

Abstract
Engineered cell-based therapies comprise a promising, emerging biomedical technology. Broad utilization of this strategy will require new approaches for implementing sophisticated functional programs, such as sensing and responding to the environment in a defined fashion. Towards this goal, we investigated whether our self-contained receptor and signal transduction system (MESA) could be multiplexed to evaluate extracellular cues, with a focus on elucidating principles governing the integration of such engineered components. We first developed a set of hybrid promoters that exhibited AND gate activation by two transcription factors. We then evaluated these promoters when paired with two MESA receptors and various ligand combinations. Unexpectedly, although the multiplexed system exhibited distinct responses to ligands applied individually and in combination, the same synergy was not observed as when promoters were characterized with soluble transcription factors. Therefore, we developed a mechanistic computational model leveraging these observations, to both improve our understanding of how the receptors and promoters interface and to guide the design and implementation of future systems. Notably, the model explicitly accounts for the impact of intercellular variation on system characterization and performance. Model analysis identified key factors that affect the current receptors and promoters, and enabled an in silico exploration of potential modifications that inform the design of improved logic gates and their robustness to intercellular variation. Ultimately, this quantitative design-driven approach may guide the use and multiplexing of synthetic receptors for diverse custom biological functions beyond the case study considered here.
Keywords: biosensor, receptor engineering, genetic circuit, mammalian, computational model, intercellular variation
Graphical Abstract
INTRODUCTION
Engineered cell-based therapies have transformative potential for addressing unmet medical needs, such as in cancer immunotherapy1. Leveraging this progress to develop therapies for other applications will require new capabilities, including technologies that enable cells to sense defined extracellular cues and respond in a customizable fashion2. Such approaches generally require natural or engineered cell surface receptors. Given their potential to be implemented and tuned in a manner that is relatively independent of native cellular signaling and regulation, engineered receptors have proven to be particularly useful for building and refining new cell functions.
Various engineered receptors have been implemented to program cell functions. Many promising cancer immunotherapies use chimeric antigen receptors (CAR), which generally regulate and induce signaling downstream of the native T cell receptor. Within this framework, strategies have been developed for multiplexing receptors to achieve NOT logic as a means to incorporate a safety switch3, or AND logic4 to reduce off-target activation. Layered logic cascades involving CARs have also been used to implement AND logic for increased precision of tumor recognition5. Other engineered receptors signal via orthogonal pathways; for example, synNotch6 senses surface-bound extracellular ligands, and the modular extracellular sensor architecture (MESA)7–8 senses soluble extracellular ligands. In the initial report of synNotch, a multilayer split transcription factor (TF) complementation approach was shown to integrate signaling from two synNotch receptors to control a downstream promoter with AND logic. Other strategies for implementing Boolean logic include multilayer transcriptional cascades9–12, hybrid promoters with multiple TF binding sites13–14, post-transcriptional or post-translational regulation15–17, DNA recombinases18–20, or distributing tasks among genetically distinct cells that signal to each other21–22. Computational approaches have proven important for identifying effective circuit designs, through case-by-case mechanistic studies23–24 and through tools for automated design using libraries of characterized parts14, 25–26. Going forward, combining logic with cell surface receptors could enable the design and construction of cell-based biosensors for diverse diagnostic or therapeutic applications,27 and such innovations may be facilitated by the integration of experimental and computational methods. Towards this goal, the central objective of this study was to elucidate principles that govern the multiplexing of MESA receptors for a wide range of applications.
In this study, we investigate how and whether two MESA receptors, each of which is individually competent to induce signaling, may be multiplexed such that their outputs converge at a single promoter. This represents a complementary question to that which was investigated for multiplexing synNotch using split TFs and multilayer transcriptional control6. To this end, we constructed and evaluated a panel of novel promoter constructs, identified several that could synergistically induce transcription with two TF inputs, and used an experimental tuning strategy to align the magnitude of TF outputs from multiplexed MESA signaling with the specific TF input requirements for promoters, a process we call level-matching. Finally, to both improve our understanding of this system and guide the design of new receptor-promoter systems, we developed a mechanistic computational model and utilized the model to identify design strategies that can be leveraged for future applications.
RESULTS AND DISCUSSION
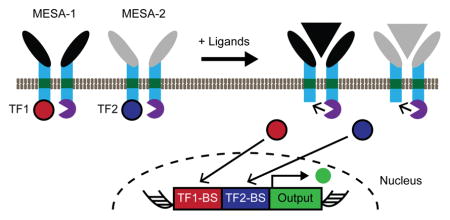
To develop a strategy for multiplexing MESA receptors, we opted to investigate a single-layer transcriptional gate (Figure 1a). Although Boolean logic gates are relatively well-studied13, 28, whether extracellular sensors can be multiplexed effectively in a single-layer transcriptional gate was not known. The MESA receptors are comprised of two separate transmembrane proteins—a target chain (TC) and a protease chain (PC)—each containing an extracellular ligand-biding domain that mediates chain dimerization upon ligand binding, a single-pass transmembrane domain, and an intracellular domain for signaling. On the intracellular face, when the protease on the PC binds to the protease recognition sequence adjacent to the TC’s TF, an enzymatic trans-cleavage event releases the TF, leaving an unaltered PC and a cleaved TC. The soluble TF can then enter the nucleus and regulate the expression of a target gene. Since MESA receptors can be designed to release a variety of defined TFs as outputs7–8, we first sought to develop a promoter that could be activated by specific combinations of two TFs. We chose to use the well-studied TFs tTA29 and Gal4-VP16 (hereafter referred to as Gal4)30, which drive transcription from reporter constructs comprising five to seven repeated DNA motifs (termed TetO (T7) and UAS (U5), respectively) upstream of a minimal promoter. Because multiple TF molecules must be recruited to drive transcription in the T7 and U5 systems29–30, we hypothesized that some hybrid promoters composed of both TetO and UAS sequences might exhibit logic. We anticipated that such hybrid constructs would exhibit AND logic by requiring the recruitment of both tTA and Gal4 to induce gene expression above a threshold (Supporting Information Figure S1); however, it is also possible that some such constructs might exhibit OR logic.
Figure 1. Design and evaluation of hybrid reporters.

(a) In the proposed strategy for multiplexing MESA, two receptors each sense a distinct ligand, undergo dimerization and enzymatic trans-cleavage, and release a transcription factor (TF1, TF2) that enters the nucleus and induces target gene expression. A hybrid promoter is regulated by both TF1 and TF2 to enable logical evaluation of the ligands. In the proposed single-layer transcriptional AND gate, the reporter is expressed if and only if both ligands (gray and black triangles) are sensed. (b) Hybrid promoters were designed using the modular TetO (red) and UAS (blue) binding domains for tTA and Gal4, respectively. Canonical single-TF promoters are illustrated for comparison. (c) Hybrid constructs were evaluated by cotransfection of plasmids (0.5 μg per plasmid) for constitutively expressed soluble tTA and Gal4 and quantification of reporter expression (fluorescence) by flow cytometry. Relative reporter expression was calculated independently for each promoter by dividing YFP mean fluorescence intensity (MFI) with either or both TFs by the MFI without TF. Experiments were conducted in biological triplicate, and error bars represent one standard deviation. (d) For three reporters from (b) termed H1, H2, and H3 as indicated, promoter activity was characterized across tTA and Gal4 plasmid dose combinations, and data were analyzed as in (c).
Development of hybrid promoters that exhibit synergistic activation by two TFs
To evaluate this hybrid promoter strategy, we first constructed a panel of promoters with different numbers and sequential arrangements of TetO and UAS motifs upstream of a CMV minimal promoter and a fluorescent reporter gene. We evaluated the performance of these constructs by measuring reporter expression (fluorescence) in response to tTA and Gal4 individually and together (Figure 1b). As hypothesized, within the limited set of constructs tested, multiple promoters functioned as AND gates. In these cases, the highest reporter expression was observed with both tTA and Gal4, and this was higher than the sum from individual TFs (Figure 1c). Three promoters (pT2U2T2U2, pU2T4U2, pT2U3) showed AND behavior and high reporter expression, whereas others had undesirable properties such as low expression (pT2U2, pU2T2), high single-TF responsiveness (pT2U4T2, pU2T4), or high background in the absence of TFs (pU2T2U2T2). Interestingly, incorporating an additional UAS domain to the proximal end of the relatively unresponsive promoter pT2U2 to generate pT2U3 led to much higher AND gate expression without substantially increasing the responsiveness to individual TFs, yet the same result was not observed when two TetO sites were incorporated to generate pT2U2T2. We identified three AND gate promoters that displayed relatively high synergy and inducible reporter expression (pT2U2T2U2, pT2U3, pU2T4U2,) and carried these forward for further evaluation. For clarity, these hybrid constructs are hereafter denoted as H1, H2, and H3, respectively.
Since the initial panel was characterized with a high TF dose, we next evaluated the three selected promoters at varied TF doses. We hypothesized that such information could be useful, because (1) the amount of MESA-bound TF that is released both with and without ligand depends on the doses of TC and PC transfected and consequently expressed, (2) the percentage of MESA-bound TF that is released with ligand could be less than 100%, and (3) receptors and soluble proteins could differ in expression due to production and/or degradation rates. We evaluated reporter expression across plasmid dose combinations for tTA and Gal4 (Figure 1d and Supporting Information Figure S2). The results showed that all three promoters exhibited synergy. Here, we define synergy as a property that is inherent to a promoter, independent of the amounts of TFs present. A promoter is identified as synergistic if, during its experimental characterization using TFs at sub-saturating levels, the background-subtracted reporter expression in the presence of both TFs is greater than the sum of background-subtracted reporter expression in the presence of each TF individually. Since the hybrid promoters exhibited low activity in the absence of TFs, the background was negligible. Separately, we define synergistic activation (or the synergistic regime) of a hybrid promoter as an implementation in which TFs are present at levels that evidence promoter synergy. In Figure 1d, synergistic activation for each promoter was most apparent at plasmid doses of around 0.05 to 0.1 μg of both TFs. Responsiveness and synergy have a basis in physical features, some of which can be deduced by comparing the promoters in Figure 1c–d. For example, the responsiveness to one TF versus the other is influenced by which has the more promoter-proximal binding site, and responsiveness scales nonlinearly with the number of consecutive binding sites for a TF. We note that, at least within the subset of designs we sampled, there may exist a trade-off between responsiveness and synergy that makes attaining either one of these properties more feasible than attaining both; for example, H1 is more responsive than H2 at low TF doses but H2 synergistic activation appears steeper than H1.
Additionally, we observed that the three promoters were more responsive to Gal4 than to tTA, with respect to transfected mass of TF-encoding plasmids. H2 and H3 had low activation at low doses and moderate activation at high doses, whereas H1 was more responsive to each TF and had higher activity overall. Since H1 and H2 exhibited distinct activity profiles, we chose to carry these two constructs forward to investigate which, if either, could be level-matched with MESA to achieve AND gate behavior.
Evaluating multiplexed receptor signaling with hybrid promoters
To investigate whether the H1 or H2 could enable MESA multiplexing, we opted to use two previously developed receptors that are responsive to either rapamycin or VEGF ligands (Rap-MESA8 and VEGF-MESA7, respectively). Rap-MESA was modified to release Gal4, and thus we first verified that this Rap-MESA exhibited ligand-inducible signaling (Supporting Information Figure S3). No modifications were made to the VEGF-MESA, which releases tTA. In the field of genetic circuit design, it is known that ideal circuit performance requires the output of upstream components to match the input requirements of downstream components31–32. For our application, we reasoned that level-matching requires that the amounts of TFs released from MESA upon ligand treatment match the amounts required for synergistic activation of the hybrid promoter. We hypothesized that level-matching could be achieved using a strategy in which we first identify the dose of each MESA required to activate the hybrid promoter in a ligand-inducible fashion when the complementary soluble TF is in excess. In this strategy, the expression of each receptor is tuned independently, in a manner that is related to the input requirements of the specific promoter. However, for any given promoter, it is also possible that the input requirements may be incompatible with the amount of TF that can be released by ligand-induced signaling in tuning the receptor dose.
To investigate this strategy, we leveraged our prior observation that the doses and ratio of TC and PC are important considerations for background signaling and ligand-inducible signaling. In general, the TC dose should be greater than or equal to the PC dose to achieve ideal fold-difference (F.D.; the measurement with ligand divided by the measurement without ligand)7 in reporter expression. Therefore, for each MESA, we varied the plasmid doses of TC and PC, and provided the complementary soluble TF in excess along with a hybrid reporter (Figure 2a and Supporting Information Figure S4). For both H1 and H2, we found that for Rap-MESA, a 1:1 plasmid ratio of TC and PC led to the best F.D., and that for VEGF-MESA, higher doses of TC compared to PC led to greater F.D. These conditions resulted in a moderate F.D. (+/− ligand). However, based upon the observed responses to soluble TFs alone (Figure 1), we reasoned that expressing the complementary soluble TF in excess produced elevated “background” reporter expression in the absence of ligand. Given this observation, we hypothesized that background signaling would be lower in the context of multiplexed receptors (when neither TF is provided in excess), such that AND behavior could be better than the F.D. in this step of MESA tuning would suggest.
Figure 2. Multiplexed receptor implementation.

(a) A complementation assay was conducted for each receptor-promoter pair, in which ligand-induced F.D. was determined across TC and PC dose combinations with the complementary soluble TF expressed constitutively and in relative excess. In the heatmaps (see Supporting Information Figure S4 for details), yellow boxes outline conditions with the highest measured F.D. Data were analyzed as in Figure 1. (b) MESA doses identified based on (a) were used to implement multiplexed receptors. Relative DsRed reporter expression was calculated independently for each promoter by dividing the DsRed mean fluorescence intensity (MFI) with MESA by the MFI without MESA, such that cells transfected with reporter only would have a value of one on this scale. Experiments were conducted in biological triplicate, and error bars represent one standard deviation. An ANOVA statistical test was utilized to compare the two-ligand case to all other cases (*p ≤ 0.05, **p ≤ 0.01).
Next, we investigated whether Rap-MESA and VEGF-MESA could be multiplexed using doses identified during MESA tuning (indicated by yellow boxes in Figure 2a). Using these conditions, we observed low reporter expression in the absence of ligands or with either ligand alone. Notably, with both ligands, H1 produced significantly elevated expression, demonstrating distinguishable two-ligand induced signaling (Figure 2b). Such outcomes were observed across several VEGF-MESA plasmid doses, providing evidence that each MESA ligand-inducibly signals independently. That is, induction of one MESA with its ligand does not induce signaling via the other receptor; if it did, the one-ligand and two-ligand cases would be indistinguishable. However, H2 outcomes were indistinguishable, as were H1 outcomes with a low Rap-MESA dose (Supporting Information Figure S5). Furthermore, inducible expression above the background was low for even the best performing implementations, which did not exhibit the same synergistic activation that was observed when promoters were evaluated with constitutively expressed soluble TFs. To explain this discrepancy, we hypothesized that receptor signaling and promoter input requirements were not yet level-matched. The initial promoter characterization showed that H1 was more TF-responsive than H2 (Figure 1d); therefore, we concluded that the amounts of TFs released with ligand treatment activated H1 more than H2, but that in both cases activation was low because the amounts of TFs released from receptors were insufficient for promoter synergy. Moreover, this explanation suggests that adjusting the MESA tuning strategy would not circumvent the failure of H2, and that such limitations are an inherent property of this receptor-promoter pairing. However, we hypothesized that modifications to the MESA tuning strategy for H1 could potentially yield improved AND behavior. Given the large potential design space involved, and the challenges associated with gaining systematic understanding from empirical tuning alone, we decided to employ computational modeling to facilitate interpretation of our observations, elucidate factors that impact receptor and hybrid promoter performance, and potentially guide future designs.
Precise promoter characterization using model-guided single-cell analysis
To further characterize this system and potentially determine how to better implement multiplexed receptors and engineered promoters, we developed a computational model that describes multiplexed receptor signaling and hybrid promoter activity (Supporting Information, Model Development Figure M1). Guiding our approach was a key experimental observation that only a small percentage of cells (ranging from about 20% to less than 1%, depending on the experiment) exhibited distinguishable promoter activation (were “ON”) in the presence of both TFs, compared to the condition with the reporter alone. As a result, mean reporter expression was generally much greater for the ON subpopulation than for the whole population. The percentage of cells in the ON state also generally varied with experimental setup: experiments with two constitutively soluble TFs and the reporter (three components on three plasmids) had ON percentages that correlated with TF plasmid doses and reached about 20% at the highest doses; experiments with one receptor, one constitutively soluble TF, and the reporter (four components on four plasmids) and with two receptors and the reporter (five components on five plasmids) had successively lower ON percentages that still correlated with plasmid doses (Supporting Information, Model Development Table M1). We hypothesized that the small ON percentage might arise from two types of intercellular variation: (1) each cell may receive a different number of molecules of each plasmid, which could potentially restrict level-matching to a subset of cells, and/or (2) cells may exhibit inherent differences in transcription rate, translation rate, and/or transfection efficiency (the efficiency with which a plasmid, once taken up, enters the nucleus and becomes transcription-competent), which comprise sources of variation that are distinct from the amounts of plasmids received. Although these two types of variation cannot be readily distinguished in experimental data, together they determine the effective initial conditions for the dose-dependent amount of each transfected gene that each cell can express. We therefore developed a model to investigate the effects of intercellular variation, and incorporated the combined effects of these two types of intercellular variation by assigning different amounts of each plasmid to each cell.
We represented heterogeneity by modeling a population of cells in which key metrics of variability matched those observed in experiments. To that end, we developed a method to generate in silico populations with the statistical features observed in a cotransfection experiment, for any specified number of plasmids (Figure 3a and Supporting Information, Model Development Figure M2). This statistical representation is consistent with a recent analysis of how levels of gene expression can be distributed among cells in a population33. From our in silico population (provided as a file in the Supporting Information), we can interrogate individual cells or calculate population-level metrics, such as mean reporter expression, for subsequent analyses and comparisons with experiments. With this framing, we first used principal component analysis (PCA) to explain sources of variation in gene expression (Supporting Information, Model Development Section II). The first principal component explained 84–90% of the variation (depending on the number of different cotransfected plasmids, from five to two, respectively) and corresponds to an axis along which plasmid amounts vary but their ratio is constant. Thus, in a cotransfection with equal amounts of two plasmids A and B, most cells will take up similar amounts of each. Some cells will take up more of plasmid A than plasmid B, or vice versa, and this effect explains the remaining 10% of the variation.
Figure 3. A model that accounts for cell variation to explain heterogeneous promoter activity.

(a) A statistical model was formulated and trained on experimental data to account for inherent intercellular variation in transcription rate, translation rate, and transfection efficiency. The marginal distribution was modeled using a Gaussian mixture model (GMM). The resulting in silico population exhibits the expected covariance between plasmids for a multi-plasmid transfection (inferred from constitutive expression of fluorescent proteins in experimental cases). Principal component analysis identified two sources of variability: the major contributor (ranging from 90% for two plasmids to 84% for five plasmids) is inherent variation, and the minor contributor is variation due to cotransfection of multiple plasmids. The Pearson correlation coefficient r in the cross-section is 0.8 on a linear scale and 0.9 on a log10 scale. (b) A dynamical model for TF expression and hybrid promoter activity in a transfected cell population was formulated and trained on mean average data in Figure 1d for various tTA and Gal4 plasmid dose combinations. (c) The promoter model maps from a three-dimensional plasmid transfection distribution onto a one-dimensional reporter expression distribution. The distributions depict the reporter expression for hybrid promoters H1 and H2, when quantified for the population mean (i.e., mean reporter expression for all transfected cells) and mean-transfected cell (i.e., a cell that receives the mean amount of each plasmid).
This result predicts that consolidating the components (genes) encoding receptor chains and hybrid promoters onto fewer plasmids would not substantially affect intercellular variation or increase the percentage of cells in the ON state. However, the system is sensitive to the number of different components; we consider the following argument: for a system of n different components, there exists an n-dimensional space that represents the amount of each component expressed per cell. Within an experiment, each cell occupies a coordinate in this space. There also exists in this space a functional region, which may be unknown a priori, and that corresponds to various combinations of the amounts of each component that yield desirable ligand-inducible promoter activation, quantified by F.D. As n increases, such as by replacing one constitutively soluble TF for the two chains of a receptor, it is possible that the overlap in n-dimensional space between the functional region and the region occupied by a cell population will change. From this perspective, one way to frame the goal of implementing an engineered function that is robust to intercellular variation is to choose component doses, and ultimately system designs, that yield high overlap between the functional region and the region that is populated by cells, where the latter can be a function of how components are delivered to cells. To this end, here we define the robustness of a system as the extent to which a performance metric (e.g., F.D.) is maintained as component doses are varied across the ranges one would observe in a plasmid cotransfection experiment. Thus, as system robustness increases, more cells in a population exhibit the desired function. Robustness is therefore distinct from performance, which is the F.D. as calculated for a single cell (at least in theory) or as measured for a population mean average (as is typically done in experimental practice). Having established this mathematical and conceptual model for describing intercellular variation, we next addressed the mechanisms by which this system operates.
To begin to capture the multiplexed MESA system, we started by developing a dynamical model for the hybrid promoters H1 and H2. Transcription was formulated as fractional activation: . The parameters wT and wG are responsiveness to tTA and Gal4, respectively, and ρ is synergy. In silico populations were initialized using the intercellular variation model and calibrated to data in Figure 1d (Figure 3b, and in Supporting Information, Model Development, Figure M3 and Tables M2–4). The calibrated parameter values indicate that between the two promoters, H1 is the more responsive and H2 is the more synergistic. H1 is 29× more responsive to Gal4 than tTA, H2 is 33× more responsive to Gal4 than tTA, the tTA response is 6.3× greater for H1 than H2, the Gal4 response is 5.5× greater for H1 than H2, and synergy is 21× greater for H2 than H1. Notably, even though the calibration utilized only the mean reporter measurements from experiments to generate parameter values, the model successfully predicted trends in the observed heterogeneity, including for the fraction of ON cells and the mean reporter expression within this subpopulation (Figure 3b). These accurate predictions validate our statistical approach for describing intercellular variation and provide confidence that our whole-population modeling approach can account for how intercellular variation affects system performance.
An important feature of the dose-response landscapes was that the mean reporter expression (averaged across all transfected cells) was consistently greater than the reporter expression for a cell receiving the mean amount of each plasmid, i.e., the mean-transfected cell (Figure 3c and Supporting Information, Model Development Figures M4–7). Cells that received greater than average plasmid amounts had even greater than average reporter expression, and the resulting reporter distributions were right-skewed. One implication is that the initial promoter characterization in Figure 1d was in part driven by outlier cells that received relatively high amounts of plasmids in each condition. Since the characterization of not only the hybrid promoters, but also other genetic circuits with nonlinear behaviors, could potentially be driven by outlier cells, we posit that a model-guided investigation of single-cell outcomes in which intercellular variation is captured explicitly may improve the interpretation of such experimental results. Indeed, this approach has been used to substantial effect in the characterization of other engineered biological systems34–35.
Elucidating properties of multiplexed receptor performance using a mechanistic model
Having established a quantitative framework for describing the transcriptional layer, we next incorporated the MESA receptors. A dynamical model for multiplexed MESA signaling was formulated at a level of mechanistic granularity that includes salient interactions for various receptor complexes (Figure 4a). Key features dictated by prior knowledge, including known receptor-ligand interactions and findings from previous experiments with MESA7–8, are: receptors are synthesized intracellularly, exocytosed to the plasma membrane, and degrade from both compartments; rapamycin can diffuse intracellularly, but VEGF cannot; VEGF-MESA can heterodimerize or homodimerize, and Rap-MESA can heterodimerize but not homodimerize; crosstalk in non-ligand mediated signaling is possible, because both MESA use the same PC protease and TC cleavage recognition sequence; and chains that recognize the same ligand can form stable dimers, but chains that recognize different ligands cannot (Supporting Information, Model Development Figure M8 and Table M5). Calibration of this mechanistic model to data in Figure 2 provided estimates for the synthesis of receptors (relative to soluble proteins), background signaling, receptor degradation, ligand-binding to each MESA, and stable chain dimerization (Supporting Information, Model Development Tables M6–7.
Figure 4. A dynamical model that links MESA receptor signaling to promoter activity.

(a) This illustration summarizes the species and reactions in the MESA model. There are 28 types of reactions, which are grouped into nine categories (named) and four modalities (boxed). Reactions that occur for both MESA (Rap-MESA and VEGF-MESA) are bolded, categories that release a soluble TF are highlighted (yellow arrow), and the modality for canonical ligand-induced signaling is highlighted (yellow box). For the four modalities: (1) background signaling is the only one that occurs in the absence of ligand, (2) ligand-binding and dimerization involve ligand but do not directly result in signaling, (3) dimerization signaling is the canonical ligand-induced pathway, and (4) the remaining categories involve, but are not directly mediated by, the ligand and are subject to crosstalk. (b) Data that were used to determine F.D. in Figure 2a are compared to simulated outcomes for a similar in silico experiment with VEGF-MESA, constitutive soluble Gal4, and promoter H1, with VEGF (lower panel) and without VEGF (upper panel). Since reporter expression is quantified in units that differ between experiments and simulations, experimental data (originally in flow cytometry-specific units) were linearly scaled to enable a more direct visual comparison with simulation results. (c) Time course H1 reporter trajectories across TC and PC doses are shown for the mean-transfected cell, +/− each ligand treatment (V, VEGF; R, Rap; VR, VEGF and Rap). In the left panel, VEGF-MESA doses are varied while Rap-MESA dose is constant, and in the right panel, Rap-MESA doses are varied while VEGF-MESA dose is constant. Simulations are grouped into five outcome cases (represent by box shading and outline color) based on the rank-ordered expression with each ligand treatment. (d) Three cases from (c) are examined in more detail. The left panel shows the absolute reporter expression, and the right panel shows ligand-induced reporter expression after the background (without ligand) is subtracted, to illustrate the additive ligand-induced response to these ligands. There exists a trade-off for two-ligand induced signaling, in which adjustments to the MESA plasmid dose that increase the F.D. compared to one ligand also decrease F.D. compared to the other ligand.
The model formulation is idealized, as it makes relatively few assumptions and avoids overfitting peaks and valleys that deviate from the main experimental trends. By smoothening (or discounting) individual outlier data points, such an analysis improves the overall interpretability of the dose-response landscape. When comparing observed vs. simulated ligand-inducible VEGF-MESA signaling in the presence of excess Gal4, the simulations were consistent with observed trends (Figure 4b). For a constitutively soluble TF and a MESA receptor (TC and PC) transfected at the same plasmid dose, the TF tended to contribute more to promoter activity. A model-guided interpretation for this outcome is that (1) rapid production of soluble TFs (relative to receptors) offsets rapid degradation, leading to high accumulation (Supporting Information, Model Development Figure M9), and (2) for the receptors, not all target chains are cleaved following ligand treatment. We next analyzed the multiplexed receptors at different plasmid doses, and in doing so identified a trade-off for multiplexing: tuning receptor levels to increase the difference in two-ligand induced reporter expression with respect to one ligand consistently led to a decrease in the difference with respect to the other ligand. To highlight this effect, we grouped reporter trajectories into different outcome cases (Figure 4c and Supporting Information, Model Development Figure M10). Examining three examples in more detail (Figure 4d) shows that across different receptor doses, reporter expression was additive: the two-ligand induced increase in reporter expression above background equaled the sum of both single-ligand induced increases above background. The additivity indicates that promoter activity had a linear dependence on the TFs in these experiments, rather than the synergy that would be expected for the AND gate promoters based upon characterizations with constitutively soluble TFs (Figure 1). We note that within any population, some individual cells may perform better than others, and that altering time points for ligand treatment and reporter measurement may modestly increase F.D. (Supporting Information, Model Development Figures M11–12). However, overall, the findings support the conclusion that the amounts of TFs released from receptors were below the amounts required to leverage promoter synergy, and so the system exhibited an additive rather than synergistic response to ligand combinations.
To better understand how multiplexed receptors integrate with the hybrid promoter, we next systematically investigated the role of level-matching, i.e., the relationship between the amounts of TFs that are released by receptors under different ligand treatments, and the amounts that are required to activate promoter synergy uniquely in the case when both ligands are sensed. This relationship is challenging to observe or elucidate through experiments alone, yet it is also key for explaining multiplexed receptor performance. Since nonlinear TF profiles from ligand-induced signaling depend on more components and are more complex than their linear counterparts from experiments with constitutively expressed soluble TFs, we could not visualize level-matching with a dose-response landscape as in Figure 3b. Therefore, we opted to introduce a new graphical representation, which we term yo-yo plots, in which multiple timecourse TF trajectories with various ligand combinations are represented simultaneously without using a time axis, and reporter expression across the timecourse (the “string”) and at the endpoint (the “yo-yo”) is indicated using a color scale (Figure 5a). Using this approach, we examined how free TFs were released over time under conditions corresponding to those used in level-matching experiments (Figure 5b). The analysis confirmed our expectation that mean reporter outcomes were driven by outlier dynamics. Furthermore, it shows how outlier effects became magnified as each constitutively soluble TF was replaced by the two chains for a receptor. Across experiments, reporter expression for the population mean was consistently greater than for the mean-transfected cell, which poses a challenge to achieving level-matching by simply tuning dosages of system components. Moreover, these analyses confirmed that across the various MESA doses evaluated in experiments, the amounts of TFs released by the receptors were below the levels required to induce synergistic activation of the H1 promoter. Altogether, this case study illustrates how model-guided analysis of combined sensor (MESA) and processor (promoter) modules can identify quantitative properties that benefit and that limit system performance. This approach may also be inverted to guide the selection of components with properties that achieve specific performance goals.
Figure 5. Level-matching between receptor signaling and the promoter.

(a) Level-matching is depicted by yo-yo plots, which represent the trajectories of free TF and reporter variables together and without using a time axis. Reporter expression across the time course (the “string”) and at the circled endpoint (the “yo-yo”, corresponding to the time point for experimental measurements) is indicated using a color scale. Each profile begins at the origin, proceeds through state space depending on plasmid doses and treatment with either, both, or no ligand, and concludes at the circled coordinate. (b) Quantitative outcomes for each ligand treatment are shown for varied plasmid doses in three scenarios: (1) two constitutively soluble TFs, (2) one constitutively soluble TF and one MESA receptor, and (3) two MESA receptors. TFs are in comparable arbitrary units (a.u.), and reporter expression is color-coded by reporter-specific a.u. Profiles in which the circled coordinate differs from the maximum coordinate along a given axis indicate that the trajectory of the corresponding TF peaks and decreases during the timecourse. Diagonal lines indicate that the trajectories of both TFs are changing proportionately, curved lines indicate that both are changing and in a way that is not proportional, and vertical and horizontal lines indicate that one is changing while the other has reached a steady state. In the second scenario, only the TF released from MESA (and not the constitutively soluble TF) is plotted, and a slight downward curvature for the 36 h time course is shown for clarity. In the third scenario, ideal level-matching for AND gate functionality would be conferred by TF trajectories that lead to much higher reporter expression with both ligands compared to either or no ligand. In such a scenario, the upper-right yo-yo would be the only one of the four that is able to leverage the synergistic regime of the hybrid promoter.
Promoter and receptor engineering strategies to improve AND gate functionality
Given the bounds on AND gate performance discussed above, we further leveraged this case study to explore whether model-guided design could identify promoter and receptor properties that better achieve performance goals, relative to the existing constructs examined above. In this prospective analysis, we considered parameter values that could reasonably be implemented physically, even if it is not yet possible to predict which specific physical modification would result in a specific new parameter value. One benefit of using a mechanistic model, as opposed to a more abstract formalism, is that the parameters do ultimately correspond to physical features that in future investigations could be tuned by making corresponding biomolecular design choices. For example, background signaling could be decreased by mutating the protease active site, protease cleavage sequence, or transmembrane domain, or ligand binding could be modulated by mutating or replacing the ECD. Thus, a computational exploration of potentially realizable scenarios could guide subsequent investigations.
To anchor our parameter exploration, we specified hypothetical constructs in which properties were varied relative to an experimentally observed base case. H1 served as a base case (promoter #1) for testing hypothetical promoters in silico that vary in responsiveness to each TF and/or in synergy (Figure 6a). Properties of promoters (cases #3–9) are reported relative to promoter #2 (H1=), in which tTA responsiveness was set equal to Gal4 responsiveness. For a controlled comparison between promoters #3–9, we chose transcriptional weights that yielded dose-response landscapes that differ from the base case but that resemble each other’s maximal activity within the range of TF doses examined (Figure 6b). Importantly, this range more closely matches the inferred range of TFs released from MESA signaling (Figure 5), and synergistic activation for the new promoters occurs within this experimentally relevant range.
Figure 6. Promoter engineering to improve AND gate performance.

(a) Hypothetical promoters were produced in silico and vary in responsiveness to each TF and/or in synergy. Multipliers for transcriptional weight parameters in cases #3–9 are in comparison to case #2, in which tTA responsiveness is set equal to Gal4 responsiveness. Case #2 is a responsiveness-balanced version of H1 (base case, #1). (b) Promoters were characterized by reporter expression for the population mean (top row) and the mean-transfected cell, using doses of constitutively soluble TFs that match the inferred range of TFs released in MESA signaling. (c) Multiplexed MESA performance with each promoter was assessed by a sweep of 1,000 receptor plasmid dose combinations (0 to 0.5 μg per plasmid), each of which is represented by a single data point on each plot. Plots report three performance metrics: two-ligand induced F.D. calculated with respect to (1) treatment with VEGF alone (x-axis), (2) treatment with rapamycin (y-axis), and (3) no ligand (color-coded). In each case, these metrics are calculated for the mean-transfected cell in a population. Better AND gates are realized towards the upper-right region of each plot. All three F.D. metrics cannot be maximized simultaneously, as evidenced by the absence of outcomes in the upper-right-most corner, since choosing plasmid doses that maximize any one metric comes at the expense of decreasing one or both others. Therefore, the best possible AND gate functionality requires each metric to be maximized only to an extent, and in a way that balances the trade-off with the others. A representative ideal instance for each case is indicated by a box and is examined further in (d) and (e). The best promoter overall (#6) is outlined in yellow. (d) A comparison of reporter expression for instances identified by the boxes in (c), still using the mean-transfected cell. (e) Effects of cell variation on the three F.D. metrics. X-axis numbers are multipliers for the relative amounts of plasmids received by cells (determined without the variance from the minor principal component that is due to cotransfection), such that a value of “1” is the mean-transfected cell. The multipliers 1/16, 1/4, 1, and 4 correspond to the 23rd, 45th, 62nd, and 85th percentiles, respectively, for amounts of plasmids received by a cell in a transfected population, as determined from the intercellular variation model. Each line represents the F.D. outcomes from 31 different simulations of increasing plasmid dose, from left to right. Greater robustness to intercellular variation (in context of the specific plasmid doses for each of the nine cases) is indicated by increases in F.D. across a wider range of x-axis values.
To assess how each promoter affects MESA multiplexing, and to avoid bias toward any dose-specific implementation, we conducted a four-dimensional sweep of receptor doses. The outcome for the mean-transfected cell from each population is presented as one data point in the plot for each promoter (Figure 6c). F.D. for the two-ligand case relative to each one-ligand case is indicated by the position along the horizontal and vertical axes, and F.D. for the two-ligand case relative to the no-ligand case is indicated by the color scale. Perimeters define the performance bounds of the promoters, and the most ideal AND behavior is realized along the upper-right region, where the two-ligand F.D. is greater than each one-ligand F.D. and the trade-off with respect to each individual ligand (as described in Figure 4d) has been balanced. For each case, a selected ideal outcome is denoted by a box. As was observed with the soluble TF sweeps, simply setting the responsiveness of each TF to be the same (#2) conferred minimal changes; however, large improvements in AND functionality were realized by increasing promoter synergy either alone (#6) or in combination with TF responsiveness (#7–9). To interpret the selected ideal F.D. outcomes in Figure 6c, we examined reporter expression (Figure 6d). Compared to promoter #1, reporter expression with different ligand treatments increased by ~50× with promoter #2, and by up to another order of magnitude with promoters #3–9. However, improved AND gate functionality did not necessarily follow from changes that conferred the largest increases in expression (e.g., promoter #4); rather, improvements arose from the largest difference in increased expression between the two-ligand case and one-ligand cases (e.g., promoter #6).
To evaluate the effects of intercellular variation, we calculated F.D. across a range of amounts of total plasmid that is received by transfected cells (Figure 6e). The maximum F.D. for each promoter was achieved by a subpopulation, and the location of this window differed between promoters. Interestingly, while the base case was the lowest performing, it was the most robust to variation in plasmid dose, with a relatively flat profile. Promoters #3–9 exhibited distinct maxima for F.D. at specific plasmid doses and exceeded the maximum F.D. of promoter #1, indicating that for these hypothetical promoters, obtaining transfected cells with intermediate amounts of plasmids (given the specified dose) would confer maximal performance. Alternatively, any strategy that reduces intercellular variation in expression levels, such as genomic integration of MESA expression constructs, may improve the performance of such promoters, although this appears to be less promising for the existing H1 promoter. Additional promoters were examined with larger effect-magnitudes for the same types of changes as in Figure 6a, but these changes had diminishing returns on performance, and further improvements were modest (Supporting Information, Model Development Figure M13). Altogether, this systematic analysis provides new insights into how future promoters may be designed, evaluated, and utilized in combination with MESA receptors, depending on the performance objectives sought.
We next investigated how MESA may be modified to tune performance in multiplexing applications. Mirroring our approach for exploring promoter variations, hypothetical but physically realistic modifications to receptor kinetics and biomolecular design were introduced individually and in combination in silico to generate a panel of distinct cases (Figure 7a). For each case, system performance (F.D. +/− ligand) was compared to the base case, comprising our existing MESA receptor, which was paired with either the base case promoter (promoter #1) or the best hypothetical promoter from Figure 6 (promoter #6) (Figure 7b). Although making changes to the receptor while retaining the base case promoter had little impact on performance, larger improvements were possible with promoter #6. The greatest effects were conferred when multiple modifications were implemented together: orthogonal cleavage recognition sequences, slower basal cleavage, and faster ligand-induced receptor dimerization (receptor case #9). Examining the reporter expression shows how each modification affected F.D. (Figure 7c). Importantly, not all intuitively attractive changes improved system performance, and so this analysis helps identify promising strategies. For example, decreasing the receptor degradation rate in receptor case #2 could increase the amount of TF that can be released, but this increases the background and one-ligand expression more than it increases two-ligand expression. In contrast, modifications such as receptor cases #6 and #9 increase the two-ligand induced signaling while maintaining or driving down background. That is, the amounts of TFs released when both ligands are sensed can activate the new promoter synergistically, whereas TF levels in the presence of neither or only one ligand drive much less promoter activation. We also found that although engineering both the promoter and receptors in combination still resulted in a scenario in which maximal F.D. was observed within windows of plasmid dose (Figure 7d), AND behavior remained robust across about an order of magnitude in cell variation, representing a large subpopulation. Altogether, these results help to identify potential strategies that could be explored for engineering receptors and promoters to achieve level-matching in this particular case of implementing single-layer transcriptional logic. Moreover, the calibrated model developed in this study could be similarly leveraged to prospectively evaluate the future use and design of MESA for implementing other cell functions. More broadly, this approach highlights the utility of quantitative analyses that capture intercellular variation to guide the design of engineered systems that confer desired performance objectives.
Figure 7. Receptor engineering to improve AND gate performance.

(a) Hypothetical modifications to MESA receptor kinetics and design features were produced in silico, in reference to the MESA base case (#1). Receptor cases #2–5 are modifications to #1, and #6–9 are combinations of the modifications in #3–5. (b) Multiplexed MESA performance was assessed and plotted as described in Figure 6c. Outcomes are shown for two promoters: base case (promoter #1, upper row) and a high-performing promoter from Figure 6 (promoter #6, lower row). For promoter #6, a representative ideal instance for each receptor modification is indicated by a box and is examined further in (c) and (d), as described for similar investigations of promoter variations in Figure 6. The best receptor overall (#9) is outlined in yellow.
CONCLUSIONS
In this study, we explored how MESA receptors may be multiplexed to implement cellular logic in response to environmental ligands. The specific logic investigated here—a single-layer transcriptional AND gate—serves as a useful test case for evaluating our strategy for tuning MESA signaling using both experimental and computational techniques to achieve level-matching with a downstream promoter. We anticipate that implementing other types of cellular logic may be achieved using a similar approach, that other engineered receptor/promoter systems may exhibit similar phenomenology, and that the construction of such custom biological functions may be realized by using in silico analysis to examine the system components and constraints and guide design choices. Furthermore, other logical programs may impose less stringent requirements for promoter or receptor design criteria. For example, an OR gate in which each MESA releases the same TF to induce a single-TF promoter should require less tuning. Since the output of each MESA receptor can be readily exchanged, our results suggest that other regulators such as zinc finger TFs36 or Cas9-based TFs37–38 could also be used to program cell functions via receptor multiplexing. Ultimately, MESA multiplexing comprises a new functional modality for engineering customized mammalian cellular programs for a variety of applications.
MATERIAL AND METHODS
DNA constructs
Hybrid reporter constructs were assembled using oligonucleotides and standard molecular biology techniques and cloned into a pBI-YFP reporter (described previously8). For some experiments, the YFP reporter gene was replaced with DsRedExpress2 (here, DsRed) using standard restriction enzyme cloning. Development of VEGF-MESA and Rap-MESA was described previously7–8. To develop a Rap-MESA with the Gal4 TF, tTA on the original target chain was replaced with Gal4 (derived from a Notch1-Gal4 fusion construct generously gifted by Steven Blacklow39) using standard PCR and restriction enzyme cloning. Additionally: 1) for each chain, a 20 amino acid flexible glycine-serine linker was incorporated between the rapamycin binding extracellular domain (ECD) and the transmembrane domain, and 2) the transmembrane domain was modified to mirror the design of the more recently developed VEGF-MESA receptors7; though these design features are not required for Rap-MESA function, our recent work suggests such features generally confer more stable MESA expression on the cell surface, and thus a similar MESA architecture was implemented to facilitate receptor dose tuning experiments. DNA constructs and primers for cloning are listed in the Supporting Information.
Cell culture and transfection
HEK 293FT cells (Life Technologies/Thermo) were maintained at 37°C and 5% CO2 in Dulbecco’s modified growth medium supplemented with 10% FBS, 1% penicillin-streptomycin, and 2 mM L-glutamine (Life Technologies). Transient transfections were performed in 24-well plates seeded at 7.5 × 104 cells in 0.5 mL of media. At 6–8 h post-seeding, cells were transfected using the CaCl2-HEPES-buffered saline (HeBS) method. All experiments included a BFP transfection control to determine transfection efficiency. At 12 h post-transfection, media was changed on all cells and, if applicable, rapamycin and/or VEGF were added by pre-mixing in the media to a final concentration of 100 nM rapamycin in 0.5% DMSO (Santa Cruz Biotechnology) or 250 ng/mL recombinant mouse VEGF-164 (BioLegend).
Flow cytometry
At 36 h post-transfection, samples were suspended in PBS with 2 mM EDTA and 5% bovine serum albumin. Approximately 1 × 104 single, live cells per sample were analyzed on a LSRII flow cytometer (BD Biosciences) that runs FACSDiva software. Data were further analyzed and compensated utilizing FlowJo Software (Tree Star). Live, single cells were identified by scatter, and BFP-positive cells were gated as “transfected”. Relative reporter expression (for DsRed or YFP) was calculated as described in the Figure 1 caption.
Supplementary Material
Acknowledgments
K.A.S. was supported in part by National Institutes of Health T32 Training Grant GM 008449 through Northwestern University’s Biotechnology Training Program. R.M.H. was supported in part by the Malkin Scholars Program from the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. J.J.M. was supported in part by the Northwestern University Graduate School Cluster in Biotechnology, Systems, and Synthetic Biology, which is affiliated with the Biotechnology Training Program, and by an award from the Cornew Innovation Fund, administered by the Chemistry of Life Processes Institute (to N.B. and J.N.L). This work was supported by the Northwestern University Flow Cytometry Facility and a Cancer Center Support Grant (NCI CA060553). This project was supported by the Defense Advanced Research Projects Agency, Award number W911NF-11-2-0066 (to J.N.L.). No funding sources had any involvement in the writing of this manuscript. We thank Taylor Dolberg, Patrick Donahue, Amy Hong, Cameron McDonald, Alexis Prybutok, and Albert Xue for helpful discussions.
ABBREVIATIONS
- CAR
chimeric antigen receptor
- ECD
extracellular domain
- F.D
fold-difference
- MESA
Modular Extracellular Sensor Architecture
- MFI
mean fluorescence intensity
- PC
protease chain
- Rap
rapamycin
- TC
target chain
- TF
transcription factor
- tTA
tetracycline transactivator
- VEGF
vascular endothelial growth factor
Footnotes
Author Contribution
K.A.S. and R.M.H. performed, designed, and analyzed experiments. J.J.M. and J.N.L. developed and analyzed the computational model. K.A.S., R.M.H., and J.N.L conceived of the study. K.A.S., R.M.H., J.J.M., N.B., and J.N.L. wrote the manuscript, and prepared the figures.
Notes
K.A.S., R.M.H., and J.N.L. are inventors on U.S. patent applications covering inventions described in this manuscript.
Supporting Information includes supplemental figures (with figures denoted by the prefix S), model development (with figures and tables denoted by the prefix M), model files, primers used for hybrid reporter construction, and plasmid sequence files of all constructs used in this study. This information is available free of charge at http://pubs.acs.org.
References
- 1.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–81. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarz KA, Leonard JN. Engineering cell-based therapies to interface robustly with host physiology. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–5. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164(4):770–9. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell. 2016;164(4):780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarz KA, Daringer NM, Dolberg TB, Leonard JN. Rewiring human cellular input-output using modular extracellular sensors. Nat Chem Biol. 2017;13(2):202–209. doi: 10.1038/nchembio.2253. [DOI] [PubMed] [Google Scholar]
- 8.Daringer NM, Dudek RM, Schwarz KA, Leonard JN. Modular Extracellular Sensor Architecture for Engineering Mammalian Cell-based Devices. ACS Synthetic Biology. 2014;3(12):892–902. doi: 10.1021/sb400128g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson JC, Voigt CA, Arkin AP. Environmental signal integration by a modular AND gate. Mol Syst Biol. 2007;3 doi: 10.1038/msb4100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Kitney RI, Joly N, Buck M. Engineering modular and orthogonal genetic logic gates for robust digital-like synthetic biology. Nat Commun. 2011;2 doi: 10.1038/ncomms1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moon TS, Lou C, Tamsir A, Stanton BC, Voigt CA. Genetic programs constructed from layered logic gates in single cells. Nature. 2012;491:249–253. doi: 10.1038/nature11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brödel AK, Jaramillo A, Isalan M. Engineering orthogonal dual transcription factors for multi-input synthetic promoters. Nat Comm. 2016;7 doi: 10.1038/ncomms13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer BP, Fischer C, Fussenegger M. BioLogic gates enable logical transcription control in mammalian cells. Biotechnol Bioeng. 2004;87 doi: 10.1002/bit.20142. [DOI] [PubMed] [Google Scholar]
- 14.Marchisio MA, Stelling J. Automatic design of digital synthetic gene circuits. PLoS Comp Biol. 2011;7:e1001083. doi: 10.1371/journal.pcbi.1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Win MN, Smolke CD. Higher-order cellular information processing with synthetic RNA devices. Science. 2008;322:456–460. doi: 10.1126/science.1160311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausländer S, Ausländer D, Müller M, Wieland M, Fussenegger M. Programmable single-cell mammalian biocomputers. Nature. 2012;487:123–127. doi: 10.1038/nature11149. [DOI] [PubMed] [Google Scholar]
- 17.Lohmueller JJ, Armel TZ, Silver PA. A tunable zinc finger-based framework for Boolean logic computation in mammalian cells. Nucleic Acids Res. 2012;40:5180–5187. doi: 10.1093/nar/gks142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D. Amplifying genetic logic gates. Science. 2013;340:599–603. doi: 10.1126/science.1232758. [DOI] [PubMed] [Google Scholar]
- 19.Siuti P, Yazbek J, Lu TK. Synthetic circuits integrating logic and memory in living cells. Nat Biotechnol. 2013;31(5):448–452. doi: 10.1038/nbt.2510. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg BH, Pham NTH, Caraballo LD, Lozanoski T, Engel A, Bhatia S, Wong WW. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat Biotechnol. 2017;35:453–462. doi: 10.1038/nbt.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regot S, Macia J, Conde N, Furukawa K, Kjellén J, Peeters T, Hohmann S, de Nadal E, Posas F, Solé R. Distributed biological computation with multicellular engineered networks. Nature. 2011;469:207–211. doi: 10.1038/nature09679. [DOI] [PubMed] [Google Scholar]
- 22.Tamsir A, Tabor JJ, Voigt CA. Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature. 2011;469:212–215. doi: 10.1038/nature09565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramalingam KI, Tomshine JR, Maynard JA, Kaznessis YN. Forward engineering of synthetic bio-logical AND gates. Biochem Eng J. 2009;47:38–47. [Google Scholar]
- 24.Schreiber J, Arter M, Lapique N, Haefliger B, Benenson Y. Model-guided combinatorial optimization of complex synthetic gene networks. Mol Syst Biol. 2016;12:899. doi: 10.15252/msb.20167265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaman F, Bhatia S, Adler A, Densmore D, Beal J. Automated selection of synthetic biology parts for genetic regulatory networks. ACS Synth Biol. 2012;1:332–344. doi: 10.1021/sb300032y. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen AAK, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, Ross D, Densmore D, Voigt CA. Genetic circuit design automation. Science. 2016;352:aac7341. doi: 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- 27.Schukur L, Geering B, Charpin-El Hamri G, Fussenegger M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci Trans Med. 2015;7:318ra201. doi: 10.1126/scitranslmed.aac4964. [DOI] [PubMed] [Google Scholar]
- 28.Gaber R, Lebar T, Majerle A, Ster B, Dobnikar A, Bencina M, Jerala R. Designable DNA-binding domains enable construction of logic circuits in mammalian cells. Nat Chem Biol. 2014;10(3):203–8. doi: 10.1038/nchembio.1433. [DOI] [PubMed] [Google Scholar]
- 29.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547–51. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335(6190):563–4. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 31.Prindle A, Hasty J. Making gene circuits sing. P Natl Acad Sci USA. 2012;109(42):16758–16759. doi: 10.1073/pnas.1214118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egbert RG, Klavins E. Fine-tuning gene networks using simple sequence repeats. P Natl Acad Sci USA. 2012;109(42):16817–16822. doi: 10.1073/pnas.1205693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beal J. Biochemical complexity drives log-normal variation in genetic expression. Engineering Biology. 2017;1(1):55–60. [Google Scholar]
- 34.Davidsohn N, Beal J, Kiani S, Adler A, Yaman F, Li Y, Xie Z, Weiss R. Accurate predictions of genetic circuit behavior from part characterization and modular composition. ACS Synth Biol. 2015;4(6):673–81. doi: 10.1021/sb500263b. [DOI] [PubMed] [Google Scholar]
- 35.Beal J, Wagner TE, Kitada T, Azizgolshani O, Parker JM, Densmore D, Weiss R. Model-driven engineering of gene expression from RNA replicons. ACS Synth Biol. 2015;4(1):48–56. doi: 10.1021/sb500173f. [DOI] [PubMed] [Google Scholar]
- 36.Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, Joung JK, Collins JJ. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 2012;150(3):647–58. doi: 10.1016/j.cell.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeder ML, Linder SJ, Cascio VM, Fu YF, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordon WR, Vardar-Ulu D, L’Heureux S, Ashworth T, Malecki MJ, Sanchez-Irizarry C, McArthur DG, Histen G, Mitchell JL, Aster JC, Blacklow SC. Effects of S1 cleavage on the structure, surface export, and signaling activity of human Notch1 and Notch2. PLoS One. 2009;4(8):e6613. doi: 10.1371/journal.pone.0006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.