

Abstract
The reaction of rhodium-bound carbenes with strained bicyclic methylene aziridines results in a formal [3+1] ring expansion to yield highly-substituted methylene azetidines with excellent regio-and stereoselectivity. The reaction appears to proceed through an ylide-type mechanism, where the unique strain and structure of the methylene aziridine promotes a ring-opening/ring-closing cascade that efficiently transfers chirality from substrate to product. The resultant products can be elaborated into new azetidine scaffolds containing vicinal tertiary-quaternary and even quaternary-quaternary stereocenters.
Keywords: aziridine, carbene, azetidine, rearrangement, enantiospecific
COMMUNICATION
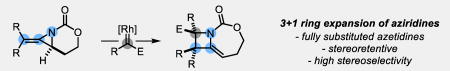
A tale of two rings: A one carbon ring expansion of methylene aziridines to methylene azetidines occurs upon reaction with a rhodium-bound carbene. This 3+1 ring expansion efficiently delivers substituted azetidines in high yields and stereoselectivities.
Azetidines are four-membered saturated nitrogen heterocycles underexplored in comparison to their aziridine, pyrrolidine and piperidine counterparts.1 However, azetidines have found great utility in medicinal chemistry to rigidify alkyl-substituted amines, to explore new chemical space, and as synthetic building blocks (Scheme 1A).1–4 While azetidines occur infrequently in natural products, Kato et. al. recently disclosed the synthetic bicyclic azetidine BRD7929, a promising and potent antimalarial believed to have a novel mechanism of action.5
Scheme 1.

Background and reaction design.
The increased interest in azetidines requires new synthetic approaches to access desired scaffolds with varied substitution and stereochemistry. Traditional methods employ cyclization of a linear precursor via a 4-exo-tet substitution, but are limited in scope.4 Other strategies accomplish ring and stereocenter construction simultaneously by uniting two fragments; however, this often leads to difficulty in establishing the correct regio- and stereochemistry (Scheme 1B). The [2+2] reactions have been well-explored6 while the corresponding [3+1] reactions are less common, although N-atom transfer from an oxaziridine to a donor-acceptor cyclopropane was recently disclosed, as well as two examples of nucleophilic aziridine opening with sulfoxonium or nitrogen ylides.7,8 Despite these advances, stereoselective access to highly substituted, densely functionalized azetidines remains difficult. We hypothesized that an umpolung [3+1] approach could address these challenges by engaging strained aziridines with electrophilic one-carbon sources to trigger a ring-opening, ring-closing cascade to yield the azetidines (Scheme 1C).9–12 While concerted [1,2]-Stevens rearrangements of aziridines are thermally forbidden, a step-wise reaction could, in principal, achieve a one-carbon ring expansion.9c,d Reports of analogous reactivity with epoxides and oxetanes are rare,10 and display regioselectivity issues and inefficient ring-closure, presumably due to the promiscuity of the ring-opened ylide intermediate. We proposed bicyclic methylene aziridines, easily available from propargyl alcohols (Table 1), would be good substrates for [3+1] reactions to generate substituted azetidines.13 The bicyclic structure forces the nitrogen lone pair orthogonal to the carbonyl, rendering it moderately nucleophilic. Additionally, ring strain should provide a driving force for ring-opening, with the carbamate tether ensuring regioselective ring-closure. Gratifyingly, initial experiments with a donor/acceptor diazoacetate (Scheme 1C) under dirhodium catalysis delivered endocyclic, bicyclic methylene azetidine in high yield and stereoselectivity. Notably, this result: a) validates our proposal that electrophilic one-carbon sources can be engaged in ring-expansion of strained, small heterocycles, b) delivers densely functionalized azetidines with substitution patterns not accessible by other methods and c) expands the reactivity profile of bicyclic methylene aziridines.
Table 1.
Scope of [3+1] ring expansion.

|
Standard conditions: Slow addition of the diazoester (1.3 equiv) to aziridine (1.0 equiv) over 2 h. Numbers refer to isolated yields.
2.0 equiv of the diazoesters were used.
4 equiv of the diazoesters were used.
4.0 equiv of the iodonium ylide were added as two portions over 15 min.
2.5 equiv of the diazoester were used at 40 °C.
The [3+1] reaction scope was explored, first using model substrate 1a (R1 = C5H11; R2 = H) with an array of aryl/ester diazo compounds 2a–g (Table 1) and catalytic Rh2(OAc)4. Reaction of phenyl diazoacetate 2a with 1a furnished azetidine 3aa in 90% yield with >20:1 dr. Electron-rich and electron-poor aryl diazoacetates were successful, providing azetidines 3ab–3ae in high yields and selectivity. Ortho substitution on the aryl rings of 2f and 2g was tolerated, although increased loading of the diazoacetate was necessary. Pyridinyl diazo 2h delivered azetidine 3ah in moderate yield. Changing the ester group of the carbene precursor was also well-tolerated. Increasing the steric bulk of the ester in 2i resulted in a noticeably cleaner reaction with an equally impressive dr. Use of a Troc ester in 2j required no slow addition, giving excellent dr in 3aj.14 The [3+1] reaction was also successful when the aryl of the diazo compound was replaced with a Me in 2ak. Dimerization of the diazoacetate was solved using multiple equivalents of 2k to provide 3ak in good dr. Diester azetidine 3al was formed efficiently using an iodonium ylide, as the corresponding diazo gave product mixtures. Acceptor carbene precursors, including ethyl diazoacetate, have been unsuccessful thus far.
Diazoacetates with vinyl substitution surprisingly gave no higher-order [3+n] ring expansions.15 While higher temperatures were required, treatment of 1a with silyl enol ether 2n resulted in exclusive [3+1] addition, delivering azetidine 3an in 40% yield as a single diastereomer, despite the propensity of silyl enol ether Rh-bound carbenes to act as three-carbon synthons.16 Styrenyl derivative 2o also cleanly gave azetidine 3ao; the lower dr was attributed to lesser steric differentiation between a styrene and methyl ester, compared to the phenyl variant 2a.
The methylene aziridine scope was explored next. E isomers (>10:1 E:Z) containing Me, iPr, cyclopropyl and remote phenyl substituents were well-tolerated. The relative configuration of 3ba was confirmed via a single-crystal X-ray structure (details in the Supporting Information), showing the major diastereomer maintains a syn relationship between the ester and the methyl group. Despite numerous attempts, reaction of phenyl diazo 2a with 1e was unsuccessful, likely due to slow reaction of substrates containing Z-substituents with bulky carbenes; indeed, switching to less-hindered styrenyl diazo 2o delivered the fully-substituted methylene azetidine 3eo in an impressive 94% yield. We then sought to explore diastereocontrol in the synthesis of 3fo to set adjacent quaternary carbons, a challenging feat.17 Silver-catalyzed nitrene transfer enabled access to aziridine 1f as a 4:1 mixture of inseparable E/Z isomers.18 Subjecting this mixture to 2o and Rh2(OAc)4 delivered 3fo in a moderate 3:1 dr and good yield. The diastereomers were separable, yielding the syn-Me/CO2Me isomer of 3fo in 54% yield and 15:1 dr.
The alkene of the methylene azetidine represents a valuable handle for divergent elaborations of the product. Hydrogenation of 3aa gave azetidine 4a in high stereoselectivity (Scheme 2). Baran’s iron-catalyzed C-C coupling19 successfully yielded 4b in good dr, an impressive radical coupling due to forging a second quaternary carbon on the azetidine ring.
Scheme 2.

Product derivatizations.
To explore the mechanism, enantioenriched 1a was treated with 2a (Scheme 3A); the reaction showed excellent chirality transfer from the aziridine stereocenter prior to its ablation. A control reaction excluding the Rh catalyst gave no conversion, validating the role of the Rh-bound carbene. TEMPO addition gave no change in selectivity and the cyclopropane ring of 1g did not ring-open in delivering azetidine 3ga (Table 1), rendering radical intermediates unlikely.
Scheme 3.

Stereochemical probe and proposed mechanism.
Our proposed mechanism (Scheme 3B) occurs through aziridinium ylide formation via attack of the aziridine nitrogen on the Rh-bound carbene to form 5. Ring-opening to 6, followed by bond rotation and ring closure yields 3a in an overall ring-opening/ring-closing cascade. Support for formation of ylide 5 over competing alkene cyclopropanation is provided by the known ability of the nitrogen to react with potent electrophiles or Lewis acids.13a Additionally, intermolecular cyclopropanation of unactivated, sterically hindered tri- or tetrasubstituted alkenes is difficult with donor/acceptor rhodium carbenes, typically yielding C-H activation instead.20
The assumption that the ylide 6 retains its stereochemistry by existing as a C-bound Rh enolate that does not racemize via isomerization through an O-bound Rh species,21 coupled with our transfer of chirality experiments, suggests that aziridinium formation occurs with high dr using aryl diazoacetates. The crystal structure of 3ba and the nOe data of 3aa, 3ak, 3an, and 3ao (SI for details) indicate the same major diastereomer for 3aa–eo, where the ester is syn to the aziridine R group.22 This configuration can be rationalized via a steric-based model wherein the approaching Rh-bound carbene orients its larger R group (aryl, styrenyl, etc. vs. the ester) in the back, convex pocket of the bowl-shaped methylene aziridine, avoiding steric interactions with the alkene.
Given the strain present in 5, we propose that ring-opening to give the (Z)-2-amidoallyl cation zwitterion 6 is governed by A1,3 strain. Diastereoselective ring-closure from the back face of 6 delivers 3a with the observed stereochemistry. Sterics likely play a role, where the smaller ester substituent aligns syn to the aziridine R group. While we currently favor this ring-opening/ring-closing pathway, an alternative mechanism in which ylide 5 does not fully open to the 2-amido allyl cation, but begins to form the azetidine C-C bond formation prior to full planarization of the nitrogen in a concerted and asynchronous fashion, cannot be ruled out. A comprehensive mechanistic study is underway to address these nuances.
In conclusion, we have outlined a new [3+1] ring expansion strategy that harnesses unique features of bicyclic methylene aziridines, in combination with Rh-supported carbenes, to deliver tri- and tetra-substituted methylene azetidines. This reaction demonstrates an impressive use of ring strain, as it forges new C-C and C-N bonds centered around a quaternary center in good selectivity and highlights the potential of methylene aziridines as a unique entry point to nitrogen heterocycles. We anticipate this work will spur design of new ring expansion reactions using small-ring heterocycles.
Supplementary Material
Acknowledgments
Dr. Charlie Fry and Dr. Heike Hofstetter of the University of Wisconsin are thanked for NMR assistance, Dr. Martha Vestling for MS data, and Anastasiya Vinokur for XRD data collection and processing. JMS thanks the NIH R01 GM11412 and the ACS-PRF No. 53146-ND1. The NMR facilities at UW-Madison are funded by the National Science Foundation (NSF; CHE-9208463, CHE-9629688) and National Institutes of Health (NIH; RR08389-01). The mass spectrometry facilities are funded by the National Institutes of Health (NIH;1S10OD020022-1).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Singh GS, D’hooge M, De Kimpe N. In: Comprehensive Heterocyclic Chemistry III. Katritzky A, Ramsden C, AAA E, Taylor R, editors. Vol. 2. Elsevier Science; York: 2008. pp. 1–110. [Google Scholar]; b) Rousseau G, Robin S. In: Modern Heterocyclic Chemistry. Alvarez-Builla J, Vaquero JJ, Barluenga J, editors. Vol. 4. Wiley-VCH: Weinheim; 2011. pp. 163–268. [Google Scholar]; c) Ghorai M, Bhattacharyya A, Das S, Chauhan N. Topics Heterocyclic Chem. 2016;41:49–142. [Google Scholar]
- 2.a) Brown A, Ellis D, Wallace O, Ralph M. Bioorg. Med. Chem. Lett. 2010;20:1851. doi: 10.1016/j.bmcl.2010.01.143. [DOI] [PubMed] [Google Scholar]; b) Han Y, Han M, Shin D, Song C, Hahn H-G. J. Med. Chem. 2012;55:8188. doi: 10.1021/jm3008294. [DOI] [PubMed] [Google Scholar]; c) Han M, Song C, Jeong N, Hahn H-G. ACS Med. Chem. Lett. 2014;5:999. doi: 10.1021/ml500187a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Carreira EM, Fessard TC. Chem. Rev. 2014;114:8257. doi: 10.1021/cr500127b. [DOI] [PubMed] [Google Scholar]; b) Marson CM. Chem. Soc. Rev. 2011;40:5514. doi: 10.1039/c1cs15119c. [DOI] [PubMed] [Google Scholar]
- 4.a) Brandi A, Cicchi S, Cordero FM. Chem. Rev. 2008;108:3988. doi: 10.1021/cr800325e. [DOI] [PubMed] [Google Scholar]; b) Couty F, Evano G. Synlett. 2009:3053. [Google Scholar]; c) Bott TM, West FG. Heterocycles. 2012;84:223. [Google Scholar]; d) Antermite D, Degennaro L, Luisi R. Org. Biomol. Chem. 2017;15:34. doi: 10.1039/c6ob01665k. [DOI] [PubMed] [Google Scholar]; e) Callebaut G, Mangelinckx S, Kiss L, Sillanpää R, Fülöp F, De Kimpe N. Org. Biomol. Chem. 2012;10:2326. doi: 10.1039/c2ob06637h. [DOI] [PubMed] [Google Scholar]; f) Kenis S, D’hooghe M, Verniest G, Reybroeck M, Thi TAD, The CP, Pham TT, Tornroos KW, Tuyen NV, De Kimpe N. Chem. Eur. J. 2013;19:5966. doi: 10.1002/chem.201204485. [DOI] [PubMed] [Google Scholar]; g) Jensen KL, Nielsen DU, Jamison TF. Chem. Eur. J. 2015;21:7379. doi: 10.1002/chem.201500886. [DOI] [PubMed] [Google Scholar]
- 5.Kato N, et al. Nature. 2016;538:344. doi: 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Sakamoto R, Inada T, Sakurai S, Maruoka K. Org. Lett. 2016;18:6252. doi: 10.1021/acs.orglett.6b03003. [DOI] [PubMed] [Google Scholar]; b) Takizawa S, Arteaga FA, Yoshida Y, Suzuki M, Sasai H. Org. Lett. 2013;15:4142. doi: 10.1021/ol401817q. [DOI] [PubMed] [Google Scholar]
- 7.a) Zhang H-H, Luo Y-C, Wang H-P, Chen W, Xu P-F. Org. Lett. 2014;16:4896. doi: 10.1021/ol5024079. [DOI] [PubMed] [Google Scholar]; b) Han J-Q, Zhang H-H, Xu P-F, Luo Y-C. Org. Lett. 2016;18:5212. doi: 10.1021/acs.orglett.6b02430. [DOI] [PubMed] [Google Scholar]; c) Ghosh A, Mandal S, Chatteraj PK, Banerjee P. Org. Lett. 2016;18:4940. doi: 10.1021/acs.orglett.6b02417. [DOI] [PubMed] [Google Scholar]
- 8.a) Malik S, Nadir UK. Synlett. 2008:108. and references therein. [Google Scholar]; b) Garima, Srivastava VP, Yadav LDS. Green Chem. 2010;12:1460. [Google Scholar]
- 9.For reactions of aziridines with metal-bound carbenes see: Hata Y, Watanabe M. Tetrahedron Lett. 1972;13:3827.Hata Y, Watanabe M. Tetrahedron Lett. 1972;13:4659.Clark JS, Hodgson PB, Goldsmith MD, Blake AJ, Cooke PA, Street LJ. J. Chem. Soc., Perkins Trans. 2001;1:3325.Rowlands GJ, Barnes WK. Tetrahedron Lett. 2004;45:5347.
- 10.For reviews on aziridinium ions in synthesis see: Couty F, David ORP. In: Synthesis of 4- to 7-membered Heterocycles by Ring Expansion. D’hooghe M, Ha H-J, editors. Vol. 1. Springer; Cham: 2016. pp. 1–47.Ghorai MK, Bhattacharyya A, Das S, Chauhan N. In: Synthesis of 4- to 7-membered Heterocycles by Ring Expansion. D’hooghe M, Ha H-J, editors. Vol. 1. Springer; Cham: 2016. pp. 49–142.Dolgen J, Yadav NN, De Kimpe N, D’hooghe M, Ha H-J. Adv. Synth. Catal. 2016;358:3485.Mack DJ, Batory LA, Njardarson JT. Org. Lett. 2012;14:378. doi: 10.1021/ol203129d.
- 11.For intramolecular carbene-mediated ring expansion of nitrogen heterocycles see: Bach R, Harthong S, Lacour J. In: Comprehensive Organic Synthesis. Knochel P, Molander GA, editors. Vol. 3. Elsevier Science; York: 2014. pp. 992–1037.
- 12.Intermolecular carbene-mediated ring expansion of nitrogen heterocycles: Bott TM, Vanecko JA, West FG. J. Org. Chem. 2009;74:2832. doi: 10.1021/jo9001323.Sharma A, Guenee L, Naubron J-V, Lacour J. Angew. Chem. 2011;123:3761. doi: 10.1002/anie.201100134. Angew. Chem. Int. Ed.2011, 50, 3677.
- 13.For previous work on bicyclic methylene aziridines see: Boralsky LA, Marston D, Grigg RD, Hershberger JC, Schomaker JM. Org. Lett. 2011;13:1924. doi: 10.1021/ol2002418.Rigoli JW, Boralsky LA, Hershberger JC, Marston D, Meis AR, Guzei IA, Schomaker JM. J. Org. Chem. 2012;77:2446. doi: 10.1021/jo3000282.Weatherly CD, Rigoli JW, Schomaker JM. Org. Lett. 2012;14:1704. doi: 10.1021/ol300269u.Adams CS, Boralsky LA, Guzei IA, Schomaker JM. J. Am. Chem. Soc. 2012;134:10807. doi: 10.1021/ja304859w.Rigoli JW, Guzei IA, Schomaker JM. Org. Lett. 2014;16:1696. doi: 10.1021/ol5003576.Adams CS, Grigg RD, Schomaker JM. Chem. Sci. 2014;5:3046.Burke EG, Schomaker JM. Angew. Chem. 2015;127:12265. doi: 10.1002/anie.201504723. Angew. Chem. Int. Ed.2015, 54, 12097.Gerstner N, Adams CS, Tretbar M, Schomaker JM. Angew. Chem. 2016;128:13434. doi: 10.1002/anie.201606195. Angew. Chem. Int. Ed.2016, 55, 13240.Adams CS, Weatherly CD, Burke EG, Schomaker JM. Chem. Soc. Rev. 2014;43:3136. doi: 10.1039/c3cs60416k.
- 14.Guptill DM, Davies HML. J. Am. Chem. Soc. 2014;136:17718. doi: 10.1021/ja5107404. [DOI] [PubMed] [Google Scholar]
- 15.Selected recent examples of vinylogous reactivity of vinyl rhodium-bound carbenes: Davies HML, Saikali E, Clark TJ, Chee EH. Tetrahedron Lett. 1990;31:6299.Davies HML, Hu B, Saikali E, Bruzinksi PR. J. Org. Chem. 1994;59:4535.Lian Y, Davies HML. Org. Lett. 2012;14:1934. doi: 10.1021/ol300632p.Qin C, Davies HML. J. Am. Chem. Soc. 2013;135:14516. doi: 10.1021/ja4069003.
- 16.Leading review on three-carbon carbene synthons: Xu X, Doyle MP. Acc. Chem. Res. 2014;47:1396. doi: 10.1021/ar5000055.and references therein. Select recent examples: Del Bel M, Rovira A, Guerrero CA. J. Am. Chem. Soc. 2013;135:12188. doi: 10.1021/ja4054866.Guzman PE, Lian Y, Davies HML. Angew. Chem. 2014;126:13299. doi: 10.1002/anie.201406440. Angew. Chem. Int. Ed.2015, 54, 13715.Lee DJ, Ko D, Yoo EJ. Angew. Chem. 2015;127:13919. doi: 10.1002/anie.201506764. Angew. Chem. Int. Ed.2015, 54, 13715.Zhu C, Xu G, Sun J. Angew. Chem. 2016;128:12046. Angew. Chem. Int. Ed.2016, 55, 11867.Jing C, Cheng Q-Q, Deng Y, Arman H, Doyle MP. Org. Lett. 2016;18:4550. doi: 10.1021/acs.orglett.6b02192.
- 17.a) Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Christoffers J, Baro A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH: Weinheim; 2005. [Google Scholar]; c) Trost BM, Jiang C. Synthesis. 2006:369. [Google Scholar]
- 18.Rigoli JW, Weatherly CD, Vo BT, Neale S, Meis AR, Schomaker JM. Org. Lett. 2013;15:290. doi: 10.1021/ol303167n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Lo JC, Kim D, Pan C-M, Edwards JT, Yabe Y, Gui J, Qin T, Gutierrez S, Giacoboni J, Smith MW, Hollan PL, Baran PS. J. Am. Chem. Soc. 2017;139:2484. doi: 10.1021/jacs.6b13155. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lo JC, Gui J, Yabe Y, Pan C-M, Baran PS. Nature. 2014;516:343. doi: 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Computations detailing the nature of rhodium ylides as carbon bound in protic mechanisms: Liang Y, Zhou H, Yu Z-X. J. Am. Chem. Soc. 2009;131:17783. doi: 10.1021/ja9086566.Li Z, Boyarskikh V, Hansen JH, Autschbach J, Museav DG, Davies HML. J. Am. Chem. Soc. 2012;134:15497. doi: 10.1021/ja3061529.Li Z, Parr BT, Davies HML. J. Am. Chem. Soc. 2012;134:10942. doi: 10.1021/ja303023n.Xie Q, Song X-S, Qu D, Guo L-P, Xie Z-Z. Organometallics. 2015;34:3112.Pospech J, Lennox AJJ, Beller M. Chem. Commun. 2015;51:14505. doi: 10.1039/c5cc03903g.Liu Y, Luo Z, Zhang JZ, Xia F. J. Phys. Chem. A. 2016;120:6485. doi: 10.1021/acs.jpca.6b05735.
- 22.nOe data on 3fo indicates formation of the opposite diastereomer expected – work on understanding this selectivity is underway.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.