry of metal ions. As part of these investigations we also prepared several hybrid ligands that contained urea groups and other functionality such as amides or pyridylcarboxyamido units.35,36 We have now extended this investigation to include hybrid tripodal ligands H41tol and H52tol that contain both urea and sulfonamide groups and prepared their corresponding CoII—OH complexes. Combining urea and sulfonamides allows for the systematic modulation of the secondary coordination sphere through variation in the intramolecular H-bonding networks that surround the CoII—OH units. Our earlier work showed that incorporation of urea groups into tripodal ligands provides excellent H-bond donors to metal-oxido/hydroxido units, whereas sulfonamide groups serve as H-bond acceptors. For instance, the symmetric urea-based tripod [H3buea]3− forms stable monomeric M—oxido complexes (M = Mn, Fe) and M′–OH complexes (M′ = Mn, Fe, Co, Zn, In).3,4,24,37–41 The accessibility to these high valent complexes was attributed to the highly anionic ligand field that was provided by the deprotonated nitrogen donors from [H3buea]3− and oxido/hydroxido ligands. In contrast to [H3buea]3−, the symmetrical sulfonamide-based ligand (N, N′, N″-(nitrilotris(ethane-2,1-diyl))tris(2,4,6-trimethylbenzene-sulfonamido)) [MST]3− (Figure 1) utilizes the sulfonamido oxygen atoms as H-bond acceptors. This ligand system has been shown to stabilize metal-aqua complexes with FeII and CoII ions.19,42 Additionally, the [MST]3− platform has been shown to facilitate binding of secondary metal ions, such as Ca2+, Sr2+, and Ba2+ ions, through the sulfonamide oxygen atoms.34,43 Several heterobimetallic complexes supported by the [MST]3− ligand scaffold with two different transition metal ions bridged by a single hydroxide have also been isolated and characterized, including complexes with FeII/FeIII, MnII/FeIII, and MnII/MnIII cores.43,44 Two new hybrid ligands, H41tol and H52tol were designed that have differing combinations of sulfonamide or urea based donors. Together with [H3buea]3−and [TST]3− (a ligand similar to [MST]3− but with tolyl groups replacing the mesityl groups on the sulfonamido moieties) these two new tripods complete a series of ligands which the both the primary and secondary coordination spheres can be systematically varied.
The preparation of H41tol followed a four-step route that began with treating tris(2-aminoethyl)amine (tren) with di-tert-butyl dicarbonate to afford the mono-boc protected species 3 (76% yield) which was converted to the disulfonamide 4 (84% yield) after addition of 2 equiv of 4-toluenesulfonyl chloride (Scheme S1). Compound 4 was deprotected (95% yield) with trifluoroacetic acid (TFA) and then treated with tert-butyl isocyanate to afford H41tol in 82% yield after purification. The preparation of H52tol was achieved from the diurea compound 6 and 1 equiv of tosyl chloride to give H52tol in 70% yield (Scheme 1).
Scheme 1.

Preparative route to H52tol.
Scheme 2 outlines the synthesis of the four CoII complexes. The CoII—OH complex of [TST]3− was synthesized by deprotonating the ligand with 3 equiv of KH under an argon atmosphere and then adding solid Co(OAc)2 to produce a dark blue solution and a white solid (KOAc, 2 equiv). After filtration to remove the KOAc, the solution was treated with 1 equivalent of H2O that induce color change to pink. Potassium hydride (1 equiv) was added to the mixture and stirred until gas evolution ceased to give a purple solution. K2[CoIITST(OH)] was purified via recrystallization using vapor diffusion of Et2O in DMA solution of the salt. The remaining Co(II)–OH complexes were prepared in a similar manner with the following modifications: deprotonation of [H1tol]3−, [H22tol]3−, and [H3buea]3− were achieved with 4 equiv of KH, followed by addition of solid Co(OAc)2 that produced a blue solution and solid KOAc (2 equiv) which was removed by filtration. K[CoII(OH)(H1tol)], K[CoII(OH)(H22tol)], and K[CoII(OH)(H3buea)] were formed upon the addition of 1 equiv of H2O and recrystallized using the method described for K[CoIITST(OH)]. All the CoII–OH complexes are high spin as determined from their room temperature magnetic moments that range from 4.10 and 4.49 μB and their perpendicular-mode EPR spectra measured at 77 K that contain a prominent feature at g = 3.9.
Scheme 2.

Preparative routes to CoII–OH complex. The synthesis of [CoIIH3buea(OH)]2− has already been reported.24 Reagents: (a) 4 KH; (b) Co(OAc)2; (c) H2O; (d) 3 KH; (e) Co(OAc)2; (f) H2O; (g) KH.
Structural Properties
We have examined the molecular structures of the CoII–OH complexes using X-ray diffraction methods (Figure 2, Tables 1 & 2). Single crystals of [CoIIH1tol(OH)]2− were not obtained and the structure of [CoIIH3buea(OH)]2− has been report previously (Figure S1;24,40 it is discussed here in comparison with the other complexes. The molecular structures of [CoIITST(OH)]2−, [CoIIH22tol(OH)]2−, and [CoIIH3buea(OH)]2− reveals that each complex had a trigonal bipyramidal coordination geometry around the Co center with the trigonal plane being defined by the three deprotonated nitrogen donors. There are statistically significant differences between the Co1–N bond lengths in the trigonal plane between the different N-donors with those involving deprotonated urea generally being shorter. For instance, an average Co1–Ntrig bond distance of 2.057(2) Å was found in [CoIIH3buea(OH)]2− that increased to 2.118(6) Å in [CoIITST(OH)]2−. A trend in the Co1–O1 bond lengths reflects the type of intramolecular H-bonding networks present in each complex. The H-bonds within [CoIIH3buea(OH)]2− arise from the urea NH donors interacting with O1 to produce the longest Co1–O1 bond distance at 2.051(3) Å. The Co1–O1 bond length contracts to 2.015(2) Å in [CoIIH22tol(OH)]2− which contains one sulfonamido group that serves as an H-bond acceptor. The shortest Co1–O1 bond length at 1.953(4) Å was observed in [CoIITST(OH)]2−: this complex contains only a single intramolecular H-bond in which the hydroxido ligand is the H-bond donor and a sulfonamide oxygen atom (O2) is the H-bond acceptor. This lone H-bond has an O1…O2 distance of 2.808(6) Å that is significantly shorter than that found in [CoIIH22tol(OH)]2−.
A trend was also observed for the displacement of the cobalt center from the trigonal plane. Each Co center is positioned out of the trigonal plane toward O1. The greatest displacement of 0.440 Å was seen in [CoIITST(OH)]2− while the other distances shorten with an increase in the number of NH…O1 H-bonds: 0.351 Å in [CoIIH22tol(OH)]2− and 0.346 Å in [CoIIH3buea(OH)]2−.
Our group45 and others6 have found correlations between M–OHn bond lengths and the number of intramolecular H-bonds. Specifically, the correlation involves the hydroxido (or aquo) ligands being the H-bond acceptor and NH groups being the H-bond donors. A lengthening of the M–O(H) bond occurs as the number of intramolecular H-bonds increases because they remove electron density from the hydroxido ligand. In the series of CoII–OH complexes in this study, we have introduced H-bond acceptors so that now the OH group can also function as an H-bond donor; this type of H-bond should lead to a shortening of the Co1–O1 bond distance because the hydroxido ligand becomes more anionic. The contraction observed for the Co1–O1 bond lengths in going from [CoIIH3buea(OH)]2− to [CoIIH22tol(OH)]2− is thus attributed to the hydroxido ligand in [CoIIH22tol(OH)]2− acting as an H-bond donor. This change was attenuated because [CoIIH22tol(OH)]2− still has two NH groups that H-bond to the O1. In the absence of urea groups, the Co1–O1 bond distance in [CoIITST(OH)]2− is significantly shorter and produces the strongest H-bond involving the hydroxido ligand. The differing intramolecular H-bond networks between the complexes also appears to affect the N1-Co1-O1 angles, which is the smallest in [CoIlTST(OH)]2− and gradual increases with the number of urea groups (Table 2).
Vibrational Properties
The vibrational properties of the complexes in the solid state were investigated using Fourier transform infrared (FTIR) spectroscopy (Figure 3, Table 3). In particular, we probed the vibrations of the O–H bonds (ν(OH)) in the hydroxido ligands to further understand the H-bonding interactions. [CoIlTST(OH)]2− has the lowest energy ν(OH) at 3574 cm−1 and also the broadest peak with a full-width-at-half height value of 24 cm−1. For [CoIIH1tol(OH)]2− a split band was observed at 3581 and 3602 cm−1 and for [CoIIH22tol(OH)]2− a single peak was found at 3625 cm−1; these features are narrower than that observed in [CoIlTST(OH)]2−. Note that we are still unable to detect the ν(OH) in [CoIIH3buea(OH)]2−.24,40
Figure 3.
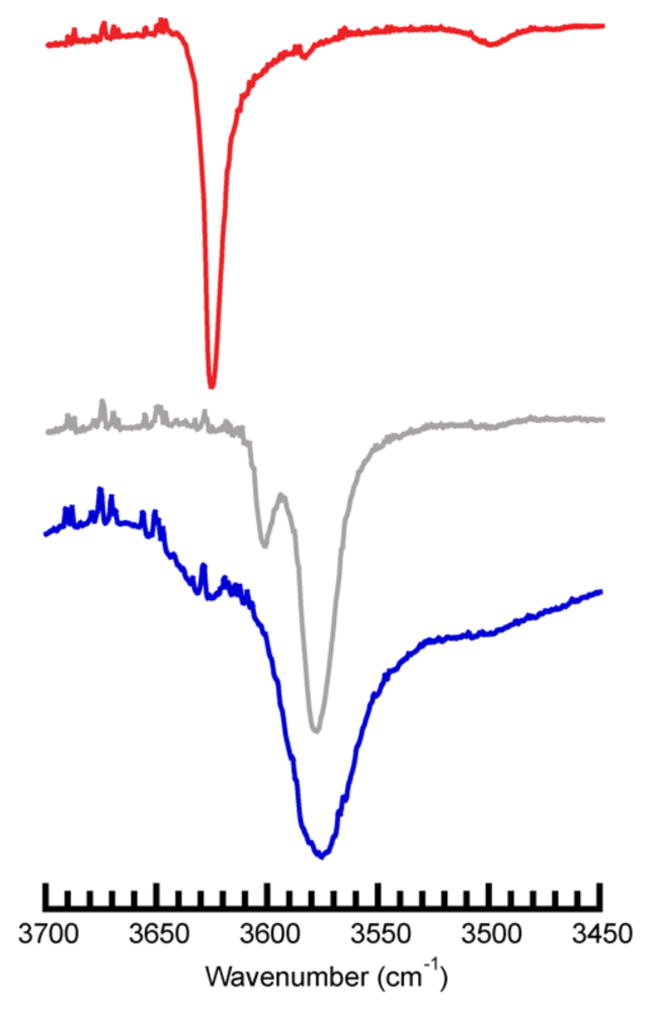
Solid-state FTIR spectra of [CoIIH22tol(OH)] (
 ), [CoIIH1tol(OH)] (
), [CoIIH1tol(OH)] (
 ), K2[CoIITST(OH)] (
), K2[CoIITST(OH)] (
 ).
).
Table 3.
Selected Physical Properties for the CoII—OH Complexes.
| λmax (nm), (E, cm−1) | ν(O—H) (FWHM, cm−1) | E1/2 (Ea) (V vs [FeCp2]+/0) | ΔE (V), ia/ic | |
|---|---|---|---|---|
| [CoIITST(OH)]2− | 890, (11,200) | 3574 (24) | N/A (−0.52) | – |
| [CoIIH1tol(OH)]2− | 820, (12,200) | 3602, 3581 (16) | −0.905 (−0.58) | 0.485, 1.04 |
| [CoIIH22tol(OH)]2− | 767, (13,000) | 3625 (9) | −0.828 (−0.71) | 0.380, 1.12 |
| [CoIIH3buea(OH)]2− | 710, (14,100) | N/A | −1.28 (−1.17) | 0.215, 1.07 |
We have previously found that the energies of the ν(OH) bands are affected by H-bonding networks and correlate with the M–O(H) bond distances. In our earlier systems, the hydroxido ligand served as the H-bond acceptor and the HNurea groups as the donors. The series of the CoII–OH complexes described here are different in that all except [CoIIH3buea(OH)]2− have hydroxido ligands that can serve as H-bond donors; nevertheless, similar trends were observed. For example, our vibrational analysis showed that [CoIlTST(OH)]2− has the lowest energy ν(OH) band and a commensurate short Co1–O1 bond length was observed (Table 2). These findings are consistent with a strong intramolecular H-bond from the hydroxido ligand to a sulfonamido oxygen atom (OH…O). In addition, the ν(OH) bands broaden as the H-bond strengthens (CoII–OH…O); similar broadening effects have been observed in organic systems (e.g., NH…O=CR2) that are attributed to an increase in the strength of intramolecular H-bonds.46
Electronic Absorbance Properties
The electronic absorbance spectra for the CoII–OH complexes measured in DMA (Figures 4 & S2, Table 3) are characterized by bands at λmax ~ 470, 550, and 600 nm with extinction coefficients less than 150 M−1 cm−1. In addition, each spectrum contained a d-d band between λmax ~ 700 – 900 nm that is also present in previously reported CoII—OH complexes with trigonal bipyramidal geometry.24 This low-energy band can be used to evaluate the relative contributions of the deprotonated urea and sulfonamide donors to the ligand field. For a high-spin CoII center in C3v symmetry, ligand field theory predicts that this band arises from the 4A1 → 4E transition whose energy is sensitive to the types of donors within the equatorial plane (Figure S3).47 We considered only σ-interactions in this qualitative analysis and assumed that π-interactions involving the axial ligands remain relatively constant within the series of complexes. The analysis found that deprotonated urea donors are stronger field ligands than deprotonated sulfonamide groups. The low-energy absorbance band for ureayl-based system [CoIIH3buea(OH)]2− is observed at 14,100 cm−1 (710 nm) which shifts to 11,200 cm−1 (890 nm) in sulfonamido-based complex [CoIITST(OH)]2−. The two complexes that contain hybrid ligands, [CoIIH1tol(OH)]2− and [CoIIH22tol(OH)]2− have transitions at 12,200 cm−1 (820 nm) and 13,000 cm−1 (767 nm), respectively. The incremental increase of approximately 1000 cm−1 observed for this band correlates with the number of deprotonated urea donors in the CoII–OH complexes and most likely reflects their increased basicity compared to deprotonated sulfonamido groups.
Figure 4.
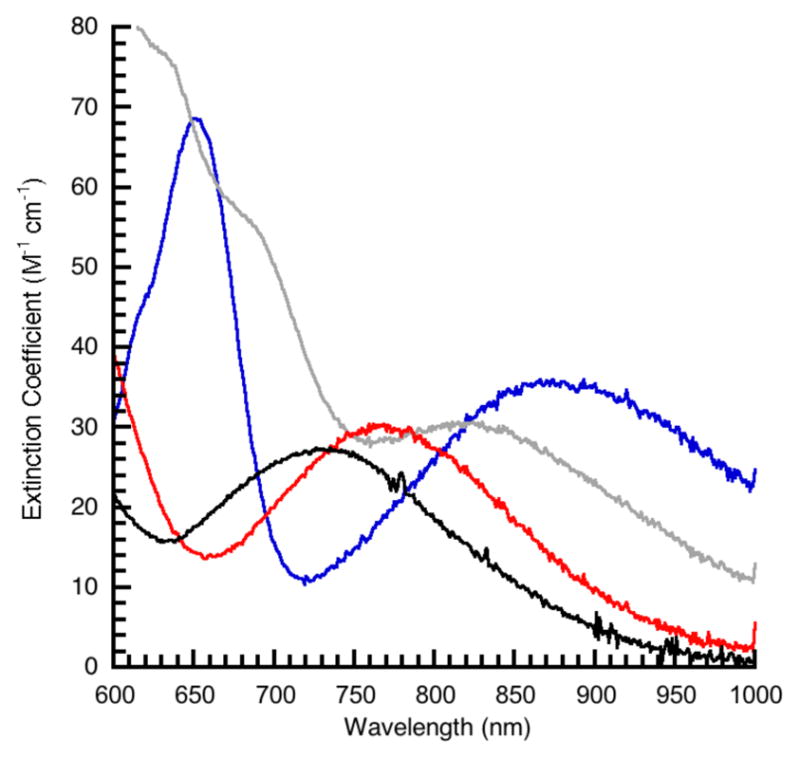
Electronic absorbance spectra for [CoIIH3buea(OH)]2− (—), [CoIIH22tol(OH)]2− (
), [CoIIH1tol(OH)]2− (
), [CoIITST(OH)]2− (
) recorded in DMA.
Electrochemical Properties
Cyclic voltammetry (CV) was used to evaluate the redox properties of the Co–OH complexes. CV experiments done at room temperature in DMA showed that each complex had a one-electron event that we assigned to the CoIII/CoII redox process (Figure 5). The process was quasi-reversible for all the complexes except [CoIITST(OH)]2− which exhibited an irreversible oxidation; because of this result, only the anodic potentials (Ea) will be compared. The Ea values for the CoII–OH complexes range from −0.52 to −1.17 V vs FeCp2+/FeCp2, illustrating that this series of tripodal ligands can effectively modulate the redox potentials of Co center. Consistent with our findings that deprotonated urea groups are stronger donors, [CoIIH3buea(OH)]2− and [CoIIH22tol(OH)]2− are the best at stabilizing the CoIII state by have the lowest Ea values of −1.17 and −0.71 V. The 0.46 V difference in potentials between these complexes is significantly larger than the 0.13 V difference observed between and [CoIIH22tol(OH)]2− and [CoIIH1tol(OH)]2−, even though the complexes within each pair only differ by one sulfonamido arm. An even smaller change of 0.06 V is observed when comparing Ea values for [CoIIH1tol(OH)]2− and [CoIITST(OH)]2−. These smaller changes in Ea values could reflect the contributions from the hydroxido ligand which become a stronger donor as the number of urea ligand diminishes (see structural properties).
Figure 5.
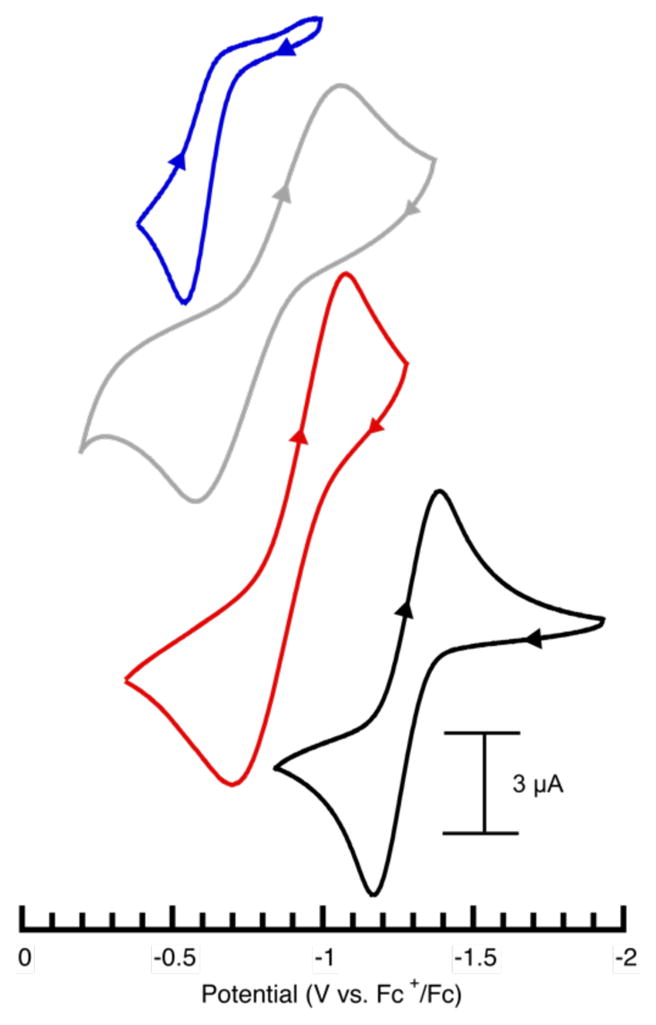
Cyclic voltammograms of [CoIIH3buea(OH)]2− (—), [CoIIH22tol(OH)]2− (
), [CoIIH1tol(OH)]2− (
), [CoIITST(OH)]2− (
) recorded in DMA. Measurements were done at room temperature under Ar, with a scan rate of 100 mV.s−1.
Oxidized Species
The results from the electrochemical experiments suggested that the CoIII–OH analogs could be accessed at negative potentials. However, the stabilities of the oxidized products are not the same throughout the series, as exemplified by the irreversibility of the cyclic voltammogram for [CoIITST(OH)]2−. These results prompted us to further examine the stability of the CoIII–OH species generated via chemical oxidation. Based on our CV data, ferrocenium was used as the oxidant and reactions were monitored spectrophotometrically using the diagnostic spectral features we reported for trigonal bipyramidal CoIII–OH (Figure S4).42 Our previous studies have established that [CoIIIH3buea(OH)]− is stable enough at room temperature to allow for its isolation as a crystalline solid.24 Similar experiments with the oxidized form of [CoIIH22tol(OH)]2− produced a species with an absorbance spectrum that is characteristic of a CoIII–OH complex with local C3 symmetry (Figure S4). This species slowly decayed in solution at room temperature with t1/2 ~ 5 d but we were still unable to isolate it as a pure solid (Figure S5A). The oxidized form of [CoIIH1tol(OH)]2− had limited stability at room temperature with a t1/2 = 16 min at 25°C (Figure S5B). In addition, both [CoIIIH1tol(OH)]− and [CoIIITST(OH)]− were only stable at temperature less than −50°C. [CoIITST(OH)]2− decayed too quickly for us to measure its half-life at room temperature but measured a t1/2 = 13 min at −20°C (Figure S5B).
The stability observed for the CoIII–OH complexes is reminiscent of results we reported for a related series of complexes.18 In this previous work, CoIII–OH complexes were prepared with urea/amidate tripodal ligands in which the number of ureayl H-bond donors were varied from three to zero. The stabilities of the CoIII–OH complexes diminished with fewer urea groups that we attributed to a reduction in the number of intramolecular H-bonds. In the present study, the cause for the differing stabilities of the CoIII–OH complexes is less straightforward; for example, [CoIIIH3buea(OH)]− and [CoIIIH22tol(OH)]− contain the same number of intramolecular H-bonds but have substantially different stabilities. One possibility is that the “type” of intramolecular H-bond involving the hydroxido ligand is an important factor (that is, whether the hydroxido ligand servers as an H-bond donor, acceptor, or both). In [CoIIIH22tol(OH)]− the hydroxido ligand presumably serves as both an H-bond donor and acceptor (as it does in [CoIIH22tol(OH)]2−). In addition, we showed that there are substantial differences in the one-electron oxidation potentials for the CoIII–OH complexes that also correlates with their stabilities. The observed stabilities of these CoIII–OH most likely result from the combination of influences involving both coordination spheres.
Summary and Conclusions
We have described preparation routes to the hybrid tripodal ligands, [H1tol]3− and [H22tol]3− containing urea and sulfonamide groups. Together with the symmetrical ligand [TST]3− and [H3buea]3−, they form a series of ligands that we used to probe the effects of H-bond donors/acceptors on the properties of Co–OH complexes. The molecular structures for three of the CoII–OH complexes showed trigonal bipyramidal primary coordination sphere which is expected for complexes with these types of tripodal ligands. The structural data illustrated the effects of placing an H-bond acceptor within the secondary coordination: we observed a shortening of the Co–O1 bond length in moving from [CoIIH3buea(OH)]2− to [CoIIH22tol(OH)]2−, then [CoIITST(OH)]2− that reflects the changes in the H-bonding network that surrounds the Co–OH unit. This effect was corroborated with FTIR spectral studies that showed commensurate changes in the energies of the frequencies associated with the Co–O1 bonds. The effect on the electronic structure induced from changes in the primary coordination sphere for the CoII–OH complexes was assessed using a qualitative ligand field approach that examined changes in energy for the 4A1 → 4E transition. Based on this analysis, deprotonated urea groups are stronger field ligands than deprotonated sulfonamide and each additional ureayl within the equatorial plane increases the transition energy by ~1000 cm−1. We also observed shifts in the one-electron oxidation potentials that, again, agree with the changes in both coordination spheres. The CoIII–OH analogs had varying stabilities at room temperatures that appear to correlate with the number of urea groups within the tripodal ligands. These results highlight the instability of trigonal bipyramidal CoIII–OH at room temperature and suggests their potential use in chemical oxidation.42 However, we were unable to identify a single specific causes for the observed trends in stabilities of the CoIII–OH species. This finding illustrates the subtle interplay between the primary and secondary coordination spheres in determining the properties of metal complexes.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health USA (GM050781) for financial support and Professor A. Heyduk for helpful discussions.
Footnotes
Supporting Information. Final crystallographic data in cif format, Figures S1–S5, the route to prepare H41tol (Scheme S1), and complete absorbance data (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shook RL, Borovik AS. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shook RL, Borovik AS. The Effects of Hydrogen Bonds on Metal-Mediated O2activation and Related Processes. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borovik AS. Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 4.Cook SA, Borovik AS. Molecular Designs for Controlling the Local Environments around Metal Ions. Acc Chem Res. 2015;48:2407–2414. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook SA, Hill EA, Borovik AS. Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry. 2015;54:4167–4180. doi: 10.1021/acs.biochem.5b00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Natale D, Mareque-Rivas JC. The Combination of Transition Metal Ions and Hydrogen-Bonding Interactions. Chem Commun. 2008:425–437. doi: 10.1039/b709650j. [DOI] [PubMed] [Google Scholar]
- 7.Mareque-Rivas JC, Prabaharan R, Martin de Rosales RT. Relative Importance of Hydrogen Bonding and Coordinating Groups in Modulating the Zinc-Water Acidity. Chem Commun. 2004:76–77. doi: 10.1039/b310956a. [DOI] [PubMed] [Google Scholar]
- 8.Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo-Copper(II) Complex with Tripodal Pyridylamine Ligands. Angew Chemie Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Mareque-Rivas JC, Salvagni E, Parsons S. Investigating the Effect of Hydrogen Bonding Environments in Amide Cleavage Reactions at Zinc(II) Complexes with Intramolecular Amide Oxygen Co-Ordination. Dalt Trans. 2004:4185–4192. doi: 10.1039/b414223c. [DOI] [PubMed] [Google Scholar]
- 10.Berreau LM, Makowska-Grzyska MM, Arif Atta M. Modeling the Active Site Chemistry of Liver Alcohol Dehydrogenase: Mononuclear Zinc Methanol and N, N-Dimethylformamide Complexes of a Nitrogen/Sulfur Ligand Possessing an Internal Hydrogen Bond Donor. Inorg Chem. 2001;40:2212–2213. doi: 10.1021/ic001190h. [DOI] [PubMed] [Google Scholar]
- 11.Kendall AJ, Zakharov LN, Gilbertson JD. Synthesis and Stabilization of a Monomeric Iron(II) Hydroxo Complex via Intramolecular Hydrogen Bonding in the Secondary Coordination Sphere. Inorg Chem. 2010;49:8656–8658. doi: 10.1021/ic101408e. [DOI] [PubMed] [Google Scholar]
- 12.Moore CM, Quist DA, Kampf JW, Szymczak NK. A 3-Fold-Symmetric Ligand Based on 2-Hydroxypyridine: Regulation of Ligand Binding by Hydrogen Bonding. Inorg Chem. 2014;53:3278–3280. doi: 10.1021/ic5003594. [DOI] [PubMed] [Google Scholar]
- 13.Szymczak NK, Oelkers AB, Tyler DR. Detection of Hydrogen Bonding in Solution: A 2H Nuclear Magnetic Resonance Method Based on Rotational Motion of a Donor/acceptor Complex. Phys Chem Chem Phys. 2006;8:4002–4008. doi: 10.1039/b608147a. [DOI] [PubMed] [Google Scholar]
- 14.Blacquiere JM, Pegis ML, Raugei S, Kaminsky W, Forget A, Cook SA, Taguchi T, Mayer JM. Synthesis and Reactivity of Tripodal Complexes Containing Pendant Bases. Inorg Chem. 2014;53:9242–9253. doi: 10.1021/ic5013389. [DOI] [PubMed] [Google Scholar]
- 15.Matson EM, Bertke JA, Fout AR. Isolation of Iron(II) Aqua and Hydroxyl Complexes Featuring a Tripodal H-Bond Donor and Acceptor Ligand. Inorg Chem. 2014;53:4450–4458. doi: 10.1021/ic500102c. [DOI] [PubMed] [Google Scholar]
- 16.Matson EM, Park YJ, Fout AR. Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J Am Chem Soc. 2014;136:17398–17401. doi: 10.1021/ja510615p. [DOI] [PubMed] [Google Scholar]
- 17.Park YJ, Matson EM, Nilges MJ, Fout AR. Exploring Mn-O Bonding in the Context of an Electronically Flexible Secondary Coordination Sphere: Synthesis of a Mn(III)-Oxo. Chem Commun. 2015;51:5310–5313. doi: 10.1039/c4cc08603a. [DOI] [PubMed] [Google Scholar]
- 18.Lucas RL, Zart MK, Murkerjee J, Sorrell TN, Powell DR, Borovik AS. A Modular Approach toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J Am Chem Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]
- 19.Lau N, Ziller JW, Borovik AS. Sulfonamido Tripods: Tuning Redox Potentials via Ligand Modifications. Polyhedron. 2015;85:777–782. doi: 10.1016/j.poly.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blake MP, Kaltsoyannis N, Mountford P. Heterobimetallic Complexes Containing Ca–Fe or Yb–Fe Bonds: Synthesis and Molecular and Electronic Structures of [M{CpFe(CO)2}2 (THF)3 ]2 (M = Ca or Yb) J Am Chem Soc. 2011;133:15358–15361. doi: 10.1021/ja207487j. [DOI] [PubMed] [Google Scholar]
- 21.Schwarz AD, Herbert KR, Paniagua C, Mountford P. Ligand Variations in New Sulfonamide-Supported Group 4 Ring-Opening Polymerization Catalysts. Organometallics. 2010;29:4171–4188. [Google Scholar]
- 22.DiVittorio KM, Hofmann FT, Johnson JR, Abu-Esba L, Smith BD. Facilitated Phospholipid Translocation in Vesicles and Nucleated Cells Using Synthetic Small Molecule Scramblases. Bioorg Med Chem. 2009;17:141–148. doi: 10.1016/j.bmc.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Motekaitis RJ, Murase I, Martell AE. The Synthesis of Binucleating Polyaza Macrocyclic and Macrobicyclic Ligands and the Dioxygen Affinities of Their Cobalt Complexes. Tetrahedron. 1995;51:77–88. [Google Scholar]
- 24.Hammes BS, Young VG, Jr, Borovik AS. Hydrogen-Bonding Cavities about Metal Ions: A Redox Pair of Coordinatively Unsaturated Paramagnetic Co-OH Complexes. Angew Chemie Int Ed. 1999;38:666–669. doi: 10.1002/(SICI)1521-3773(19990301)38:5<666::AID-ANIE666>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 25.Evans DF. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J Chem Soc. 1959:2003–2005. [Google Scholar]
- 26.Bruker AXS, I. APEX2 Version 201411-0. Madison, WI: 2014. [Google Scholar]
- 27.Bruker AXS, I. SAINT Version 834a. Madison, WI: 2013. [Google Scholar]
- 28.Sheldrick GM. SADABS. Bruker AXS, Inc; Madison: 2014. [Google Scholar]
- 29.Sheldrick GM. SHELXTL. Bruker AXS, Inc; Madison: 2014. [Google Scholar]
- 30.Shirin Z, Borovik SA, Young GV., Jr Synthesis and Structure of a MnIII(OH) Complex Generated from Dioxygen. Chem Commun. 1967;1997 [Google Scholar]
- 31.Ray M, Golombek AP, Hendrich MP, Young VG, Borovik AS. Synthesis and Structure of a Trigonal Monopyramidal Fe(II) Complex and Its Paramagnetic Carbon Monoxide Derivative. J Am Chem Soc. 1996;118:6084–6085. [Google Scholar]
- 32.Ray M, Hammes BS, Yap GPA, Rheingold AL, Borovik AS. Structure and Physical Properties of Trigonal Monopyramidal Iron(II), Cobalt(II), Nickel(II), and Zinc(II) Complexes. Inorg Chem. 1998;37:1527–1532. [Google Scholar]
- 33.Ray M, Golombek AP, Hendrich MP, Yap GPA, Liable-Sands LM, Rheingold LA, Borovik AS. Structure and Magnetic Properties of Trigonal Bipyramidal Iron Nitrosyl Complexes. Inorg Chem. 1999;38:3110–3115. [Google Scholar]
- 34.Park YJ, Ziller JW, Borovik AS. The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS. A Monomeric MnIII–Peroxo Complex Derived Directly from Dioxygen. J Am Chem Soc. 2008;130:8888–8889. doi: 10.1021/ja802775e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shook RL, Peterson SM, Greaves J, Moore C, Rheingold AL, Borovik AS. Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature. J Am Chem Soc. 2011;133:5810–5817. doi: 10.1021/ja106564a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi T, Gupta R, Lassalle-Kaiser B, Boyce DW, Yachandra VK, Tolman WB, Yano J, Hendrich MP, Borovik AS. Preparation and Properties of a Monomeric High-Spin Mn(V)-Oxo Complex. J Am Chem Soc. 2012;134:1996–1999. doi: 10.1021/ja210957u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta R, MacBeth CE, Young VG, Borovik AS. Isolation of Monomeric MnIII/II–OH and MnIII–O Complexes from Water: Evaluation of O–H Bond Dissociation Energies. J Am Chem Soc. 2002;124:1136–1137. doi: 10.1021/ja016741x. [DOI] [PubMed] [Google Scholar]
- 39.MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. Utilization of Hydrogen Bonds To Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 40.MacBeth CE, Hammes BS, Young VG, Borovik AS. Hydrogen-Bonding Cavities about Metal Ions: Synthesis, Structure, and Physical Properties for a Series of Monomeric M–OH Complexes Derived from Water. Inorg Chem. 2001;40:4733–4741. [PubMed] [Google Scholar]
- 41.Sickerman NS, Henry RM, Ziller JW, Borovik AS. Preparation and Structural Properties of InIII–OH Complexes. Polyhedron. 2013;58:65–70. doi: 10.1016/j.poly.2012.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS. Assembly and Properties of Heterobimetallic CoII/III/CaII Complexes with Aquo and Hydroxo Ligands. J Am Chem Soc. 2012;134:17526–17535. doi: 10.1021/ja304525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park YJ, Cook SA, Sickerman NS, Sano Y, Ziller JW, Borovik AS. Heterobimetallic Complexes with MIII-(μ-OH)-MII Cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): Structural, Kinetic, and Redox Properties. Chem Sci. 2013;4:717–726. doi: 10.1039/C2SC21400H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sano Y, Weitz AC, Ziller JW, Hendrich MP, Borovik AS. Unsymmetrical Bimetallic Complexes with MII–(μ-OH)–MIII Cores (MIIMIII = FeIIFeIII, MnIIFeIII, MnIIMnIII): Structural, Magnetic, and Redox Properties. Inorg Chem. 2013;52:10229–10231. doi: 10.1021/ic401561k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukherjee J, Lucas RL, Zart MK, Powell DR, Day VW, Borovik AS. Synthesis, Structure, and Physical Properties for a Series of Monomeric Iron(III) Hydroxo Complexes with Varying Hydrogen-Bond Networks. Inorg Chem. 2008;47:5780–5786. doi: 10.1021/ic800048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gellman SH, Dado GP, Liang C, Adam BR. Conformation-Directing Effects of a Single Intramolecular Amide-Amide Hydrogen Bond : Variable-Temperature NMR and IR Studies on a Homologous Diamide Series. J Am Chem Soc. 1991;113:1164–1173. [Google Scholar]
- 47.Lever AB. Inorganic Electronic Spectroscopy. 2. Elsevier Science B.V; New York: 1984. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.