

Abstract
As a result of therapy-induced apoptosis, peripheral blood monocytes are recruited to tumors, where they become tumor-associated macrophages (TAMs). To date, few studies have investigated noninvasive molecular imaging for assessment of macrophage infiltration in response to therapy-induced apoptosis. Here, noninvasive assessment of changes in tumor accumulation of TAMs was proposed as a new way to measure early tumor response to anticancer therapy. Three different nanoparticles, QD710-Dendron quantum dots (QD710-D), Ferumoxytol, and PG-Gd-NIR813, were used for near-infrared fluorescence imaging, T2-weighted magnetic resonance imaging, and dual optical/T1-weighted MR imaging, respectively, in the MDA-MB-435 tumor model. Treatment with Abraxane induced tumor apoptosis and infiltrating macrophages. In spite of markedly different physicochemical properties among the nanoparticles, in vivo imaging revealed increased uptake of all three nanoparticles in Abraxane-treated tumors compared with untreated tumors. Moreover, imaging visualized increased uptake of QD710-D in MDA-MB-435 tumors but not in drug-resistant MDA-MB-435R tumors grown in the mice treated with Abraxane. Our results suggest that infiltration of macrophages due to chemotherapy-induced apoptosis was partially responsible for increased nanoparticle uptake in treated tumors. Noninvasive imaging techniques in conjunction with systemic administration of imageable nanoparticles that are taken up by macrophages are a potentially useful tool for assessing early treatment response.
Keywords: Macrophage, Chemotherapy response, Nanoparticle, Optical Imaging, Magnetic Resonance Imaging
1. Introduction
The tumor microenvironment contributes to tumor initiation, progression, metastasis, and resistance to therapy. Macrophages are found in all tissue types including tumors. Their origin is bone marrow with an intermediary step in their continuum of differentiation as peripheral blood monocytes. Young monocytes are capable of inducing inflammation due to their ability to secrete various cytokines, as they mature/differentiate in tissues, they adjust to the environment. In tumors, tumor-associated macrophages (TAMs) primarily exhibit the M2 phenotype. Referred to as alternatively activated macrophages, these TAMs are believed to contribute to tumor growth and progression and to promote tumor cell survival, proliferation, and dissemination [1, 2]. In many cancers, high macrophage infiltration correlates with poor outcome [3-5].
Historically, studies examining tissue and cellular responses to chemotherapy and radiation therapy predominantly focused on the eradication of proliferating malignant cells. However, increasing evidence suggests that these treatments also significantly alter the tumor microenvironment, particularly with respect to tumor-infiltrating immune cells, including TAMs. Most chemotherapeutic agents kill cancer cells by inducing apoptosis [6-8]. Recent studies have shown that cells undergoing apoptosis express specific “eat me” signals (e.g. phosphatidylserine) that facilitate recognition and ingestion by macrophages [9, 10]. As a result, both chemotherapy and radiation therapy have been shown to result in tumor infiltration of macrophages [11-14].
Given the recruitment of TAMs in response to apoptosis, assessment of TAMs has been considered as a new way to measure early tumor response to anticancer therapy. At present, the gold standard for the assessment of TAMs is immunohistochemical and histological examination of excised tumor specimens. Such techniques, however, are limited in the assessment of TAM infiltration over time and cannot be used when biopsy samples are not available.
We hypothesized that TAMs could be imaged noninvasively to monitor early tumor response to anticancer therapy. Specifically, since TAMs are phagocytes and very efficient at internalizing particles, we hypothesized that TAMs could be imaged by using nanoparticles that would be readily phagocytosed or internalized. To test this hypothesis, we used three different nanoparticles with markedly different physicochemical properties and had previously been evaluated for selective visualization of TAMs in cancer: QD710-Dendron quantum dots (QD710-D), Ferumoxytol, and PG-Gd-NIR813 (Fig. 1) [15-17]. QD710-D is a new type of dendron-coated InP/ZnS core/shell QD. The core size of QD710-D is about 5 nm in diameter, with a hydrodynamic diameter of about 11.8 nm. QD710-D emit a fluorescent signal in the near-infrared (NIR) region, have high chemical stability and photostability in biological media, and exhibit low systemic toxicity [18]. Ferumoxytol is an ultrasmall superparamagnetic iron oxide nanoparticle approved by the US Food and Drug Administration (FDA) as an iron supplement for intravenous treatment of iron deficiency in patients [19-21]. Ferumoxytol is composed of superparamagnetic iron oxide and polyglucose sorbitol carboxymethyl ether coated on its surface. It has a mean hydrodynamic diameter of 30 nm and displays a prolonged blood pool circulation time [22-24]. Ferumoxytol has also been evaluated as a contrast agent for T2-weighted magnetic resonance imaging (MRI) [25-28]. PG-Gd-NIR813 is a dual magneto-optical imaging probe consisting of poly(L-glutamic-acid) (PG) conjugated with DTPA-Gd and a NIR fluorescence (NIRF) dye NIR813. PG-Gd has been shown to be an excellent blood-pool MRI agent in both mice and rhesus monkeys for T1-weighted MR imaging [29]. PG-Gd-NIR813 has also been shown to accumulate in macrophages of solid tumors [17, 30].
Figure 1.
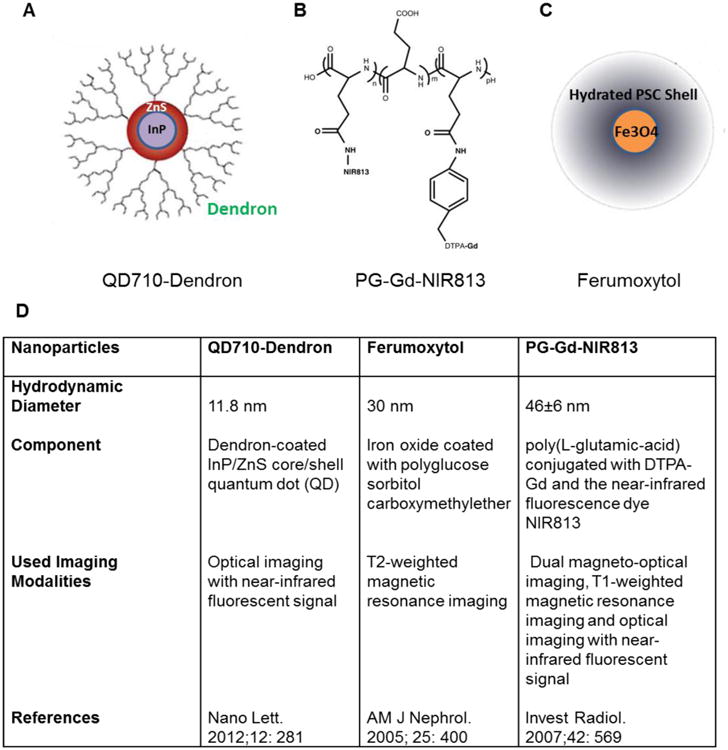
Structure and character of three nanoparticles: (A) QD710-Dendron, (B) PG-Gd-NIR813, and (C) Ferumoxytol. (D) Characteristics of these three nanoparticles.
2. Materials and methods
2.1. Chemicals and reagents
Abraxane (10% paclitaxel and 90% human serum albumin) was purchased from Abraxis Oncology (Bridgewater, NJ). The kits for the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) cell proliferation assay and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay were purchased from Promega (Madison, WI). QD710-Dendron quantum dots (QD710-D) was provided by NN-Labs (Fayetteville, AR). Ferumoxytol (Feraheme) was purchased from AMAG Pharmaceuticals Inc. (Waltham, MA). PG-Gd-NIR813 was synthesized as reported before [17]. Rat anti-mouse CD169 antibody was purchased from AbD Serotec (Raleigh, NC). FITC-labeled rat anti-mouse CD11b antibody was purchased from BD Biosciences (San Jose, CA). Alexa Fluor 594-conjugated goat anti-rat secondary antibody was purchased from Invitrogen Molecular Probes (Grand Island, NY). Mounting medium with DAPI was purchased from Vector Laboratories (Burlingame, CA). FITC-labeled anti-CD45, PE-labeled anti-CD11b, eFluor660-labeled anti-CD169, Percp.Cy5.5-labeled anti-F4/80, and fixable viability dye eFluor780 used for flow cytometry experiments were purchased from eBioscience (San Diego, CA). Isoflurane was purchased from Abbott Laboratories (Abbott Park, IN). D-luciferin was purchased from Biosynth International, Inc. (Itasca, IL).
2.2. Cell lines
Human MDA-MB-435 (M4A4) breast carcinoma cells were purchased from the American Type Culture Collection (Manassas, VA) and were reconfirmed with Short Tandem Repeat (STR) DNA fingerprinting method by the Characterized Cell Line Core Facility of The University of Texas MD Anderson Cancer Center when the cells were used in this study. The MDA-MB-435 cells were maintained in Leibovitz's L-15 medium (Invitrogen) supplemented with 10% fetal bovine serum at 37°C.
The resistant subline MDA-MB-435R was generated in our laboratory by continuous exposure of MDA-MB-435 cells to paclitaxel. Briefly, the parent MDA-MB-435 cells were exposed to increasing concentrations of paclitaxel, from 1 nM to 25 nM, and the cells were maintained in continuous culture with 25 nM paclitaxel for 30 days.
Mouse RAW264.7 macrophages were purchased from American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum at 37°C and 5% CO2. The cells were used within 6 months after receipt.
2.3. Tracking of cellular uptake of QD710-D in vitro
RAW264.7 murine macrophages and MDA-MB-435 human breast cancer cells (1×105 cells for each cell line) were separately seeded in cell culture petri dishes for 24 h. Then, QD710-D was added (final concentration, 1 nM), and cells were incubated for 1 h or 20 h. After the washing steps, the cells were stained with FITC-labeled rat anti-mouse CD11b antibody. All fluorescence images were acquired under the same conditions and displayed under the same color scale using a Zeiss fluorescence microscope (Zeiss Axio Observer.Zl, Carl Zeiss MicroImaging GmbH, Gȍttingen, Germany).
2.4. Tracking of cellular uptake of Ferumoxytol by Prussian blue staining
RAW264.7 murine macrophages and MDA-MB-435 human breast cancer cells (1×105 cells for each cell line) were separately seeded in cell culture petri dishes for 24 h. Then, Ferumoxytol was added (final concentration, 200 μg/ml), and cells were incubated for 1 h or 24 h. After the washing steps, the cells were fixed with a 1% paraformaldehyde and stained with Prussian blue staining kit (Ocean Nanotech, CA). Images were acquired by a Zeiss microscope.
2.5. Tracking of cellular uptake of PG-Gd-NIR813 by T1-weighted MRI
RAW264.7 murine macrophages and MDA-MB-435 human breast cancer cells (5×106 cells for each cell line) were separately seeded in cell culture dishes for 24 h. Then, PG-Gd-NIR813 was added (final concentration, 25 μM Gd) [31], and cells were incubated for 1 h or 24 h. After the washing steps, the cells were collected in 200 μl Eppendorf tubes and mixed with 1% Agarose (BD, Franklin lakes, NJ). After the agarose solidified, T1-weighted MRI was acquired on a 7.0-Tesla Bruker Biospec scanner (Fremont, CA).
2.6. Animal models
All animal experiments were performed in compliance with the guidelines for the care and use of research animals established by Stanford University's Animal Studies Committee and The University of Texas MD Anderson Cancer Center. Female athymic nude mice (nu/nu) were purchased from Harlan (Indianapolis, IN) at 6-8 weeks of age. MDA-MB-435 human breast cancer cells were harvested and washed with sterile phosphate-buffered saline (PBS), suspended and adjusted the cell concentration to 5×107 cells/ml in PBS. Viable cells (5×106) in PBS (100 μl) were inoculated into the mammary fat pad or subcutaneously into the shoulder. Tumor growth was monitored by digital caliper with measuring perpendicular diameters of the tumor. The tumor volume was estimated by the following formula: tumor volume = a × (b2)/2, where a and b are the tumor length and width, respectively, in millimeters.
2.7. Treatment of mice with MDA-MB-435 breast tumors
When mice bearing MDA-MB-435 tumors grew around 100–150 mm3, mice were randomly divided into two groups (n = 8 per group). Mice in group 1 were not treated and served as a control. Mice in group 2 received 5 daily intravenous injections of Abraxane at an equivalent paclitaxel dose of 30 mg/kg per injection. The mouse body weight and tumor volume were measured every 3 days before animals were euthanized.
In a separate study, MDA-MB-435 tumor-bearing mice were randomly divided into two groups (n = 5 per group). Mice in group 1 were not treated and served as a control. Mice in group 2 received a single intravenous injection of Abraxane at an equivalent paclitaxel dose of 30 mg/kg. The mouse body weight and tumor volume were measured every 3 days before animals were euthanized.
2.8. In vivo NIRF optical imaging with QD710-D
Mice bearing subcutaneous MDA-MB-435 tumors were used in the in vivo imaging studies with QD710-D. Mice were either not treated or treated with an intravenous injection of Abraxane at an equivalent paclitaxel dose of 30 mg/kg (n = 3/group). Three days later, mice in the untreated control and treatment groups were anesthetized with 1%–2% isoflurane in O2 and injected intravenously with QD710-D (200 pmol/mouse). Two-dimensional NIRF images were acquired at various time points after injection using a Xenogen IVIS-200 optical in vivo imaging system (Caliper Life Sciences, Hopkinton, MA).
To compare response to treatment between chemosensitive and chemoresistant tumor cells, each mouse was inoculated in the mammary fat pads with the parent MDA-MB-435 cells on one side and the resistant MDA-MB-435R cells on the contralateral side. When the tumor size reached 150 mm3, mice were either not treated or treated with an intravenously injection of Abraxane at an equivalent paclitaxel dose of 30 mg/kg. Three days later, QD710-D were injected intravenously and NIRF images acquired as described in the preceding paragraph.
2.9. In vivo T2-weighted MR imaging with Ferumoxytol
Mice bearing MDA-MB-435 tumors were either not treated or treated with Abraxane at a single equivalent paclitaxel dose of 30 mg/kg. Three days later, Ferumoxytol was injected through the tail vein (50 mg equivalent Fe/kg of mouse). T2-weighted fast spin-echo images were acquired on a 7.0-Tesla Bruker Biospec scanner before and at 1 h and 24 h after contrast agent injection, with the following parameters: TE, 38 ms; TR, 1800 ms; thickness, 0.75 mm; FOV, 4 × 3 cm; NEX, 4.0; Matrix, 256 × 192; and echo train length (RATE factor), 8.
2.10. In vivo T1-weighted MR/NIRF optical imaging with PG-Gd-NIR813
Three days after Abraxane treatment (single dose, 30 mg/kg equivalent paclitaxel), PG-Gd-NIR813 was injected through the tail vein (0.2 mmol Gd/kg, 14.4 μmol NIR813/kg of mouse). Mice in Abraxane-treated and untreated groups were imaged at multiple time points using the IVIS-200 system, and T1-weighted MR imaging. T1-weighted images were acquired on a 7.0-Tesla Bruker Biospec scanner (Fremont, CA) before and at 1 h, 24 h, and 48 h after contrast agent injection. The following acquisition parameters were used: TE (echo time), 9.5 ms; TR (repetition time), 900 ms; thickness, 0.75 mm; FOV (field of view), 4 × 3 cm; NEX (number of excitations), 2; and Matrix, 256 × 192.
2.11. TUNEL staining
Tissue slices (10 μm thick) were stained for TUNEL according to the manufacturer-provided protocol. After staining, slides were mounted with DAPI-containing medium, and fluorescent images were obtained under a fluorescence microscope. For the double staining of TUNEL and macrophage marker CD169 on tumor tissue, slides were blocked with 10% goat serum in PBS for 15 min at room temperature after TUNEL staining and then incubated with rat anti-mouse CD169 antibody (1:100) for 1 h at room temperature. After being washed three times for 5 min with PBS, slides were incubated with Alexa Fluor 594-conjugated goat anti-rat secondary antibody (1:2000) for 0.5 h at room temperature, and sections were washed with PBS three times for 5 min. After staining, slides were mounted with Vectashield DAPI-containing mounting medium and examined under a fluorescence microscope.
2.12. Flow cytometric analysis
To prepare single-cell suspensions for flow cytometry, harvested tumor tissues were cut into small fragments and digested with 0.25% collagenase type I (Life Technologies), 0.02% hyaluronidase (Sigma Roche) and 0.01% DNase I (Sigma Roche) in RPMI-1640 containing 10% FBS for 1 h at 37°C in water bath while shaking. Next, cell suspensions were passed through 100-micron cell strainers to remove aggregates, single-cell suspensions were counted, and viability was determined by trypan blue dye. Single-cell suspensions were incubated with FITC-labeled CD45, PE-labeled CD11b, eFluor660-labeled CD169, and fixable viability dye eFluor780 for 30 min on ice. Cells were washed twice with PBS, and then fixed with 1% paraformaldehyde. These tumor tissue single-cell samples were detected with LSR Fortessa X-20 Analyzer (BD Biosciences), and the data were analyzed with FlowJo software (Treestar).
2.13. Statistical analysis
Statistical significance was determined by one-way ANOVA. P values < 0.05 were considered statistically significant.
3. Results
3.1. Abraxane treatment delayed tumor growth and caused apoptotic cell death in MDA-MB-435 tumors
Five daily doses of Abraxane at an equivalent paclitaxel dose of 30 mg/kg per dose significantly delayed the growth of orthotopic MDA-MB-435 breast tumors compared with the growth of the untreated tumors at each time point from days 5 to 22 after initiation of treatment (P < 0.001 for each time point) (Fig. 2A). A single dose of Abraxane at an equivalent paclitaxel dose of 30 mg/kg also significantly delayed tumor growth compared with the growth of the untreated tumors (P < 0.05 for each time point from days 4 to 16 after injection) (Fig. 2B).
Figure 2.
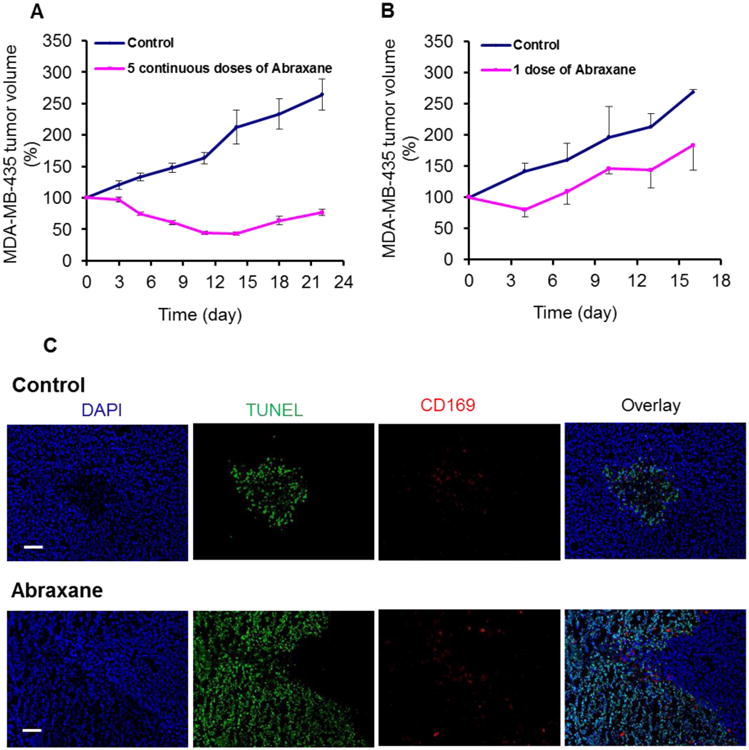
Effect of Abraxane on mice bearing MDA-MB-435 breast tumors. (A) Growth curves of MDA-MB-435 tumors treated with 5 daily doses of Abraxane or untreated control. Day 0 indicates the day that the first of the 5 daily doses was administered. (mean ± SE, n = 8/group). (B) Growth curves of MDA-MB-435 tumors treated with 1 dose of Abraxane or untreated control. Day 0 indicates the day that Abraxane was administered. (mean ± SE, n = 5/group). (C) Staining for the apoptosis marker TUNEL (green) and the activated macrophage marker CD169 (red) in untreated and Abraxane-treated MDA-MB-435 tumors (DAPI nuclear stain, blue) (original magnification ×100; scale bar, 100 μm).
TUNEL assay showed that a single dose of Abraxane at 30 mg/kg markedly increased the apoptotic response in MDA-MB-435 tumors at 5 days after treatment compared with the untreated control (Fig. 2C). The apoptotic cells showed nuclear shrinkage, nuclear fragmentation, and chromatin condensation (Supplementary Fig. S1). To evaluate whether Abraxane-induced apoptosis correlated with the number of infiltrating TAMs, MDA-MB-435 tumors from untreated and single-dose Abraxane-treated mice were double-stained for TUNEL (green) and activated macrophage marker CD169 (red). As shown in Figure 2C, most TAMs were distributed in the tumor region with patches of apoptotic cells. On the basis of these results, the single-dose treatment regimen was used in the subsequent imaging studies.
3.2. Macrophages but not tumor cells internalized nanoparticles in vitro
After incubation with QD710-D nanoparticles for 20 h, RAW264.7 mouse macrophages exhibited a strong fluorescent signal. In contrast, MDA-MB-435 breast cancer cells did not take up QD710-D under the same conditions (Fig. 3A). Similar data were observed after 1 h of incubation (data not shown). Moreover, RAW264.7 macrophages but not MDA-MB-435 cells showed uptake of Ferumoxytol at 1 h and 24 h after incubation (Fig. 3B). T1-weighted MRI showed a significant increase in the MR signal for RAW264.7 macrophages incubated with PG-Gd-NIR813 for 1 h and 24 h, whereas no change in MR signal was observed for MDA-MB-435 breast cancer cells incubated with PG-Gd-NIR813 for the same time periods (Fig. 3C, 3D).
Figure 3.
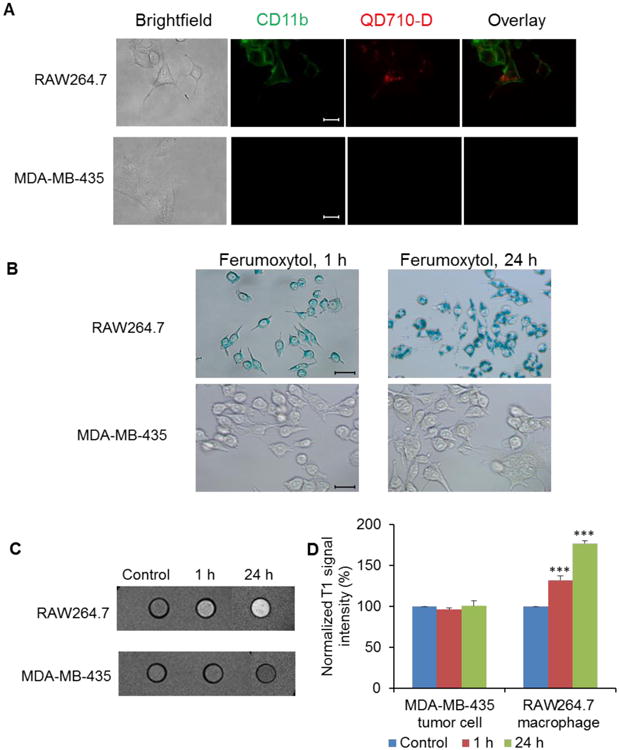
Macrophages, but not tumor cells, internalized nanoparticles in vitro. (A) QD710-D uptake after QD710-D incubation with RAW264.7 macrophages or MDA-MB-435 tumor cells for 20 h (original magnification ×600; scale bar, 20 μm). (B) Ferumoxytol uptake was evaluated by Prussian blue staining after Ferumoxytol incubation with RAW264.7 macrophages or MDA-MB-435 tumor cells for 1 h and 24 h (original magnification ×400; scale bar, 20 μm). (C) PG-Gd-NIR 813 uptake was assessed by T1-weighted MRI after PG-Gd-NIR 813 incubation with RAW264.7 macrophages or MDA-MB-435 tumor cells for 1 h and 24 h. (D) Quantitative analysis of T1-weighted MRI signals intensity of PG-Gd-NIR 813 uptakes by RAW264.7 macrophages or MDA-MB-435 tumor cells at 1 h and 24 h. (*** P < 0.001)
To address whether different macrophage phenotypes could take up nanoparticles, TAMs isolated from MDA-MB-435 tumor tissues [32] were exposed to interferon-γ / lipopolysaccharide to induce M1-polarized TAMs, and to interleukin-4 to induce M2-polarized TAMs. Both M1 and M2 TAMs effectively took up Ferumoxytol nanoparticles (Supplementary Fig. S2). Similarly, both M1- and M2-polarized bone marrow-derived macrophages (BMDMs) effectively phagocytosed Ferumoxytol nanoparticles (Supplementary Fig. S3). Taken together, macrophages could efficiently take up nanoparticles independent of their functional states.
3.3. Noninvasive optical imaging and MRI indicated increased uptake of nanoparticles in tumors of Abraxane-treated mice
Figure 4A shows representative NIRF optical images of mice at 4 h after intravenous injection of QD710-D. The uptake of QD710-D in MDA-MB-435 tumors was much higher in mice treated with Abraxane than in untreated mice. Quantitative analysis of fluorescence signal intensity confirmed that MDA-MB-435 tumors from Abraxane-treated mice had significantly higher QD710-D uptake than did MDA-MB-435 tumors from untreated mice at all-time points analyzed after QDs injection (P < 0.01) (Fig. 4B).
Figure 4.
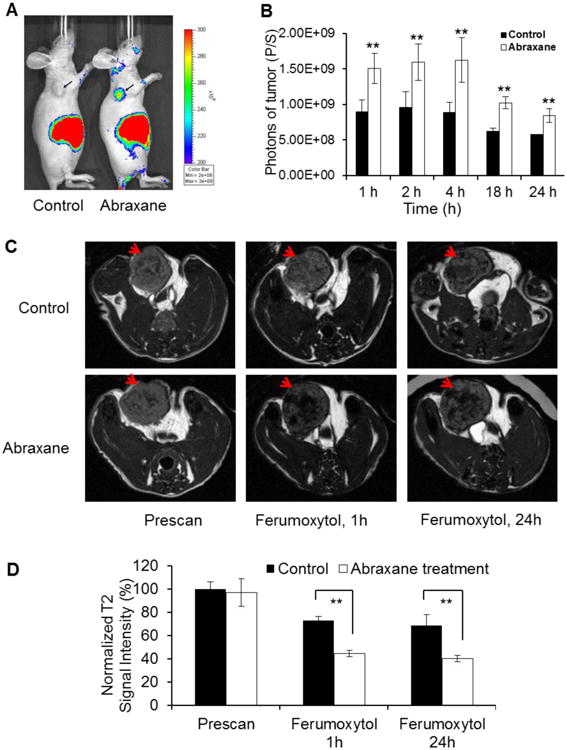
Administration of tumor-associated macrophages (TAMs) after Abraxane treatment could be monitored by nanoparticle-based imaging. (A) Representative NIRF images of Abraxane-treated and untreated MDA-MB-435 tumor–bearing mice 4 h after intravenous injection of QD710-D. (B) Quantitative analysis of QD710-D signal (photons/second) in untreated and Abraxane-treated tumors at 1, 2, 4, 18, and 24 h after administration of QD710-D (**P < 0.01). (C) T2-weighted MR imaging with Ferumoxytol for assessment of TAMs in MDA-MB-435 tumor after Abraxane treatment. T2-weighted MR images of MDA-MB-435 tumor before and 1 and 24 h after injection of Ferumoxytol. (D) Quantitative analysis of T2-weighted MRI signal intensity in tumors before and at 1 and 24 h after injection of Ferumoxytol. The signals were normalized to those of the prescan images acquired from untreated control mice (** P < 0.01). Arrows: tumor. (mean ± SD; n = 3/group).
Ex vivo NIRF optical imaging performed 4 h after injection showed similar uptake of QD710-D in untreated and treated mice for all major organs but significantly higher uptake of QD710-D in tumors from Abraxane-treated mice than in tumors from untreated mice (Supplementary Fig. S4). In both treated and untreated mice, QD710-D exhibited relatively high uptake in the liver, kidney, skin, and tumor. Interestingly, the uptake of QD710-D in the spleen was relatively low. A previous study showed that QD710-D could be cleared through the renal system, which helps explain the relatively high uptake of the nanoparticles observed in the kidney [18]. The optical signal from the skin could be attributed to nonspecific accumulation of QD710-D nanoparticles in the skin, similar to what was observed by Sykes et al. [33] with another quantum dot nanoparticle system.
We next investigated whether Ferumoxytol, a T2-MRI contrast agent, could also detect changes after Abraxane treatment. In Abraxane-treated mice, all MDA-MB-435 tumors showed significant enhancement on T2-weighted MR imaging at 1 h and 24 h after injection of Ferumoxytol compared with images acquired before the injection of the contrast agent. However, in untreated mice, MDA-MB-435 tumors did not show significant enhancement after Ferumoxytol injection compared with images acquired before Ferumoxytol (Fig. 4C). Significantly lower T2 signal intensity was observed in tumors of Abraxane-treated mice than in tumors from untreated control mice at either 1 h or 24 h after Ferumoxytol injection (Fig. 4D).
The dual-modality optical/MR imaging probe PG-Gd-NIR813 was used to evaluate the macrophage infiltration and response to treatment. NIRF optical imaging of mice with MDA-MB-435 tumors showed that PG-Gd-NIR813 had higher uptake in Abraxane-treated tumors than in untreated tumors at 48 h after contrast agent injection (Fig. 5A). Quantitative analysis of images showed a significantly higher tumor-to-muscle fluorescence intensity ratio in the Abraxane-treated mice (4.99 ± 0.66) than in the untreated mice (3.03 ± 0.06) at 48 h after contrast agent injection (P < 0.01) (Fig. 5B). These data were confirmed by ex vivo NIRF optical imaging of major organs and tumors taken from both untreated and Abraxane-treated mice at 48 h after intravenous injection of PG-Gd-NIR813 (Supplementary Fig. S5A). Quantitative analysis showed that there was significantly higher uptake of PG-Gd-NIR813 in Abraxane-treated tumors than in untreated tumors (Supplementary Fig. S5B). T1-weighted MR images with PG-Gd-NIR813 showed a distribution pattern similar to that observed with optical imaging (Fig. 5C). Before (prescan) and at 1 h after contrast agent injection, no difference was seen in tumor T1-weighted signal intensity between tumors from untreated and Abraxane-treated mice. However, at 24 h and 48 h after contrast agent injection, signal intensity was significantly higher in tumors from Abraxane-treated mice than in tumors from untreated mice (P < 0.05) (Fig. 5D).
Figure 5.
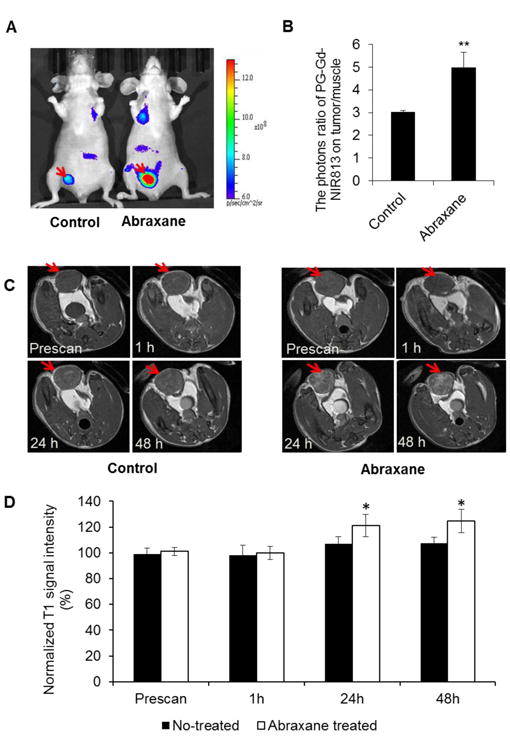
MR/NIRF optical imaging with PG-Gd-NIR813 for assessment of TAMs in MDA-MB-435 tumors after Abraxane treatment. (A) NIRF optical imaging of untreated and Abraxane-treated MDA-MB-435 tumors 48 h after injection of PG-Gd-NIR813. Arrows: tumor. (B) Quantitative analysis of photon flux ratio between tumor and muscle from NIR optical images acquired at 48 h after injection of PG-Gd-NIR813. (C) MR images of MDA-MB-435 tumors before and 1, 24, and 48 h after injection of PG-Gd-NIR813. (D) Quantitative analysis of T1-weighted MRI signals intensity in tumors before and at 1, 24, and 48 h after injection of PG-Gd-NIR813. * P < 0.05; ** P < 0.01. All data are expressed as mean ± SD (n = 3/group).
3.4. Abraxane treatment resulted in increased infiltration of macrophages in MDA-MB-435 tumors
Tumor tissues staining with CD11b antibody showed increased infiltration of leukocytes into tumors of Abraxane-treated mice (Fig. 6A). Moreover, some CD11b-positive cells co-localized with fluorescent signal from QD710-D in tumor tissue (Fig. 6B). Since macrophages possess the phagocytic function in CD11b+ cell population, these data suggest that QD710-D was taken up by TAMs.
Figure 6.
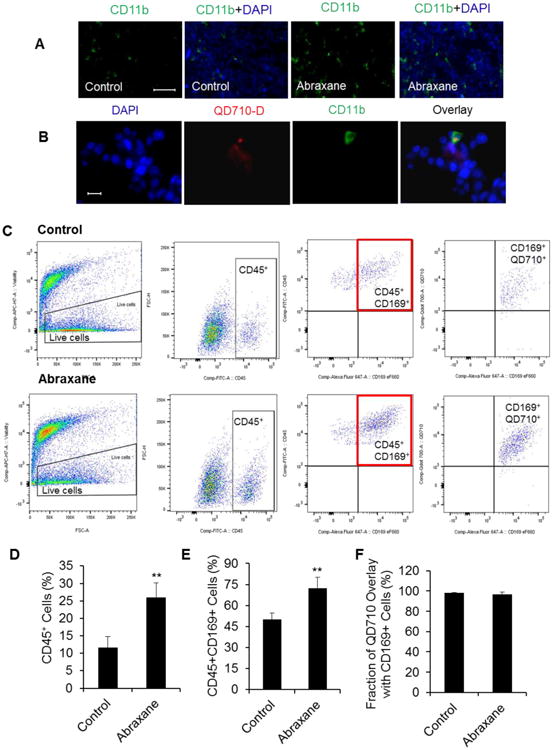
Tumor microenvironment cell population. (A) Tumor infiltrating leukocytes detected by marker CD11b (green, original magnification ×100; scale bar, 100 μm). (B) FITC-CD11b staining showing the co-localization of QD710-D in leukocytes of MDA-MB-435 tumor tissue (original magnification ×600; scale bar, 20 μm). (C) Flow cytometry of dissected single cells from MDA-MB-435 tumor tissue showed significantly higher CD45+ myeloid cells; CD45+CD169+ macrophages infiltrated into tumor tissue of the Abraxane-treated group compared with the untreated group. (D) Quantity analysis of infiltrating CD45+ myeloid cells in tumor tissue single cells (*** P < 0.001). (E) CD45+CD169+ macrophages were the main part of these infiltrated myeloid cells and were significantly higher in the Abraxane-treated group than in the untreated group (** P < 0.01). (F) QD710-D nanoparticles had good co-localization with CD169-positive macrophages (mean ± SD; n = 4/group).
To further confirm TAMs uptaking of nanoparticles, tumors from both Abraxane-treated and untreated mice that received intravenous injection of QD710-D were analyzed by flow cytometry. Results showed that there was significantly higher CD45+ myeloid cells infiltrated into tumor tissue of Abraxane-treated MDA-MB435 tumors than tumors from the untreated mice (Fig. 6C, 6D) (P < 0.001) and that there was significantly higher infiltration of CD45+CD169+ macrophages in Abraxane-treated tumors than in tumors from the untreated group (Fig. 6C, 6E) (P < 0.01). Moreover, QD710-D co-localized with CD169+ macrophages in the tumors from both treated and untreated groups: 96.5% ± 2.7% and 98.2% ± 0.5% of QDs were co-localized to CD169+ cells in the treated and untreated tumors, respectively (Fig. 6C, 6F), suggesting that QDs in the tumors were mostly taken up by TAMs.
3.5. Abraxane treatment resulted in more macrophage infiltration and higher nanoparticle accumulation in drug-sensitive MDA-MB-435 tumors than in drug-resistant MDA-MB-435R tumors
In vitro cell proliferation study showed that MDA-MB-435R cells were more resistant to paclitaxel treatment than were parental MDA-MB-435 cells after 72 h and 96 h of continuous incubation (Supplementary Fig. S6).
Flow cytometry analysis of parent MDA-MB-435 and resistant MDA-MB-435R tumors grown in female nude mice showed that there was significantly increased cell death in the parent MDA-MB-435 tumors treated with Abraxane than in untreated tumors (Fig. 7A, 7C) (P < 0.05). However, treatment with Abraxane at the same dose did not cause cell death in the resistant MDA-MB-435R tumors (Fig. 7B, 7C). In addition, for MDA-MB-435 tumors, significantly higher CD45+ myeloid cells, CD11b+ leukocytes, and F4/80+ macrophages infiltrated into the tumor tissue of Abraxane-treated mice than into the tumor tissue of untreated mice (Fig. 7A, 7D) (P < 0.001). In contrast, for resistant MDA-MB-435R tumors, there were significantly lower CD45+ myeloid cell and CD11b+ leukocyte subpopulations in tumors from Abraxane-treated mice than in tumors from untreated mice (Fig. 7B, 7D) (P < 0.05). There were no differences in population of F4/80+ macrophages between MDA-MB-435R tumors of Abraxane-treated mice and untreated mice (Fig. 7D). Moreover, there were significantly higher CD45+ myeloid cells, CD11b+ leukocytes, and F4/80+ macrophages in Abraxane- sensitive MDA-MB-435– tumors than in Abraxane-resistantMDA-MB-435R tumors (Fig. 7D) (P < 0.001). These results indicated that there were more macrophages infiltrating into chemotherapy-sensitive MDA-MB-435 tumors than into chemotherapy-resistant MDA-MB-435R tumors.
Figure 7.
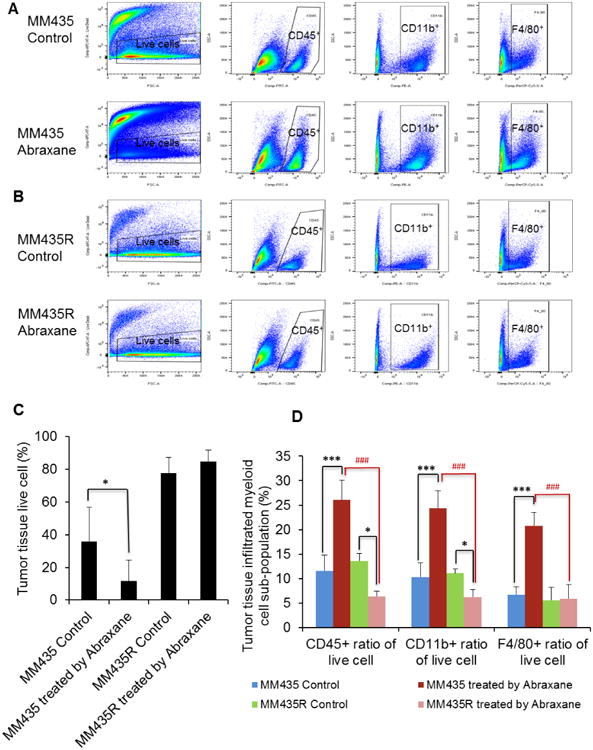
Flow cytometry analysis the tumor infiltrating immune cells on parent wild-type MDA-MB-435 and resistant MDA-MB-435R tumors. (A) Untreated or Abraxane-treated MDA-MB-435 wild-type tumor tissues were dissected into single-cell suspensions and assessed by flow cytometry with fixable viability dye eFluor780, FITC-labeled CD45, PE-labeled CD11b, and Percp.Cy5.5-labeled F4/80. (B) Untreated or Abraxane-treated resistant MDA-MB-435R tumor tissues were dissected into single-cell suspensions and assessed by flow cytometry with fixable viability dye eFluor780, FITC-labeled CD45, PE-labeled CD11b, and Percp.Cy5.5-labeled F4/80. (C) Quantity data of negative cell population with fixable viability dye eFluor780 channel showed that there was significantly increased cell death of wild-type MDA-MB-435 tumors treated with Abraxane compared with untreated tumors (* P < 0.05), but there were no differences in the cell death ratio in resistant MDA-MB-435R tumors between the Abraxane-treated and untreated groups. (D) Cell population quantity data of immunized cell infiltrated into tumor tissue showed that there were significantly higher numbers of CD45+ myeloid cells, CD11b+ leukocytes, and F4/80+ macrophages infiltrated into tumor tissue in the Abraxane-treated group than in the untreated group for wild-type MDA-MB-435 tumors (*** P < 0.001). However, there was a significantly lower populations of CD45+ myeloid cells and CD11b+ leukocytes in the Abraxane-treated group than in the untreated group for resistant MDA-MB-435R tumors (* P < 0.05), and there were no differences in F4/80+ macrophage of Abraxane-treated group compared with the untreated group for resistant MDA-MB-435R tumors. Moreover, there were significantly lower populations of CD45+ myeloid cells, CD11b+ leukocytes, and F4/80+ macrophages in Abraxane-treated resistant MDA-MB-435R tumors than in Abraxane-treated wild-type MDA-MB-435 tumors (### P < 0.001), (mean ± SD; n = 4/group).
In vivo optical images acquired after injection of QD710-D showed that after Abraxane treatment, MDA-MB-435 tumors but not resistant MDA-MB-435R tumors displayed a strong fluorescence signal (Fig. 8A). Quantitative analysis of signal intensity confirmed that in MDA-MB-435 tumors, the intensity of the fluorescence signal from QD710-D was significantly greater in tumors from Abraxane-treated mice than in tumors from untreated mice (P < 0.01). However, in resistant MDA-MB-435R tumors, there were no differences between Abraxane-treated and untreated tumors (Fig. 8B).
Figure 8.
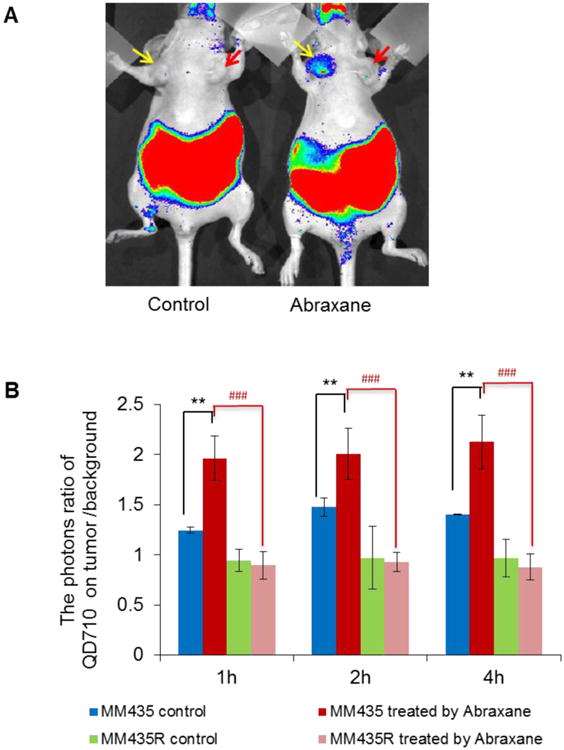
Accumulation of QD710-D in parental MDA-MB-435 and Abraxane-resistant MDA-MB-435R tumors with and without Abraxane treatment. (A) Representative optical images at 4 h after QD710-D injection in mice with MDA-MB-435 and MDA-MB-435R tumors untreated and treated with Abraxane. Yellow arrow, wild-type MDA-MB-435 tumor; red arrow, Abraxane-resistant MDA-MB-435R tumor. (B) Quantitative analysis of tumor-to-muscle intensity ratio of optical images (** P < 0.01, * P < 0.05); Abraxane-treated tumors compared with control tumors (### P < 0.001); Abraxane-treated resistant MDA-MB-435R tumors compared with Abraxane-treated wild-type MDA-MB-435 tumors (mean ± SD; n = 4/group).
MM435: MDA-MB-435; MM435R: MDA-MB-435R.
4. Discussion
In this study, we tested the hypothesis that tumor infiltration by macrophages in response to therapy-induced apoptosis can be monitored with noninvasive imaging of nanoparticles that are readily taken up by macrophages. Using noninvasive optical imaging and MRI, we found that three different types of nanoparticles with different chemical compositions showed significantly higher uptake in Abraxane-treated tumors than in untreated tumors (Fig. 4, 5). Several lines of evidence support the idea that these intensity enhancements can be attributed, at least in part, to increased infiltration of TAMs to tumors after therapy. First, in vitro studies showed that all three types of nanoparticles were taken up by macrophages but not by MDA-MB-435 cells (Fig. 3). Second, ex vivo studies of macrophage immunofluorescence staining of tumor tissues and flow cytometry of single-cell suspensions from dissected tumor tissues confirmed increased infiltration of CD11b+ and CD169+ macrophages in the tumors responding to Abraxane therapy, and QDs were almost completely co-localized with CD169+ macrophages in both treated and untreated tumors (Fig. 6). Third, treatment-induced apoptosis coincided with increased infiltration of macrophages, which were co-localized with apoptotic cells (Fig. 2). Finally, only the Abraxane-sensitive MDA-MB-435 tumors but not the Abraxane-resistant MDA-MB-435R tumors showed increased tumor uptake of QD710-D QDs after Abraxane treatment (Fig. 8). Flow cytometry analysis of single-cell suspensions from dissected tumors confirmed significantly increased population of F4/80+ macrophages in the MDA-MB-435 tumors but not in the MDA-MB-435R tumors between Abraxane-treated mice and untreated control (Fig. 7). Taken together, these data support the idea that chemotherapy-induced apoptosis resulted in increased tumor infiltration by macrophages, which are known to assist in the clearance of cellular debris. Change in the population of TAMs that had taken up nanoparticles was then visualized after therapy. These data indicate that it is feasible to monitor changes of tumor microenvironment by using imageable nanoparticles as a surrogate for the assessment of early apoptotic response.
Macrophages originate from blood monocytes, which differentiate into two distinct subtypes: classic M1 and alternative M2 macrophages. TAMs resemble M2-macrophages in that they mostly exhibit pro-tumoral functions [34-36]. Most of the studies to date have indicated a strong correlation between the presence of TAMs and poor outcome [4, 37-39]. Since both M1 and M2 macrophages possess a phagocytic function, imageable nanoparticles used in the current study could not distinguish M1 and M2 subtypes. However, since both M1- and M2-polarized TAMs and BMDMs effectively took up Ferumoxytol nanoparticles (Supplemental Fig. S2&S3), we conclude that the signal enhancement observed in our imaging studies was mainly a result of increased numbers of TAMs rather than increased uptake efficiency by each macrophage cell. Our findings indicate that a large influx of macrophages into the tumor could be seen early after initiation of treatment. However, the exact nature of these newly recruited macrophages remains to be defined. Also, the potential impact of these newly recruited TAMs on long-term tumor regression/progression after therapy is not known. If an increase in newly infiltrated TAMs correlates with poor outcome, it would be beneficial to test the combination of TAM-ablation therapy and apoptosis-inducing agents. In such a scenario, noninvasive imaging studies that reveal the timing and extent of tumor infiltration of macrophages would be valuable in guiding the planning of an optimized treatment schedule.
A limitation of this study is that we did not examine the possible role of changes in microvessel density (MVD) resulting from chemotherapy that may affect tumor uptake of nanoparticles. Previous studies have shown that tumors with high angiogenic activity (i.e. higher MVD) had higher intratumoral deposition of nanoparticles [40, 41]. Nevertheless, we note that in the MDA-MB-435 model, treatment with paclitaxel in fact decreased tumor MVD [42, 43]. These findings argue against the possibility that increased tumor uptake of nanoparticles was a result of increased angiogenic blood vessels, and further support the notion that increased number of TAMs infiltrating tumor bed was a main factor of increased tumor uptake of nanoparticles after paclitaxel chemotherapy. Clearly, further studies are needed to clarify the relative roles of apoptosis/TAMs and altered blood perfusion on increased imaging signals from nanoparticles after chemotherapy. Because most nanoparticles have high background signal in the liver, the method reported in this work might not be suitable for imaging response of liver cancer or liver metastasis to treatments. Another limitation of this study is that we did not examine uptake of nanoparticles in human tumor-associated macrophages from human tumor tissues. Availability of such data should further strengthen the translational potential of nanoparticle-based imaging agents.
To facilitate deeper understanding of the complex roles of macrophages in cancer progression and response to therapy, it is necessary to develop and validate clinically translatable noninvasive imaging techniques for tracking the migration, tumor homing capabilities, and fate of macrophages [44]. Several imaging probes have been evaluated for selective visualization of TAMs in cancer, including superparamagnetic iron oxide nanoparticles for T2-weighted MR imaging [25], gadolinium-based contrast agents for T1-weighted MR imaging [17], NIRF probes for optical imaging [15, 45, 46], and radiotracers for positron emission tomography imaging [47]. Appropriate physicochemical properties of nanoparticles promoted macrophage uptake and befitted as imageable nanoparticles [48-50]. The identification of a particular nanoparticle system for further development for clinical use is likely governed by a number of economic and regulatory issues and requires further improvements in sensitivity and selectivity [51].
Monitoring therapy response using imaging methods is an important component of personalized medicine. Positron emission tomography (PET) and single photon emission computed tomography (SPECT) with appropriate radiotracers can be used to assess changes associated with metabolic activity (e.g.,18F-FDG [52]), cell proliferation (e.g., 18F-FLT [53]), tumor hypoxia [54], and other functional measures [55]. Magnetic resonance imaging (MRI) that measures water relaxation, diffusion, and exchange can provide data related to the breakdown of macromolecules, loss of membrane integrity, change in cell size, and change in water content during cell death [56, 57]. Dynamic contrast-enhanced ultrasound (DCE-US) has been proposed for monitoring therapy-induced changes in blood perfusion [58]. Quantitative ultrasound (QUS) has been demonstrated to be capable of detecting cell death [59]. Imaging apoptotic cell death with visualizable nanoparticles on the basis of increased tumor infiltration of macrophages would be a useful addition to the toolbox of imaging techniques for early assessment of treatment response.
5. Conclusion
Tumor infiltrating macrophage changes due to chemotherapy-induced apoptosis can partly reflect the tumor early response to therapy, which can be monitored by non-invasive molecular imaging with suitable nanoparticles.
Supplementary Material
Supplementary Figure S1. Apoptotic response in MDA-MB-435 tumor cells. Top, No treatment control. Bottom, 5 days after a single dose of Abraxane at an equivalent paclitaxel dose of 30 mg/kg. Blue, DAPI nuclear staining; green, apoptotic cells. (Original magnification: ×200. scale bar 50 μm).
Supplemental Figure S2. Uptake of Ferumoxytol nanoparticles in TAMs isolated from MDA-MB-435 tumors. (A) Representative flow cytometry gating of TAMs. TAMs are defined as CD45+Ly6G-Ly6C-CD11b+F4/80+ live cells. (B) Representative microphotographs of TAMs, M1-polarized TAMs (stained with CD86, green), and M2-polarized TAMs (stained with CD206, red) (original magnification ×200, scale bar 50 μm). (C) Ferumoxytol uptakes in TAMs, M1-polarized TAMs and M2-polarized TAMs after 24 h of incubation. Nanoparticles were stained with Prussian blue (original magnification ×200; scale bar, 50 μm).
Supplemental Figure S3. Uptake of Ferumoxytol nanoparticles in bone marrow derived macrophages (BMDMs). (A) Flow cytometry characterization of M1-polarized BMDMs and M2-polarized BMDMs. M1 BMDMs express high level of CD86 biomarker, M2 BMDMs express high level of CD206 biomarker. (B)) Ferumoxytol uptakes in BMDMs, M1 BMDMs, and M2 BMDMs after 24 h of incubation. Nanoparticles were stained with Prussian blue (original magnification ×200; scale bar, 50 μm).
Supplementary Figure S4. (A) Ex vivo NIRF images of main organs removed from mice 4 h after intravenous injection of QD710-D. (B) Quantitative analysis of QD710-D signal (photons/second) in main organs removed from mice 4 h after QD710-D injection.
Supplementary Figure S5. (A) Ex vivo NIRF images of main organs removed from mice 48 h after intravenous injection of PG-Gd-NIR813. (B) Quantitative analysis of PG-Gd-NIR813 (photons/second) in main organs removed from mice 48 h after PG-Gd-NIR813 injection. *P < 0.05.
Supplementary Figure S6. Cell proliferation assay of wild-type MDA-MB-435 cells and paclitaxel-resistant MDA-MB-435R cells. (A) Results of a cell proliferation assay 72 h after Abraxane treatment. ** P < 0.01. (B) Results of a cell proliferation assay 96 h after Abraxane treatment. ** P < 0.01.
Acknowledgments
We thank Stephanie Deming and Tamara Locke for editing the article. This work was supported in part by the John S. Dunn Foundation (to CL), U.S. Department of Energy (DE-SC0008397) (to ZC), and the Cancer Center Support Grant (P30CA016672) for support of the animal studies. We thank the support from Department of Veterinary Medicine and Surgery, Small Animal Imaging Facility, and Flow Cytometry & Cellular Imaging Core Facility at UT MD Anderson Cancer Center.
Financial support: This work was supported in part by the John S. Dunn Foundation, the Office of Science (BER), U.S. Department of Energy (DE-SC0008397), and the Cancer Center Support Grant (P30CA016672) used for the Small Animal Imaging Facility and the Research Animal Support Facility.
Footnotes
Competing Interests: The authors have declared that no competing interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 2.Talmadge JE, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007;26(3-4):373–400. doi: 10.1007/s10555-007-9072-0. [DOI] [PubMed] [Google Scholar]
- 3.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. Clin Oncol. 2005;23(5):953–64. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 4.Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. J Dev Biol. 2011;55(7-9):861–7. doi: 10.1387/ijdb.113371dl. [DOI] [PubMed] [Google Scholar]
- 5.Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17(3):445–51. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 6.Makin G, Dive C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001;11(11):S22–6. doi: 10.1016/s0962-8924(01)02124-9. [DOI] [PubMed] [Google Scholar]
- 7.Pervaiz S. Anti-cancer drugs of today and tomorrow: are we close to making the turn from treating to curing cancer? Pharm Des. 2002;8(19):1723–34. doi: 10.2174/1381612023394025. [DOI] [PubMed] [Google Scholar]
- 8.Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98(9):2603–14. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 9.Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell. 2001;12(4):919–930. doi: 10.1091/mbc.12.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadeel B. Programmed cell clearance. Cell Mol Life Sci. 2003;60(12):2575–85. doi: 10.1007/s00018-003-3145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-Mc Guinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–79. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR, van der Burg SH. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73(8):2480–92. doi: 10.1158/0008-5472.CAN-12-3542. [DOI] [PubMed] [Google Scholar]
- 13.Shiao SL, Coussens LM. The tumor-immune microenvironment and response to radiation therapy. J Mammary Gland Biol Neoplasia. 2010;15(4):411–21. doi: 10.1007/s10911-010-9194-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol. 2013;4:157. doi: 10.3389/fphys.2013.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson H, Muhammad O, Daneshvar H, Nelms J, Popescu A, Vogelbaum MA, Bruchez M, Toms SA. Quantum dots are phagocytized by macrophages and colocalize with experimental gliomas. Neurosurgery. 2007;60(3):524–91. doi: 10.1227/01.NEU.0000255334.95532.DD. discussion 529-30. [DOI] [PubMed] [Google Scholar]
- 16.Thorek DL, Ulmert D, Diop NF, Lupu ME, Doran MG, Huang R, Abou DS, Larson SM, Grimm J. Non-invasive mapping of deep-tissue lymph nodes in live animals using a multimodal PET/MRI nanoparticle. Nat Commun. 2014;5:3097. doi: 10.1038/ncomms4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melancon MP, Lu W, Huang Q, Thapa P, Zhou D, Ng C, Li C. Targeted imaging of tumor-associated M2 macrophages using a macromolecular contrast agent PG-Gd-NIR813. Biomaterials. 2010;31(25):6567–73. doi: 10.1016/j.biomaterials.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Chen K, Luong R, Bouley DM, Mao H, Qiao T, Gambhir SS, Cheng Z. A novel clinically translatable fluorescent nanoparticle for targeted molecular imaging of tumors in living subjects. Nano Lett. 2012;12(1):281–6. doi: 10.1021/nl203526f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landry R, Jacobs PM, Davis R, Shenouda M, Bolton WK. Pharmacokinetic study of ferumoxytol: a new iron replacement therapy in normal subjects and hemodialysis patients. Am J Nephrol. 2005;25(4):400–10. doi: 10.1159/000087212. [DOI] [PubMed] [Google Scholar]
- 20.Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85(5):315–9. doi: 10.1002/ajh.21656. [DOI] [PubMed] [Google Scholar]
- 21.Weinstein JS, Varallyay CG, Dosa E, Gahramanov S, Hamilton B, Rooney WD, Muldoon LL, Neuwelt EA. Superparamagnetic iron oxide nanoparticles: diagnostic magnetic resonance imaging and potential therapeutic applications in neurooncology and central nervous system inflammatory pathologies, a review. J Cereb Blood Flow Metab. 2010;30(1):15–35. doi: 10.1038/jcbfm.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bullivant JP, Zhao S, Willenberg BJ, Kozissnik B, Batich CD, Dobson J. Materials characterization of Feraheme/ferumoxytol and preliminary evaluation of its potential for magnetic fluid hyperthermia. Int J Mol Sci. 2013;14(9):17501–10. doi: 10.3390/ijms140917501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balakrishnan VS, Rao M, Kausz AT, Brenner L, Pereira BJ, Frigo TB, Lewis JM. Physicochemical properties of ferumoxytol, a new intravenous iron preparation. J Clin Invest. 2009;39(6):489–96. doi: 10.1111/j.1365-2362.2009.02130.x. [DOI] [PubMed] [Google Scholar]
- 24.Gossuin Y, Gillis P, Hocq A, Vuong QL, Roch A. Magnetic resonance relaxation properties of superparamagnetic particles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(3):299–310. doi: 10.1002/wnan.36. [DOI] [PubMed] [Google Scholar]
- 25.Daldrup-Link HE, Golovko D, Ruffell B, Denardo DG, Castaneda R, Ansari C, Rao J, Tikhomirov GA, Wendland MF, Corot C, Coussens LM. MRI of tumor-associated macrophages with clinically applicable iron oxide nanoparticles. Clin Cancer Res. 2011;17(17):5695–704. doi: 10.1158/1078-0432.CCR-10-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Salanitri J, Tutton S, Dunkle EE, Schneider JR, Caprini JA, Pierchala LN, Jacobs PM, Edelman RR. Lower extremity deep venous thrombosis: evaluation with ferumoxytol-enhanced MR imaging and dual-contrast mechanism-- preliminary experience. Radiology. 2007;242(3):873–81. doi: 10.1148/radiol.2423052101. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Tutton S, Vu AT, Pierchala L, Li BS, Lewis JM, Prasad PV, Edelman RR. First-pass contrast-enhanced magnetic resonance angiography in humans using ferumoxytol, a novel ultrasmall superparamagnetic iron oxide (USPIO)-based blood pool agent. J Magn Reson Imaging. 2005;21(1):46–52. doi: 10.1002/jmri.20235. [DOI] [PubMed] [Google Scholar]
- 28.Neuwelt EA, Varallyay CG, Manninger S, Solymosi D, Haluska M, Hunt MA, Nesbit G, Stevens A, Jerosch-Herold M, Jacobs PM, Hoffman JM. The potential of ferumoxytol nanoparticle magnetic resonance imaging perfusion, and angiography in central nervous system malignancy: a pilot study. Neurosurgery. 2007;60(4):601–11. doi: 10.1227/01.NEU.0000255350.71700.37. discussion 611-2. [DOI] [PubMed] [Google Scholar]
- 29.Tian M, Wen X, Jackson EF, Ng C, Uthamanthil R, Liang D, Gelovani JG, Li C. Pharmacokinetics and magnetic resonance imaging of biodegradable macromolecular blood-pool contrast agent PG-Gd in non-human primates: a pilot study. Contrast Media Mol Imaging. 2011;6(4):289–97. doi: 10.1002/cmmi.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson EF, Esparza-Coss E, Wen X, Ng CS, Daniel SL, Price RE, Rivera B, Charnsangavej C, Gelovani JG, Li C. Magnetic resonance imaging of therapy-induced necrosis using gadolinium-chelated polyglutamic acids. Int J Radiat Oncol Biol Phys. 2007;68(3):830–8. doi: 10.1016/j.ijrobp.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang AM, Ananta JS, Zhao H, Cisneros BT, Lam EY, Wong ST, Wilson LJ, Wong KK. Cellular uptake and imaging studies of gadolinium-loaded single-walled carbon nanotubes as MRI contrast agents. Contrast Media Mol Imaging. 2011;6(2):93–9. doi: 10.1002/cmmi.410. [DOI] [PubMed] [Google Scholar]
- 32.Cassetta L, Noy R, Swierczak A, Sugano G, Smith H, Wiechmann L, Pollard JW. Isolation of Mouse and Human Tumor-Associated Macrophages. Tumor Microenvironment: Study Protocols. 2016;899:211–229. doi: 10.1007/978-3-319-26666-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sykes EA, Dai Q, Tsoi KM, Hwang DM, Chan WCW. Nanoparticle exposure in animals can be visualized in the skin and analysed via skin biopsy. Nature Communications. 2014;5 doi: 10.1038/ncomms4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 35.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 36.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 37.Clear AJ, Lee AM, Calaminici M, Ramsay AG, Morris KJ, Hallam S, Kelly G, Macdougall F, Lister TA, Gribben JG. Increased angiogenic sprouting in poor prognosis FL is associated with elevated numbers of CD163+ macrophages within the immediate sprouting microenvironment. Blood. 2010;115(24):5053–6. doi: 10.1182/blood-2009-11-253260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukocyte Biol. 2009;86(5):1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 39.Mukhtar RA, Nseyo O, Campbell MJ, Esserman LJ. Tumor-associated macrophages in breast cancer as potential biomarkers for new treatments and diagnostics. Expert Rev Mol Diagn. 2011;11(1):91–100. doi: 10.1586/erm.10.97. [DOI] [PubMed] [Google Scholar]
- 40.Perry JL, Reuter KG, Luft JC, Pecot CV, Zamboni W, DeSimone JM. Mediating Passive Tumor Accumulation through Particle Size, Tumor Type, and Location. Nano Letters. 2017;17(5):2879–2886. doi: 10.1021/acs.nanolett.7b00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mei KC, Bai J, Lorrio S, Wang JTW, Al-Jamal KT. Investigating the effect of tumor vascularization on magnetic targeting in vivo using retrospective design of experiment. Biomaterials. 2016;106:276–285. doi: 10.1016/j.biomaterials.2016.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao Q, Li ZB, Chen K, Wu Z, He L, Neamati N, Chen X. Evaluation of biodistribution and anti-tumor effect of a dimeric RGD peptide-paclitaxel conjugate in mice with breast cancer. Eur J Nucl Med Mol Imaging. 2008;35(8):1489–98. doi: 10.1007/s00259-008-0744-y. [DOI] [PubMed] [Google Scholar]
- 43.Klos KS, Zhou X, Lee S, Zhang L, Yang W, Nagata Y, Yu D. Combined trastuzumab and paclitaxel treatment better inhibits ErbB-2-mediated angiogenesis in breast carcinoma through a more effective inhibition of Akt than either treatment alone. Cancer. 2003;98(7):1377–85. doi: 10.1002/cncr.11656. [DOI] [PubMed] [Google Scholar]
- 44.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13(2):125–38. doi: 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 45.Higuchi Y, Oka M, Kawakami S, Hashida M. Mannosylated semiconductor quantum dots for the labeling of macrophages. J Control Release. 2008;125(2):131–6. doi: 10.1016/j.jconrel.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K, Bachmann MH, Ellman JA, Bogyo M. A nonpeptidic cathepsin S activity-based probe for noninvasive optical imaging of tumor-associated macrophages. Chem Biol. 2012;19(5):619–28. doi: 10.1016/j.chembiol.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nahrendorf M, Keliher E, Marinelli B, Waterman P, Feruglio PF, Fexon L, Pivovarov M, Swirski FK, Pittet MJ, Vinegoni C, Weissleder R. Hybrid PET-optical imaging using targeted probes. Natl Acad Sci U S A. 2010;107(17):7910–5. doi: 10.1073/pnas.0915163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu SS, Lau CM, Thomas SN, Jerome WG, Maron DJ, Dickerson JH, Hubbell JA, Giorgio TD. Size- and charge-dependent non-specific uptake of PEGylated nanoparticles by macrophages. Int J Nanomedicine. 2012;7:799–813. doi: 10.2147/IJN.S28531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moore A, Weissleder R, Bogdanov A., Jr Uptake of dextran-coated monocrystalline iron oxides in tumor cells and macrophages. J Magn Reson Imaging. 1997;7(6):1140–5. doi: 10.1002/jmri.1880070629. [DOI] [PubMed] [Google Scholar]
- 50.Wilhelm C, Billotey C, Roger J, Pons JN, Bacri JC, Gazeau F. Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials. 2003;24(6):1001–11. doi: 10.1016/s0142-9612(02)00440-4. [DOI] [PubMed] [Google Scholar]
- 51.Li C. A targeted approach to cancer imaging and therapy. Nat Mater. 2014;13(2):110–115. doi: 10.1038/nmat3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spaepen K, Stroobants S, Dupont P, Van Steenweghen S, Thomas J, Vandenberghe P, Vanuytsel L, Bormans G, Balzarini J, De Wolf-Peeters C, Mortelmans L, Verhoef G. Prognostic value of positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose ([18F]FDG) after first-line chemotherapy in non-Hodgkin's lymphoma: is 18F]FDG-PET a valid alternative to conventional diagnostic methods? J Clin Oncol. 2001;19(2):414–9. doi: 10.1200/JCO.2001.19.2.414. [DOI] [PubMed] [Google Scholar]
- 53.Oborski MJ, Demirci E, Laymon CM, Lieberman FS, Mountz JM. Assessment of early therapy response with 18F-FLT PET in glioblastoma multiforme. Clin Nucl Med. 2014;39(10):e431–2. doi: 10.1097/RLU.0000000000000321. [DOI] [PubMed] [Google Scholar]
- 54.Eschmann SM, Paulsen F, Reimold M, Dittmann H, Welz S, Reischl G, Machulla HJ, Bares R. Prognostic impact of hypoxia imaging with F-18-misonidazole PET in non-small cell lung cancer and head and neck cancer before radiotherapy. J Nucl Med. 2005;46(2):253–260. [PubMed] [Google Scholar]
- 55.Brindle K. New approaches for imaging tumour responses to treatment. Nat Rev Cancer. 2008;8(2):94–107. doi: 10.1038/nrc2289. [DOI] [PubMed] [Google Scholar]
- 56.Chenevert TL, McKeever PE, Ross BD. Monitoring early response of experimental brain tumors to therapy using diffusion magnetic resonance imaging. Clin Cancer Res. 1997;3(9):1457–1466. [PubMed] [Google Scholar]
- 57.Colvin DC, Loveless ME, Does MD, Yue Z, Yankeelov TE, Gore JC. Earlier detection of tumor treatment response using magnetic resonance diffusion imaging with oscillating gradients. Magn Reson Imaging. 2011;29(3):315–323. doi: 10.1016/j.mri.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamuraglia M, Bridal SL, Santin M, Izzi G, Rixe O, Paradiso A, Lucidarme O. Clinical relevance of contrast-enhanced ultrasound in monitoring anti-angiogenic therapy of cancer: current status and perspectives. Crit Rev Oncol Hematol. 2010;73(3):202–12. doi: 10.1016/j.critrevonc.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Kolios MC, Czarnota GJ. Potential use of ultrasound for the detection of cell changes in cancer treatment. Future Oncology. 2009;5(10):1527–1532. doi: 10.2217/fon.09.157. [DOI] [PubMed] [Google Scholar]
- 60.Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008;(2008):pdb prot5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Apoptotic response in MDA-MB-435 tumor cells. Top, No treatment control. Bottom, 5 days after a single dose of Abraxane at an equivalent paclitaxel dose of 30 mg/kg. Blue, DAPI nuclear staining; green, apoptotic cells. (Original magnification: ×200. scale bar 50 μm).
Supplemental Figure S2. Uptake of Ferumoxytol nanoparticles in TAMs isolated from MDA-MB-435 tumors. (A) Representative flow cytometry gating of TAMs. TAMs are defined as CD45+Ly6G-Ly6C-CD11b+F4/80+ live cells. (B) Representative microphotographs of TAMs, M1-polarized TAMs (stained with CD86, green), and M2-polarized TAMs (stained with CD206, red) (original magnification ×200, scale bar 50 μm). (C) Ferumoxytol uptakes in TAMs, M1-polarized TAMs and M2-polarized TAMs after 24 h of incubation. Nanoparticles were stained with Prussian blue (original magnification ×200; scale bar, 50 μm).
Supplemental Figure S3. Uptake of Ferumoxytol nanoparticles in bone marrow derived macrophages (BMDMs). (A) Flow cytometry characterization of M1-polarized BMDMs and M2-polarized BMDMs. M1 BMDMs express high level of CD86 biomarker, M2 BMDMs express high level of CD206 biomarker. (B)) Ferumoxytol uptakes in BMDMs, M1 BMDMs, and M2 BMDMs after 24 h of incubation. Nanoparticles were stained with Prussian blue (original magnification ×200; scale bar, 50 μm).
Supplementary Figure S4. (A) Ex vivo NIRF images of main organs removed from mice 4 h after intravenous injection of QD710-D. (B) Quantitative analysis of QD710-D signal (photons/second) in main organs removed from mice 4 h after QD710-D injection.
Supplementary Figure S5. (A) Ex vivo NIRF images of main organs removed from mice 48 h after intravenous injection of PG-Gd-NIR813. (B) Quantitative analysis of PG-Gd-NIR813 (photons/second) in main organs removed from mice 48 h after PG-Gd-NIR813 injection. *P < 0.05.
Supplementary Figure S6. Cell proliferation assay of wild-type MDA-MB-435 cells and paclitaxel-resistant MDA-MB-435R cells. (A) Results of a cell proliferation assay 72 h after Abraxane treatment. ** P < 0.01. (B) Results of a cell proliferation assay 96 h after Abraxane treatment. ** P < 0.01.