

Abstract
Asymmetric nitrene transfer reactions are a powerful tool for the preparation of enantioenriched amine building blocks. Herein, we report chemo- and enantioselective silver-catalyzed aminations that transform di- and trisubstituted homoallylic carbamates into [4.1.0]-carbamate-tethered aziridines in good yields and ee up to 92%. The effects of the substrate, silver counteranion, ligand, solvent and temperature on both chemoselectivity and ee were explored to complement the scope of traditional metal-catalyzed nitrene transfer reactions. Stereochemical models were proposed to rationalize the observed absolute stereochemistry of the aziridines, which undergo nucleophilic ring-opening to yield enantioenriched amines with no erosion in stereochemical integrity.
Keywords: nitrene, silver, aziridines, asymmetric, amines
COMMUNICATION
Asymmetric nitrene transfer reactions are a powerful tool for preparing enantioenriched amines. We have developed chemo- and enantioselective silver-catalyzed aminations that transform di- and trisubstituted homoallylic carbamates into aziridines in good yields and ee of up to 92%. Stereochemical models are proposed to rationalize the observed absolute stereochemistry of the products, which undergo nucleophilic ring-opening to yield enantioenriched amines with no erosion in stereochemical integrity.
Asymmetric, metal-catalyzed nitrogen-atom transfer reactions offer excellent opportunities to transform simple precursors into enantioenriched amines, key building blocks for pharmaceuticals, agrochemicals, natural products and ligands.[1,2] Additions of metal nitrenes to alkenes are particularly useful, as the strain in the resulting aziridines enables facile nucleophilic ring cleavage to furnish enantioenriched amines, amino acids, aminoalcohols and β-lactams.[3] Compared to asymmetric epoxidations, the corresponding aziridinations are underexplored and display narrower substrate scope. In this communication, we report an intramolecular chemo- and enantioselective silver-catalyzed nitrene transfer that furnishes di- and trisubstituted aziridines in good yields and ee. Ring-opening with diverse nucleophiles occurs at the distal aziridine carbon to yield amines with excellent retention of stereochemical information.
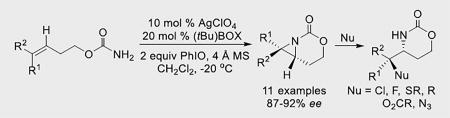
Early investigations of asymmetric metal-catalyzed nitrene transfer were pioneered by Evans and Jacobsen (Scheme 1A–B).[4,5] Cu supported by bis(oxazoline) (BOX) and diimine ligands were employed with imidoiodinanes as nitrogen sources. The biggest drawback of Cu-based catalysis is the limited scope, which is largely restricted to styrenes, terminal olefins and conjugate acceptors.[6] Chiral Ru catalysts supported by salen ligands (Scheme 1C)[7] use sulfonyl azides as nitrene precursors to furnish aziridines in up to 99% ee; however, the reaction works best with styrenes and terminal alkyl olefins. Dinuclear Rh catalysts and Zhang's porphyrin-Co complexes have also enjoyed success with certain classes of substrates.[1a,8]
Scheme 1.

Approaches to metal-catalyzed chemoselective nitrene transfers.
Surprisingly, few examples of metal-catalyzed intramolecular asymmetric aziridinations have been reported, despite their potential for synthesizing valuable synthetic building blocks. A chiral Rh catalyst, [Rh2(4S-MEOX)4], gave aziridination of styrenes in up to 76% ee in moderate yields.[9] Aziridination of homoallylic sulfamates using [Cu(MeCN)4]PF6 supported by a BOX ligand has also been described (Scheme 1D).[10] Styrenes gave ee up to 84%, but replacing Ph with alkyl groups lowers the ee; to our knowledge, no successful examples of asymmetric aziridinations of trisubstituted alkenes have been reported.
Our group has reported tunable, chemo- and site-selective nitrene transfers catalyzed by Ag(I) complexes supported by diverse N-chelating ligands.[11–16] We wondered if the flexibility exhibited by these catalysts might improve both the scope and the ee of intramolecular alkene and allene aziridinations. Such studies would provide corroboration of our hypothesis that lower-coordination Ag(I) complexes favour aziridination, while higher coordination at the metal prefers C-H bond amination instead.[11]
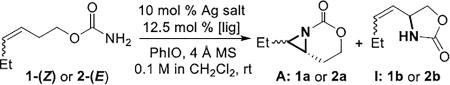
In light of the need to favor low-coordinate Ag(I) complexes, preliminary studies focused on bidentate asymmetric ligands using AgClO4 as the metal salt (see Supporting Information for full details). Chiral BOX ligands gave the best balance between chemoselectivity for aziridination and ee. Optimizations using homoallylic carbamates 1-(Z) and 2-(E) were carried out with various Ag salts and BOX ligands (Table 1, full details in the SI). L2 and L5 gave good chemoselectivity and ee with AgClO4 (entries 1, 5), while other ligands displayed either poor yield or ee (entries 2–4). Various Ag salts with L2 and L5 (entries 6–13) showed that coordinating anions, such as −OAc (entry 6), were not effective. AgOTf (entries 7–8) was superior using L5 vs. L2, while AgBF4 and AgPF6 (entries 9–12) behaved similarity, but gave lower ee compared to AgClO4. The ee with AgSbF6 (entry 13) was on par with AgClO4, but due to lower cost, AgClO4 was chosen for further study. Similar ee was seen using other non-coordinating anions with 2-(E) (entries 14–18) and L2/L5.
Table 1.
Initial optimization of the asymmetric aziridination.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | substrate | Ag salt | ligand | yield A[a] | yield I[a,b] | ee A |
| 1 | 1-(Z) | AgClO4 | L1 | 46% | 7% | 16% |
| 2 | 1-(Z) | AgClO4 | L2 | 89% | 10% | 82% |
| 3 | 1-(Z) | AgClO4 | L3 | 77% | 6% | 45% |
| 4 | 1-(Z) | AgClO4 | L4 | 81% | 11% | 23% |
| 5 | 1-(Z) | AgClO4 | L5 | 91% | 9% | (−)80% |
|
| ||||||
| 6 | 1-(Z) | AgOAc | L2 | 29% | 16% | 6% |
| 7 | 1-(Z) | AgOTf | L2 | 63% | 10% | 67% |
| 8 | 1-(Z) | AgOTf | L5 | 86% | 6% | (−)81% |
| 9 | 1-(Z) | AgBF4 | L2 | 92% | 8% | 71% |
| 10 | 1-(Z) | AgBF4[c] | L2 | 55% | 5% | 76% |
| 11 | 1-(Z) | AgBF4 | L5 | 74% | 10% | (−)67% |
| 12 | 1-(Z) | AgPF6 | L2 | 69% | 6% | 71% |
| 13 | 1-(Z) | AgSbF6 | L2 | 83% | 5% | 79% |
|
| ||||||
| 14 | 2-(E) | AgBF4 | L2 | 73% | 11% | 83% |
| 15 | 2-(E) | AgPF6 | L2 | 73% | 13% | 79% |
| 16 | 2-(E) | AgSbF6 | L2 | 79% | 8% | 83% |
| 17 | 2-(E) | AgClO4 | L2 | 79% | 17% | 85% |
| 18 | 2-(E) | AgClO4 | L5 | 90% | 10% | (−)84% |
NMR yield using mesitylene as internal standard.
I: C-H insertion byproduct.
NaBARF (BARF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) was employed as an additive.
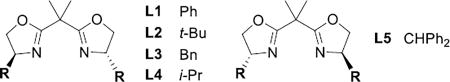
Further optimization used L2 and L5 with AgClO4 (Table S3 in the SI for details). CH2Cl2 was the optimal solvent, in contrast to Cu-catalyzed aziridination, where CH3CN was preferred.[10] Inter-estingly, addition of an extra 10 mol % of AgClO4 delivered high yields at −20 °C and increased ee to >90%. This likely results from the role the Ag salt plays in breaking up polymeric PhIO, a process that suffers at low temperatures.[17] Sc(OTf)3 could be substituted for additional AgClO4, with L5 also showing increased ee upon cooling to −20 °C (see Table S3).
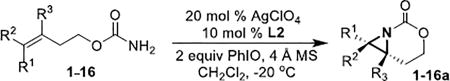
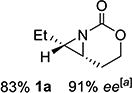
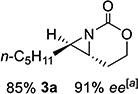
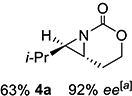
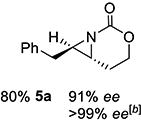
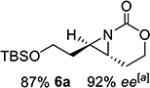
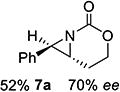
With optimized conditions in hand, the scope was explored (Table 2). Dialkylsubstituted alkenes 1-(Z) and 2-(E) (entries 1–2) gave excellent yields and >90% ee. The remainder of the mass balance in Table 2 was C-H insertion product, easily separated from the aziridine by column chromatography or recrystallization. Changing the length or steric bulk of the alkyl side chain (entries 3–4) furnished products in high ee. Benzyl and ether-containing alkyl groups were well-tolerated (entries 5–6); the ee of 5a increased to >99% after one recrystallization. In contrast to Cu-catalyzed enantioselective aziridination, styrene 7-(E) initially gave racemic aziridine 7a. Interestingly, changing the solvent to C6H6, improved the ee to 70%; decreased ee was observed in toluene and xylene. Other substrates did not show this effect, suggesting that Ag-π interactions may be responsible for the increased ee.[10,18]
Table 2.
Scope of the asymmetric aziridination.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | substrate | product | entry | substrate | product |
| 1 | 1-(Z) |
|
9 | 9-(E) |
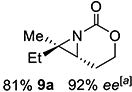
|
| 2 | 2-(E) |
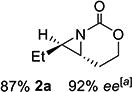
|
10 | 10-(E) |
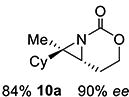
|
| 3 | 3-(E) |
|
11 | 11-(E) |
|
| 4 | 4-(E) |
|
12 | 12-(E) |
|
| 5 | 5-(E) |
|
13 | 13-(E) |
|
| 6 | 6-(E) |
|
14 | 14-(Z) |
|
| 7[c] | 7-(E) |
|
15 | 15-(Z) |
|
| 8 | 8-(E) |
|
16 | 16 allene |
|
ee was determined after ring-opening of aziridine with NaI.
ee after one recrystallization.
Reaction was run in C6H6 at room temperature.
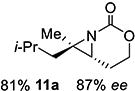
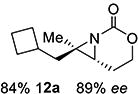
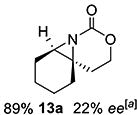
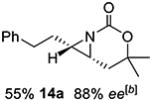
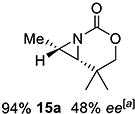
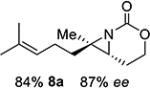
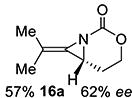
Gratifyingly, 1,1',2-trisubstituted alkenes 8-(E)–12-(E) gave good chemoselectivity and ee for 8a–12a (entries 8–12). A 1,2,2'-substitution pattern in 13-(E) (entry 13) lowered ee to 22%, although the yield and chemoselectivity remained high. Steric bulk in the tether between the alkene and carbamate of 14-(Z) (entry 14) resulted in lower yield, but good ee after one recrystallization. However, moving the bulk closer to the alkene in 15-(Z) (entry 15) had a detrimental effect on the ee. Asymmetric aziridination of allene 16 to methyleneaziridine 16a (entry 14) gave a promising 62% ee; further optimization studies are underway.[19] Although allylic carbamate substrates precludes issues of chemoselectivity, no ee was noted.
X-ray crystallography (SI for details) of 5a resulting from reaction of 5-(E) with L2 (Scheme 2) contained (R,R) absolute stereochemistry. Reaction of 14 with L2 gave (S,R)-14a, while L5 furnished the antipodes. Interestingly, the observed absolute stereochemistry of trans-5a was opposite to that seen in Cu-catalyzed aziridination of homoallylic sulfamates.[10]
Scheme 2.

Determination of the absolute stereochemistry.
The results in Table 2 point to several alkene features important to obtaining high ee with Ag-catalyzed nitrene transfer. These include at least one substituent on the distal alkene carbon, monosubstitution only at the proximal alkene carbon, distal substituents that are not conjugated to the alkene and a two-carbon tether between the carbamate and the site of unsaturation. These observations, coupled with our knowledge of the absolute aziridine stereochemistry (Scheme 2), suggested the model for stereochemical induction proposed in Figure 1. High ee is observed when substitution is present at R1, R2, or at both R1 and R2. The poor ee observed when the proximal alkene carbon R3 is substituted is rationalized by steric clashing between substrate and catalyst. Furthermore, the dramatic solvent effect observed with the styrene 7-(E) suggests that potential π-cation interactions between the Ph group and the silver complex may erode the ee in non-aromatic solvents. Using C6H6 as solvent (Table 2, entry 7) may interfere with these substrate-catalyst interactions, restoring some ee to the reaction. Other aromatic solvents (C6H5F and C6H5CF3) also restored some ee, but not as effectively as C6H6.
Figure 1.

Stereochemical model for asymmetric aziridination.
A comparison of Ag(I) vs. Cu(I) catalysts supported by L2 using both carbamates and sulfamates was carried out to better understand the differences between these two systems (Table 3). Our Ag(I) catalysts required carbamates as precursors (entry 1); a sulfamate gave only trace aziridine (entry 3). This was surprising, as we have previously shown sulfamates to be effective in other Ag-catalyzed nitrene transfers.[12,13] Closer examination by 1H NMR showed formation of an iminoiodinane intermediate, which eventually furnished the aziridine over a period of days (Figure S2 for details). We propose this arises from either increased Lewis acidity of Ag(I) compared to Cu(I), or more likely, the increased size of the Ag(I) cation (115–126 pm) vs. a Cu(I) ion (96 pm). The sulfamate oxygen and the Ph of the iminoiodinane may bind to Ag, slowing down the formation of the active silver nitrenoid species, even at rt (Figure S2). This is not an issue in Ag catalysts with fewer open coordination sites on the metal.[12,13] Attempts to utilize [Cu(MeCN)4]PF6 with carbamates gave no reaction (entry 2), while sulfamates were successful as previously reported (entry 4).[10] These results highlight the impact of differences in metal identity and nitrogen transfer reagent on reaction behavior and point to the importance of further studies to better understand such effects.
Table 3.
Comparison of carbamates and sulfamates in Cu- and Ag-catalyzed asymmetric nitrene transfer.
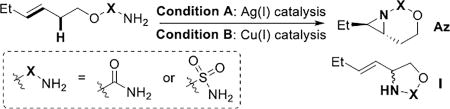
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | substrate | conditions | yield Az | yield I | recovered substrate |
ee of Az | |
| 1 | X = CO | 2-(E) | A | 87% | 10%[a] | n/d | 92% |
| 2 | X = CO | 2-(E) | B | 15%[a] | 5%[a] | 44%[a] | n/a |
| 3 | X = SO2 | 17-(E) | A | 3%[a] | n/d | 28%[a] | n/a |
| 4[b] | X = SO2 | 17-(E) | B | 83% | n/a | n/a | 80% |
Condition A: 20 mol % AgClO4, 10 mol % L2, PhIO, 4 Å MS, CH2Cl2, −20 °C.
Condition B: 5 mol % Cu(CH3CN)4PF6, 5.5 mol % L2, PhIO, 3 Å MS, CH3CN, −20 °C.
NMR yield, mesitylene internal standard.
See ref. 10.
Nucleophilic ring-opening of aziridines introduces valuable functionality into the amine products.[3] To demonstrate that the stereochemical integrity of the aziridination is maintained during subsequent manipulations, 2a was opened with diverse nucleophiles to give enantioenriched amines 18–23 (Scheme 3).[20,21] These ring-openings display reactivity complementary to that of the corresponding cyclic sulfamates, which open at the proximal aziridine carbon.[10] For example, 2a undergoes ring-opening with halides, azides, carboxylic acids, sulfides and cuprates at the distal aziridine carbon. No erosion in either dr or ee of the amine products was noted. These aziridines could also be transformed into (2-aziridinyl)acetic acids by carbamate cleavage and oxidation of the primary alcohol.[22]
Scheme 3.

Ring-openings of enantioenriched aziridines.
In conclusion, we have described the first general examples of intramolecular, asymmetric aziridination proceeding via a silver-catalyzed nitrene transfer pathway. A BOX-supported AgClO4 complex transforms di- and trisubstituted homoallylic carba-mates to [4.1.0]-carbamate-tethered aziridines in good yields and ee ~90%. Nucleophilic ring-opening occurs smoothly at the distal aziridine carbon to furnish enantioenriched amines with no erosion of stereochemical information. Future efforts will explore ligands for asymmetric Ag-catalyzed C-H amination and expand the scope of asymmetric allene aziridination.
Experimental Section
A pre-dried reaction flask was charged with AgClO4 (10.4 mg, 0.050 mmol, 20 mol %) and BOX ligand L2 (7.4 mg, 0.025 mmol, 10 mol %). CH2Cl2 (5 mL) was added and the mixture stirred vigorously for 15 min. 4 Å molecular sieves (250 mg, 1 g of sieves/mmol of substrate) were added and the mixture stirred for 5 min. The carbamate (0.25 mmol, 1 equiv) was added, the mixture stirred for 2 min, cooled to −20 °C and PhIO (110 mg, 0.50 mmol, 2 equiv) added in one portion. The reaction mixture was stirred at −20 °C until 1H NMR indicated complete alkene consumption (1–2 days). The mixture was filtered through Celite and the filtrate concentrated under reduced pressure. The crude aziridine was purified by silica gel chromatography using CH2Cl2/ethyl acetate.
Supplementary Material
Acknowledgments
This work was funded by an NSF-CAREER Award 1254397 and the Wisconsin Alumni Research Foundation to JMS. The NMR facilities at UW-Madison are funded by the NSF (CHE-1048642, CHE-0342998) and NIH S10 OD012245.
References
- 1.Selected reviews of metal-catalyzed nitrene transfer and organo-catalytic methods: Müller P, Fruit C. Chem. Rev. 2003;103:2905. doi: 10.1021/cr020043t.Collet F, Lescot C, Dauban P. Chem. Soc. Rev. 2011;40:1926. doi: 10.1039/c0cs00095g.Díaz-Requejo MM, Pérez PJ. Chem. Rev. 2008;108:3379. doi: 10.1021/cr078364y.Collet F, Dodd R, Dauban P. Chem. Commun. 2009:5061. doi: 10.1039/b905820f.Zhu Y, Wang Q, Cornwall RG, Shi Y. Chem. Rev. 2014;114:8199. doi: 10.1021/cr500064w.
- 2.Examples of Rh-catalyzed nitrene transfers: Roizen JL, Harvey ME, Du Bois J. Accts. Chem. Res. 2012;45:911. doi: 10.1021/ar200318q.Zalatan DN, Du Bois J. Top. Curr. Chem. 2010;292:347. doi: 10.1007/128_2009_19.Dequirez G, Pons V, Dauban P. Angew. Chem. Int. Ed. 2012;51:7384. doi: 10.1002/anie.201201945.Espino CG, Du Bois J. Angew. Chem. Int. Ed. 2001;40:598. doi: 10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9.Fiori KW, Du Bois J. J. Am. Chem. Soc. 2007;129:562. doi: 10.1021/ja0650450.Collet F, Lescot C, Liang CG, Dauban P. Dalton Trans. 2010;39:10401. doi: 10.1039/c0dt00283f.Espino CG, Fiori KW, Kim M, Du Bois J. J. Am. Chem. Soc. 2004;126:15378. doi: 10.1021/ja0446294.Zalatan DN, Du Bois J. J. Am. Chem. Soc. 2008;130:9220. doi: 10.1021/ja8031955.Liang C, Robert-Peillard F, Fruit C, Muller P, Dodd RH, Dauban P. Angew. Chem. Int. Ed. 2006;45:4641. doi: 10.1002/anie.200601248.Lescot C, Darses B, Collet F, Retailleau P, Dauban P. J. Org. Chem. 2012;77:7232. doi: 10.1021/jo301563j.Lebel H, Spitz C, Leogane O, Trudel C, Parmentier M. Org. Lett. 2011;13:5460. doi: 10.1021/ol2021516.
- 3.Aziridine ring-opening in synthesis: Sweeney JB. Chem. Soc. Rev. 2002;31:247. doi: 10.1039/b006015l.Chamchaang W, Pinhas AR. J. Org. Chem. 1990;55:2943.Tanner D. Angew. Chem. Int. Ed. 1994;33:599.McCoull W, Davis FA. Synthesis. 2000:1347.Wang Z, Hong W-X, Sun J. Curr. Org. Chem. 2016;20:1851.Pineschi M. Synlett. 2014;25:1817.Johnson J. In: Science of Synthesis, Stereoselective Synthesis. De Vries JG, Molander GA, Evans PA, editors. Georg Thieme; Stuttgart: 2011. pp. 759–827.Lu P. Tetrahedron. 2010;66:2549.Schneider C. Angew. Chem. Int. Ed. 2009;48:2082. doi: 10.1002/anie.200805542.Pineschi M. Eur. J. Org. Chem. 2006;22:4979.Watson I, Yu L, Yudin A. Acc. Chem. Res. 2006;39:194. doi: 10.1021/ar050038m.Hu XE. Tetrahedron. 2004;60:2701.Stankovic S, D'hooghe M, Catak S, Eum H, Waroquier M, Van Speybroeck V, De Kimpe N, Ha H-J. Chem. Soc. Rev. 2012;41:643. doi: 10.1039/c1cs15140a.Singh G, D'hooghe M, De Kimpe N. Chem. Rev. 2007;107:2080. doi: 10.1021/cr0680033.
- 4.a) Evans DA, Faul MM, Bilodeau MT, Anderson BA, Barnes DM. J. Am. Chem. Soc. 1993;115:5328. [Google Scholar]; b) Evans DA, Bilodeau MT, Faul MM. J. Am. Chem. Soc. 1994;116:2742. [Google Scholar]
- 5.a) Li Z, Conser KR, Jacobsen EN. J. Am. Chem. Soc. 1993;115:5326. [Google Scholar]; b) Li Z, Quan RW, Jacobsen EN. J. Am. Chem. Soc. 1995;117:5889. [Google Scholar]
- 6.For selected examples of Cu-catalyzed asymmetric aziridinations, see: Bergmeier SC, Lapinsky DJ. Prog. Hetero. Chem. 2013;25:47.Lebel H, Parmentier M, Leogane O, Ross K, Spitz C. Tetrahedron. 2012;68:3396.Taylor S, Gullick J, Galea N, McMorn P, Bethell D, Bulman-Page PC, Hancock FE, King F, Willock DJ, Hutchings GJ. Top. Catalysis. 2003;25:81.Suga H, Kakehi A, Ito S, Ibata T, Fudo T, Watanabe Y, Kinoshita Y. Bull. Chem. Soc. Japan. 2003;76:189.Llewellyn DB, Adamson D, Arndtsen BA. Org. Lett. 2000;2:4165. doi: 10.1021/ol000303y.Charette AB, Lebel H, Roy M-N. In: Copper-Catalyzed Asymmetric Synthesis. Alexakis A, Krause N, Woodward S, editors. Wiley-VCH; Weinheim: 2014. pp. 203–238.Cranfill DC, Lipton MA. Org. Lett. 2007;9:3511. doi: 10.1021/ol071350u.Ma L, Du D-M, Xu J. Chirality. 2006;18:575. doi: 10.1002/chir.20282.Ma L, Du D-M, Xu J. J. Org. Chem. 2005;70:10155. doi: 10.1021/jo051765y.Xu J, Ma L, Jiao P. Chem. Commun. 2004;14:1616. doi: 10.1039/b404134h.
- 7.Kim C, Uchida T, Katsuki T. Chem. Commun. 2012;48:7188. doi: 10.1039/c2cc32997b. [DOI] [PubMed] [Google Scholar]
- 8.Jin L-M, Lu H, Cui X, Wojtas L, Zhang XP. Angew. Chem. Int. Ed. 2013;52:5309. doi: 10.1002/anie.201209599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang J-L, Yuan S-X, Chan PWH, Che C-M. Tetrahedron Lett. 2003;44:5917. [Google Scholar]
- 10.Esteoule A, Duran F, Retailleau P, Dodd RH, Dauban P. Synthesis. 2007;1251 [Google Scholar]
- 11.Rigoli JW, Weatherly CD, Alderson JM, Vo BT, Schomaker JM. J. Am Chem. Soc. 2013;135:17238. doi: 10.1021/ja406654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alderson JM, Phelps AM, Scamp RJ, Dolan NS, Schomaker JM. J. Am. Chem. Soc. 2014;136:16720. doi: 10.1021/ja5094309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scamp RJ, Jirak JG, Dolan NS, Guzei IA, Schomaker JM. Org. Lett. 2016;18:3014. doi: 10.1021/acs.orglett.6b01392. [DOI] [PubMed] [Google Scholar]
- 14.Dolan NS, Scamp RJ, Yang T, Berry JF, Schomaker JM. J. Am. Chem. Soc. 2016;138:14658. doi: 10.1021/jacs.6b07981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scamp R, Rigoli J, Schomaker JM. Pure Appl. Chem. 2014;86:381. [Google Scholar]
- 16.Rigoli JW, Weatherly CD, Vo BT, Neale S, Meis AR, Schomaker JM. Org. Lett. 2013;15:290. doi: 10.1021/ol303167n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu C, Wei Y, Ji L. Syn. Commun. 2010;40:2057. [Google Scholar]
- 18.a) Price JR, White NG, Perez-Velasco A, Jameson GB, Hunter CA, Brooker S. Inorg. Chem. 2008;47:10729. doi: 10.1021/ic800959s. [DOI] [PubMed] [Google Scholar]; b) Maier JM, Li P, Hwang J, Smith MD, Shimizu KD. J. Am. Chem. Soc. 2015;137:8014. doi: 10.1021/jacs.5b04554. [DOI] [PubMed] [Google Scholar]; c) Salazar-Mendoza D, Baudron SA, Hosseini MW. Chem. Commun. 2007:2252. doi: 10.1039/b704466f. [DOI] [PubMed] [Google Scholar]; d) Habata Y, Taniguchi A, Ikeda M, Hiraoka T, Matsuyama N, Otsuka S, Kuwahara S. Inorg. Chem. 2013;52:2542. doi: 10.1021/ic302511e. [DOI] [PubMed] [Google Scholar]
- 19.a) Tehrani KA, De Kimpe N. Curr. Org. Chem. 2009;13:854. [Google Scholar]; b) Adams CS, Weatherly CD, Burke EG, Schomaker JM. Chem. Soc. Rev. 2014;43:3136. doi: 10.1039/c3cs60416k. [DOI] [PubMed] [Google Scholar]
- 20.Hayes C, Beavis PW, Humphries LA. Chem. Commun. 2006:4501. doi: 10.1039/b611662k. [DOI] [PubMed] [Google Scholar]
- 21.Liu R, Herron SR, Fleming SA. J. Org. Chem. 2007;72:5587–91. doi: 10.1021/jo0705014. [DOI] [PubMed] [Google Scholar]
- 22.a) Callebaut G, Meiresonne T, De Kimpe N, Mangelinckx S. Chem. Rev. 2014;114:7954. doi: 10.1021/cr400582d. [DOI] [PubMed] [Google Scholar]; b) Lu L, Gerstner NC, Oxtoby LJ, Guzei IA, Schomaker JM. Org. Lett. ASAP; 2017. http://dx.doi.org/10.1021/acs.orglett.7b01342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.