

Summary
Type I interferon restrains interleukin-1β (IL-1β)-driven inflammation in macrophages by upregulating cholesterol-25-hydroxylase (Ch25h) and repressing SREBP transcription factors. However, the molecular links between lipid metabolism and IL-1β production remain obscure. Here we demonstrate that production of 25-hydroxycholesterol (25-HC) by macrophages is required to prevent inflammasome activation by the DNA sensor protein absent in melanoma 2 (AIM2). We find that in response to bacterial infection or lipopolysaccharide (LPS) stimulation, macrophages upregulate Ch25h to maintain repression of SREBP2 activation and cholesterol synthesis. Increasing macrophage cholesterol content is sufficient to trigger IL-1β release in a crystal-independent but AIM2-dependent manner. Ch25h-deficiency results in cholesterol-dependent reduced mitochondrial respiratory capacity and release of mitochondrial DNA into the cytosol. Ch25h−/−Aim2−/− rescues the increased inflammasome activity observed in Ch25h−/−. Therefore, activated macrophages utilize 25-HC in an anti-inflammatory circuit that maintains mitochondrial integrity and prevents spurious AIM2 inflammasome activation.
Keywords: Oxysterol, cholesterol, inflammasome, mitochondria, macrophage, IL-1β, Ch25h, SREBP
eTOC
Cholesterol overload directly triggers mitochondrial DNA release and activation of the AIM2 inflammasome in metabolic diseases
Introduction
Cholesterol is a crucial structural component of mammalian cell membranes. However, unlike fatty acids, cholesterol cannot be used as an energy source, necessitating feedback mechanisms to control cellular cholesterol content. The molecular basis for cholesterol feedback inhibition involves the Sterol Regulatory Element Binding Protein 2 (SREBP2) pathway, whereby increased endoplasmic reticulum (ER) cholesterol content causes the chaperone protein SCAP to bind more tightly to INSIG, which results in SREBP2 sequestration in the ER (Goldstein et al., 2006). In states of low ER cholesterol, SCAP dissociates from INSIG, allowing for translocation of the SCAP-SREBP2 complex to the Golgi apparatus where SREBP2 is cleaved (Goldstein et al., 2006). This releases the transcription factor domain of SREBP2, which translocates to the nucleus and activates the transcription of genes involved in cholesterol biosynthesis and uptake.
While cholesterol can inhibit sterol biosynthesis, the oxysterol 25-hydroxycholesterol (25-HC) is even more potent than cholesterol in its capacity to inhibit SREBP2 in biochemical studies (Goldstein et al., 2006). Whereas cholesterol is sensed by SCAP, 25-HC binds directly to INSIG causing increased association with SCAP-SREBP2. However, it has been unclear whether endogenously produced 25-HC contributes to cholesterol regulation in mammalian cells. Mice deficient in the enzyme cholesterol 25-hydroxylase (Ch25h), which produces 25-HC from cholesterol, do not show defects in baseline cholesterol homeostasis, raising the question of whether this metabolite has a physiologic function (McDonald and Russell, 2010).
It has recently been appreciated that Ch25h is an interferon (IFN)-inducible gene in macrophages. Stimulation of mouse bone marrow-derived macrophages (BMDMs) with LPS or polyI:C results in IFNAR1-dependent upregulation of Ch25h and 25-HC synthesis (Cyster et al., 2014). Consistent with the key role of type I interferon (IFN) in responses to viruses, 25-HC has antiviral activity (Blanc et al., 2013; Cyster et al., 2014; Liu et al., 2013). Type I IFN additionally acts to inhibit IL-1β-driven inflammation (Cyster et al., 2014). This occurs by repression of Il1b transcription and inflammasome-dependent IL-1β processing (Guarda et al., 2011). Recent work suggests that the inhibitory action of type I IFN on IL-1β requires induction of Ch25h/25-HC (Reboldi et al., 2014).
Inflammasomes are multi-protein complexes that form within the cytosol following exposure to a wide variety of pathogen-derived stimuli (Rathinam and Fitzgerald, 2016). Inflammasome activation requires recognition of ligand by a sensor protein followed by recruitment of the adaptor protein ASC (Rathinam and Fitzgerald, 2016). This results in ASC oligomerization and the recruitment and activation of caspase-1, a protease that processes pro-IL-1β into mature IL-1β. How 25-HC represses inflammasome activation has been unclear. The effects of Ch25h-deficiency on pro-IL-1β processing could be rescued by overexpression of INSIG1 or deletion of SCAP, suggesting a contribution by the SREBP pathway, but the nature of this contribution has not been defined (Reboldi et al., 2014).
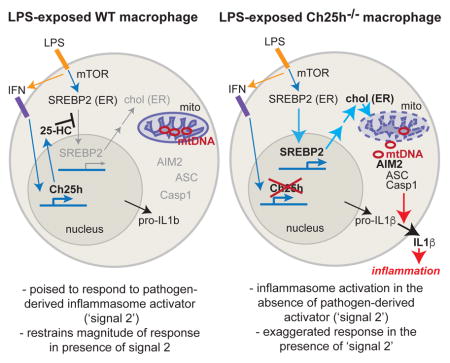
Here, we describe a metabolic circuit that links regulation of cellular cholesterol content to the prevention of inflammasome activation. We find LPS-activated macrophages shut down cholesterol biosynthesis and decrease their cholesterol content in a Ch25h-dependent manner. Enforcement of increased macrophage cholesterol content after LPS stimulation is sufficient to drive inflammasome-dependent IL-1β production. Cholesterol-dependent inflammasome activation unexpectedly requires the cytosolic DNA sensor AIM2. Metabolic studies reveal that Ch25h−/− and cholesterol-loaded macrophages have impaired mitochondrial respiration and membrane polarization. This results in increased cytosolic mitochondrial DNA (mtDNA) content and depletion of mtDNA reduces cholesterol-dependent IL-1β release. Finally, we find that Ch25h/Aim2 double-deficiency reverses inflammasome activation caused by Ch25h-deficiency. These findings suggest that macrophage upregulation of 25-HC after pathogen recognition prevents cholesterol-dependent mitochondrial dysfunction and spurious crosstalk between mtDNA and the inflammasome.
Results
Ch25h controls IL-1β release and intracellular bacterial growth through ASC-dependent inflammasomes
Ch25h−/− BMDMs overproduce IL-1β in response to Listeria monocytogenes infection and show increased control of intracellular bacterial growth (Reboldi et al., 2014). To determine whether these responses were due to increased inflammasome activity, we generated Ch25h−/−Asc−/− mice. BMDMs from these mice and controls were treated with carrier (ethanol) or 25-HC (1uM), and infected with L. monocytogenes for 24 hr. Ch25h−/−Asc+/+ BMDMs had increased production of IL-1β, which could be inhibited by addition of 25-HC as expected (Fig. 1A) (Reboldi et al., 2014). However, in the setting of ASC-deficiency, Ch25h−/− BMDMs showed no elevation in extracellular IL-1β and 25-HC addition to these cultures did not reduce IL-1β levels (Fig. 1B). We also observed decreased L. monocytogenes colony forming units (CFUs) in Ch25h−/−Asc+/+ BMDMs, which could be rescued by the addition of 25-HC (Fig. 1C). Asc−/− BMDMs showed an overall increase in L. monocytogenes CFUs and the protective effect of Ch25h-deficiency was lost (Fig. 1D).
Figure 1. Increased IL-1β production and control of L. monocytogenes intracellular growth in Ch25h−/− macrophages involves ASC-dependent inflammasome activation.

IL-1β enzyme-linked immunosorbent assay (ELISA) from supernatants (A, B) and bacterial colony forming units (CFU) from cell lysates (C, D) of control (EtOH) or 25-HC (1uM) treated Ch25h+/−Asc+/+ and Ch25h−/−Asc+/+ (A, C) or Ch25h+/−Asc−/− and Ch25h−/−Asc−/− (B, D) BMDMs after 24 hr of L. monocytogenes infection (M.O.I. 10:1). (E) IL-1β ELISA from supernatants of LPS and LPS+ATP stimulated BMDMs of the indicated genotypes. (n=3 per genotype, means+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test). See also Figure S1.
In previous work we demonstrated that Ch25h−/− BMDMs overproduced IL-1β following exposure to LPS and the NLRP3 inflammasome activator ATP (Reboldi et al., 2014). Consistent with these observations, LPS+ATP treated Ch25h−/− cells produced increased IL-1β compared to controls (Fig. 1E). However, in the setting of Asc−/−, neither Ch25h genotype produced detectable IL-1β, ruling out the possibility that the increased cytokine production by Ch25h−/− cells was due to passive loss of membrane integrity (Fig. 1E). These data support a model in which IL-1β overproduction and decreased bacterial replication in Ch25h−/− BMDMs are due to increased activity of ASC-dependent inflammasomes. These observations led us to more deeply explore how Ch25h acts to restrain inflammasome activity.
Ch25h and 25-HC enforce repression of the SREBP2 pathway and cholesterol synthesis in LPS-activated macrophages
Type I IFN has been described to repress the SREBP2 pathway in macrophages (Reboldi et al., 2014; York et al., 2015), but it is not clear to what extent this process depends on 25-HC. We therefore performed time-course analyses of cholesterol biosynthetic enzyme mRNA expression in control and Ch25h−/− BMDMs after stimulation with LPS. As expected, Ch25h transcript levels peaked ~4 hr post-LPS stimulation and subsequently declined (Fig. 2A) (Cyster et al., 2014). Similar Ch25h induction kinetics were observed following L. monocytogenes infection (Fig. S1A). The sterol biosynthetic enzymes Hmgcs1, Lss, Dhcr24 and Sqle were initially repressed 2 hr post-LPS, regardless of the genetic status of Ch25h (Fig. 2A). However, by 8 hr of LPS stimulation, Ch25h−/− BMDMs showed increased expression of sterol biosynthetic enzyme mRNAs (Fig. 2A). Overexpression of these mRNAs was also observed after 8 hr of L. monocytogenes infection (Fig. S1B). These data suggest that Ch25h is induced in macrophages by bacterial sensing, and acts to repress sterol biosynthetic enzyme expression. However, Ch25h is not required for initial shutdown of the cholesterol biosynthesis pathway, but instead enforces sustained repression.
Figure 2. Ch25h induction prevents cholesterol buildup in macrophages by enforcing repression of mTORC1-dependent SREBP activity.

(A) RT-qPCR analysis of Ch25h, Hmgcs1, Lss, Dhcr24, and Sqle in Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS. Data are standardized by comparison to Hprt, and A.U. indicates arbitrary unit (means+/−SD from 4 independent experiments). (B) LC-MS quantification of 25-HC, desmosterol, lanosterol, and 7-dehydrocholestrol in Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS for 8 hr. Y-axis indicates ng/2×106 cells. Data from two independent experiments (mean+/−SD). (C) Quantification of cholesterol content by Amplex Red fluorescence in Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS. Data from 4 independent experiments (mean+/−SD). (D) FACS-based readout of cholesterol content by Filipin staining. Representative of three independent experiments (mean+/−SD). (E) GC-MS quantification of cholesterol content in Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS for 8 hr (n=3, mean+/−SD). (F) Flow cytometry for pS6K1 (Thr389) and pS6 (S240/244) from BMDMs stimulated with LPS. (G) RT-qPCR analysis of Hmgcs1 expression in Ch25h+/− and Ch25h−/− BMDMs treated with LPS and DMSO or rapamycin. Data from three independent experiments (mean+/−SD). (H) IL-1β ELISA from supernatants of Ch25h+/− and Ch25h−/− BMDMs treated with LPS and DMSO or rapamycin, and then with ATP. Data from two independent experiments (mean+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test). See also Figure S1.
As a second approach to determine whether Ch25h represses cholesterol biosynthesis in activated macrophages, we performed liquid chromatography-mass spectrometry (LC-MS) for sterol biosynthetic intermediates after 8 hr of LPS treatment. Ch25h−/− BMDMs showed the expected reduction in 25-HC but showed increased levels of desmosterol, lanosterol, and 7-dehydrocholesterol, indicating that Ch25h acts to repress cholesterol biosynthesis pathway activity (Fig. 2B).
We next tested whether cholesterol content itself was altered in Ch25h−/− macrophages. Using a cholesterol fluorescence assay, Ch25h−/− BMDMs showed an initial increase in total cholesterol content compared to wild-type cells (Fig. 2C). LPS stimulation resulted in decreased cholesterol content in both genotypes by 4 hr, whereas only Ch25h−/− cells showed an increase in total cholesterol by 8 hr (Fig. 2C). Similarly, staining with filipin, a fluorescent cholesterol-binding antibiotic, showed higher fluorescence intensity in 8 hour LPS-activated Ch25h−/− macrophages (Fig. 2D). LC-MS analysis confirmed the increased cholesterol content (Fig. 2E). To determine whether Ch25h−/− macrophages increase their cholesterol in vivo, we injected mice I.P. with LPS and sorted peritoneal macrophages after 8 hr. LPS caused Ch25h−/− macrophages to increase their cholesterol content, whereas cholesterol levels in controls remained constant (Fig. S1C).
The increased expression of SREBP2-responsive genes between 4 and 8 hr of LPS exposure in the Ch25h−/− setting suggested that there is a latent positive drive for cholesterol synthesis that is limited by 25-HC. mTORC1 signaling has been shown to drive SREBP1 and SREBP2 activation, and is activated by TLR stimulation (Düvel et al., 2010; Porstmann et al., 2008; Wang et al., 2011). We therefore tested whether this pathway was active in LPS stimulated BMDMs. Phospho-flow cytometry for total S6K1, pS6K1 (Thr389) and pS6 (Ser240/244) showed that LPS stimulation resulted in a time-dependent increase in mTORC1 activity (Fig. 2F). We confirmed that S6 phosphorylation during LPS stimulation was mTORC1-dependent since it was prevented by rapamycin treatment (Fig. S1D). Consistent with mTORC1 activity driving SREBP2 activation, the augmented expression of Hmgcs1 mRNA in Ch25h−/− cells was largely prevented by rapamycin (Fig. 2G). Moreover, in accord with a connection between SREBP activity and IL-1β production (Reboldi et al., 2014), rapamycin reversed the overproduction of IL-1β in response to ATP in LPS stimulated Ch25h−/− macrophages, while having minimal effect on IL-1β production by Ch25h+/− cells (Fig. 2H). Taken together, these data indicate that Ch25h/25-HC are required in 8 hour LPS activated macrophages to antagonize mTORC1-induced SREBP2 pathway activity and prevent cholesterol over-accumulation.
Non-crystalline cholesterol is sufficient to promote inflammasome activation
To determine whether increased cholesterol biosynthesis can be linked to inflammasome activation, we used a retrovirus system to transduce cDNAs expressing HMG-CoA reductase (Hmgcr) and dehydrocholesterol 24-reductase (Dhcr24), enzymes in the cholesterol biosynthetic pathway, into BMDMs. Using a fluorescent peptide that acts as a reporter for caspase-1 activity, we found that over-expression of either of these cDNAs resulted in increased inflammasome activity after 8 hr of LPS stimulation (Fig. 3A). Inflammasome activation in mouse macrophages typically requires addition of a ‘second signal’ (such as ATP) to engage an inflammasome sensor protein (Rathinam and Fitzgerald, 2016). However, the effect of increased expression of Hmgcr and Dhcr24 mRNAs on caspase-1 was observed in the absence of an exogenous second signal. Attempts to measure IL-1β in the LPS-stimulated macrophage supernatant by ELISA were unsuccessful, consistent with a low amount of inflammasome activity. We therefore used an IL-17A-based IL-1β bioassay (Reboldi et al., 2014) to test whether cholesterol synthesis could drive inflammasome activation. Supernatants from BMDMs transduced with either Hmgcr or Dhcr24 had augmented capacity to stimulate IL-17A production from T cells, whereas this capacity was lost in the setting of ASC-deficiency (Fig. 3B).
Figure 3. Increased macrophage cholesterol content promotes crystal-independent inflammasome activation.

(A) FACS analysis of FLICA staining on BMDMs transduced with either MSCV-vector, -Hmgcr, or -Dhcr24 retrovirus and stimulated with LPS for 8 hr. Data from 3 independent experiments (mean+/−SD). (B) IL-1β bioactivity detected as percentage of CD4+ T cells producing IL-17A after culture for 4 days in supernatants from Asc+/+ or Asc−/− BMDMs transduced and stimulated as in A. Graph is representative of 3 independent experiments (mean+/−SD). (C) IL-1β ELISA of supernatants from BMDMs treated with 4 hr with LPS+MCD-Chol. Data from 6 independent experiments (mean+/−SD). (D) IL-1β bioactivity detected as in (B) with supernatants from BMDMs stimulated for 8 hr with either nil, LPS, LPS+MCD (10ug/ml), or LPS+MCD-Chol (10ug/ml). Data from three independent experiments (mean+/− SD). (E) IL-1β ELISA from supernatants of BMDMs treated with LPS, LPS+MCD-Chol, LPS+Alum or LPS+ATP and with DMSO or cytochalasin D. Data from 3 independent experiments (mean+/−SD). (F) IL-1β ELISA of supernatants from Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut and stimulated with either LPS, LPS+MCD-Chol, or LPS+ATP. Data from four independent experiments (mean+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test). See also Figure S2.
To determine whether cholesterol itself was involved in driving inflammasome activity, we treated BMDMs with LPS and cholesterol that had been solubilized by forming complexes with the carrier methyl-β-cyclodextrin (MCD) at a 25:1 ratio of MCD:cholesterol (MCD-chol). Strikingly, treatment of LPS activated macrophages with 100ug/ml MCD-chol, a concentration typical of other studies (Robinson et al., 2014; York et al., 2015), was sufficient to promote IL-1β production as measured by ELISA (Fig. 3C). A 10-fold lower dose of MCD-chol also triggered IL-1β production as detected using the IL-1β bioassay (Fig. 3D).
Previous work has suggested that the NLRP3 inflammasome is activated by cholesterol crystals in macrophages (Franklin et al., 2016). To determine whether our observed IL-1β release in response to MCD-chol was due to crystal formation, we noted that crystalline inflammasome activators depend on phagocytic uptake (Hornung et al., 2008). We treated LPS-activated BMDMs with cytochalasin D to inhibit actin polymerization and challenged them with MCD-chol, ATP, or alum. Whereas IL-1β release by the crystalline activator alum was blocked by cytochalasin D treatment, IL-1β release in response to MCD-chol or ATP was unaffected, providing evidence that MCD-chol is acting in a soluble fashion (Fig. 3E).
To determine whether 25-HC can prevent cholesterol-dependent IL-1β release, we reconstituted Ch25h−/− BMDMs with either wild-type Ch25h, a mutant Ch25h that cannot synthesize 25-HC (Reboldi et al., 2014), or empty vector. Overexpression of Ch25h could repress the sterol biosynthesis enzyme Sqle mRNA, whereas vector and Ch25hmut had no effect, confirming the functional status of these constructs (Fig. S2A). We found that while overexpression of Ch25h had a small capacity to diminish IL-1β release in response to ATP, it more substantially reduced the capacity of MCD-chol to stimulate IL-1β release, whereas Ch25hmut had no effect (Fig. 3F). In addition to its ability to inhibit SREBP2 activation, 25-HC could possibly protect cells from cholesterol overloading by acting as an LXR agonist. Reconstitution of LXRα/β double knockout BMDMs with wild type versus mutant Ch25h showed the same repressive effect on MCD-chol-dependent IL-1β release, suggesting that this phenomenon does not involve LXRs (Fig. S2B). These observations provide further evidence that a key function of Ch25h/25-HC induction in activated macrophages is to prevent cholesterol-dependent IL-1β production.
Cholesterol-dependent IL-1β release requires the AIM2 inflammasome
Mitochondrial reactive oxygen species (mtROS) have been implicated as activators of the NLRP3 inflammasome in response to a diverse range of stimuli (Zhou et al., 2011). Additionally, diseases associated with cholesterol dysregulation have been shown to induce mtROS (Kennedy et al., 2014). Therefore, we tested whether mtROS induction could be responsible for cholesterol-dependent IL-1β production. While LPS plus ATP treatment increased mtROS as expected (Yu et al., 2014; Zhou et al., 2011) Ch25h−/− BMDMs had heightened mtROS production after compared to control BMDMs (Fig. 4A). Retroviral reconstitution of Ch25h−/− BMDMs with Ch25h reduced mtROS, whereas Ch25hmut showed no effect (Fig. 4B). Treatment of Ch25h+/− and Ch25h−/− BMDMs with the mtROS scavenger MitoTEMPO inhibited IL-1β in both genotypes (Fig. S3A). Furthermore, treatment of uninfected BMDMs with LPS plus MCD-chol resulted in increased mtROS compared to LPS alone, consistent with data suggesting that elevated cholesterol content can cause mitochondrial dysfunction (Fig. 4C). Moreover, using a 293T NLRP3 inflammasome reconstitution system we found that SREBP2 over-expression was capable of promoting IL-1β cleavage and release (Fig. S3B, S3C, S3D). Combined, these observations suggested that IL-1β release by macrophages following cholesterol accumulation would occur via the NLRP3 inflammasome. Surprisingly, however, BMDMs lacking NLRP3 had no defect in IL-1β production in response to LPS plus MCD-chol treatment, whereas these cells failed to respond to LPS plus ATP (Fig. 4D).
Figure 4. Cholesterol-dependent inflammasome activation requires AIM2 and redundantly involves NLRP3.

(A) Mitochondrial superoxide levels determined by MitoSOX staining of Ch25h+/− and Ch25h−/− BMDMs stimulated for 8 hr with LPS, or LPS+ATP treatment. Data are representative of 6 independent experiments (mean+/−SD). (B) MitoSOX staining of Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut and stimulated as in A. Data from 2 independent experiments (mean+/−SD). (C) MitoSOX staining from BMDMs stimulated for with LPS or LPS+MCD-Chol. Data are representative of 3 independent experiments (mean +/− SD). (D-J) IL-1β ELISA of supernatants from Nlrp3+/+ and Nlrp3−/− (D), Asc+/+ and Asc−/− (E), Casp1/11+/+ and Casp1/11−/− (F), Nlrc4+/+ and Nlrc4−/− (G), Casp11+/+ and Casp11−/− (H), Aim2+/+ and Aim2−/− (I), or Nlrp3+/+Aim2+/+ and Nlrp3−/−Aim2−/− BMDMs treated as indicated. Data from 2 to 4 experiments (mean+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test). See also Figure S3.
To test whether the observed IL-1β release in MCD-chol challenged BMDMs was an inflammasome-dependent process we challenged Asc−/− and Casp-1/11−/− BMDMs with LPS and MCD-chol. Asc−/− (Fig. 4E) and Casp-1/11−/− (Fig. 4F) BMDMs showed complete abrogation of IL-1β production in response to cholesterol, establishing that this process was indeed inflammasome-dependent.
To identify the relevant sensor protein required for cholesterol-dependent IL-1β production, we challenged Nlrc4−/−, Casp11−/−, and Aim2−/− BMDMs with LPS and MCD-Chol. Nlrc4−/− (Fig. 4G) and Casp11−/− (Fig. 4H) BMDMs showed no difference in IL-1β production in response to cholesterol challenge. On the other hand, Aim2−/− BMDMs showed a striking reduction in IL-1β release under these conditions (Fig. 4I). Given that different inflammasomes can cooperate in some contexts, we tested whether the low amount of remaining inflammasome activity in Aim2−/− BMDMs involved NLRP3. Consistent with a partially redundant contribution of NLRP3 to cholesterol-induced IL-1β release, Aim2−/−Nlrp3−/− BMDMs showed a near complete loss of secreted IL-1β when compared with Casp1/11−/− BMDMs (Fig. 4J).
Neutrophilic peritonitis is a well-described model system for in vivo assessment of inflammasome activation. To determine whether cholesterol could drive inflammasome activation in vivo, we injected LPS, LPS and MCD-chol, or LPS and Alum I.P. Strikingly, MCD-chol was as efficacious as Alum in causing neutrophil accumulation in the peritoneum (Fig. 5A and 5B). To determine whether AIM2 inflammasome activation was required for neutrophilic peritonitis, lethally irradiated mice were reconstituted with wild-type or Aim2−/− BM. We found that hematopoietic AIM2-deficiency resulted in a reduction in MCD-chol induced neutrophil recruitment (Fig. 5C and 5D). We also generated chimeras using Nlrp3−/−, or Casp1/11−/− BM; consistent with our observations in BMDMs, lack of NLRP3 had no effect on neutrophil recruitment following MCD-chol challenge, whereas caspase-1 deletion resulted in the expected strong reduction (Fig. 5E).
Figure 5. AIM2 is required for cholesterol-dependent neutrophilic peritonitis.

(A) Representative FACS plots of peritoneal Ly6G+ cells from mice injected I.P. 8 hr earlier with the indicated treatments displaying percentage of Ly6G+ cells. (B) Number of peritoneal neutrophils from mice as treated in A. (C) Representative FACS plots of peritoneal Ly6G+ cells from LPS or LPS+MCD-Chol treated wild type or Aim2−/− BM chimeras. (D) Number of peritoneal neutrophils from mice of the type in C treated with LPS or LPS+MCD-chol for 8 hr. (E) Number of peritoneal neutrophils from wild type mice irradiated and transplanted with either wild type, Nlrp3−/−, or Casp1/11−/− BM, and treated as in D. Each dot represents an individual mouse and data are pooled from 3 independent experiments (mean+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test).
Ch25h-deficiency results in impaired mitochondrial metabolism
The requirement for AIM2, a DNA sensor, in cholesterol-dependent inflammasome activation led us to consider the possibility that dysregulation of cellular cholesterol homeostasis results in release of host DNA into the cytosol. Mitochondria contain multiple copies of mtDNA that encodes for components of the oxidative metabolism machinery. A number of recent studies have demonstrated that mitochondrial dysfunction can lead to mtDNA release into the cytosol (Rongvaux et al., 2014; West et al., 2015; White et al., 2014). Additionally, baseline mitochondrial cholesterol levels are low, and increased mitochondrial cholesterol accumulation is associated with impaired mitochondrial function (Kennedy et al., 2014). Due to these observations and our findings that Ch25h-deficiency and cholesterol accumulation cause mtROS production, we asked whether the altered cholesterol homeostasis caused by Ch25h-deficiency resulted in impaired mitochondrial function in macrophages. We first assessed mitochondrial respiration and abundance using Mitotracker Deep Red and Mitotracker Green dyes, respectively, by flow cytometry. Ch25h−/− BMDMs showed diminished Mitotracker Deep Red staining compared to control macrophages after 8 hr of LPS stimulation (Fig. 6A). Mitotracker Deep Red staining could be rescued by reconstituting Ch25h−/− BMDMs with Ch25h, but not Ch25hmut (Fig. 6B). Mitotracker Green staining also showed a small but statistically significant decrease in 8 hour LPS-stimulated Ch25h−/− BMDMs (Fig. 6C). This defect could also be rescued by wild-type but not mutant Ch25h (Fig. 6D). We additionally found that Ch25h−/− macrophages showed diminished Mitotracker Deep Red and Mitotracker Green staining in response to 8 hr of L. monocytogenes infection (Fig. S4A and S4B).
Figure 6. Ch25h-deficient and cholesterol loaded macrophages have impaired mitochondrial metabolism.

(A) (Left) Representative histogram of Mitotracker Deep Red staining on Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS for 8 hr. (Right) Summary mean fluorescence intensity (MFI) data from 3 experiments (mean +/− SD). Representative of 8–10 independent experiments. (B) MFI of Mitotracker Deep Red staining on Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut. Data from 4 independent experiments (mean+/−SD). (C) (Left) Representative histogram of Mitotracker Green staining on Ch25h+/− and Ch25h−/− BMDMs stimulated as above. (Right) Summary MFI data from 3 experiments (mean+/−SD). Representative of 8–10 independent experiments (mean +/− SD). (D) MFI of Mitotracker Green staining on Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut. Data from 4 independent experiments (mean+/−SD). (E) Representative histogram of TMRM staining on Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS for 8 hr. Graph shows summary MFI data from 3 experiments (mean+/−SD). (F) MFI of TMRM staining on Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut. Data from 2 independent experiments (mean+/−SD). (G) Seahorse analysis of oxygen consumption rate (OCR) over time from BMDMs pretreated for 8 hr with LPS and after the indicated additional treatments. Data are representative of 3 independent experiments (mean+/−SD). (H) Seahorse analysis of basal OCR and extracellular acidification rate (ECAR) of Ch25h+/− and Ch25h−/− BMDMs stimulated with LPS for 8 hr. Data from 3 independent experiments (mean+/−SD). (I, J) Mitotracker Deep Red (I) and Mitotracker Green (J) staining of Ch25h+/− and Ch25h−/− peritoneal macrophages 8 hr after I.P. saline or LPS injection. Data from 3 independent (n=6–7 mice per group). See also Figure S4.
To determine whether increased cholesterol content was sufficient to explain the Mitotracker staining effects in Ch25h−/− BMDMs, we treated Asc+/+ and Asc−/− BMDMs with LPS and MCD-chol and measured Mitotracker staining. Both Mitotracker Deep Red and Mitotracker Green staining were diminished by increasing cellular cholesterol content (Fig. S4C and S4D). Importantly, Asc−/− BMDMs also showed decreased Mitotracker staining, demonstrating that this effect was not secondary to increased inflammasome activation (Yu et al., 2014). As a second test of whether increased cholesterol content was sufficient to affect Mitotracker staining, we again transduced BMDMs with Hmgcr, or Dhcr24 and stimulated these macrophages with LPS. Both Mitotracker Deep Red and Mitotracker Green staining were diminished by enzyme overexpression (Fig. S4E and S4F). To determine whether cholesterol efflux could rescue Mitotracker staining, we treated macrophages with HDL or the LXR agonist GW3965 along with LPS. The diminished Mitotracker Deep Red staining in Ch25h−/− cells was rescued by both HDL and GW3965, further supporting a role for cholesterol in this outcome (Fig. S4G).
Mitochondrial dysfunction is often associated with alterations in the ion potential of the inner membrane (Shimida et al., 2012). We therefore tested whether Ch25h affected mitochondrial membrane potential using tetramethylrhodamine methyl ester (TMRM), a cationic dye that becomes sequestered in the mitochondrial matrix in amounts directly proportional to the membrane potential. LPS stimulation resulted in diminished TMRM staining in Ch25h−/− macrophages compared to controls (Fig. 6E). TMRM staining could be rescued by retroviral transduction of Ch25h−/− macrophages with active but not mutant Ch25h enzyme (Fig. 6F). Diminished TMRM staining was also observed in Ch25h−/− macrophages after L. monocytogenes infection (Fig. S4H)
As another approach to assess mitochondrial function we utilized the Seahorse assay to measure oxygen consumption by BMDMs after 8 hr of LPS stimulation. Ch25h−/− macrophages showed a decreased basal oxygen consumption rate (OCR) compared to wild-type cells (Fig. 6G and 6H). More strikingly, BMDMs lacking Ch25h showed impaired spare respiratory capacity (SRC) after FCCP treatment, suggesting that maximal mitochondrial respiration is impaired in settings of altered cholesterol homeostasis (Fig. 6G). The decrease in basal oxygen consumption rate in Ch25h−/− macrophages was also coupled to an increased extracellular acidification rate (ECAR), suggesting that these cells were metabolically reprogrammed towards aerobic glycolysis (Fig. 6H). To determine whether impaired mitochondrial respiration in the absence of Ch25h could be explained by increased cholesterol content, we treated BMDMs with MCD-chol and again performed Seahorse analysis. Consistent with our hypothesis, cholesterol loaded macrophages also showed impaired basal OCR as well as diminished SRC (Fig. S4I).
To test whether Ch25h−/− macrophages showed signs of mitochondrial dysfunction in vivo, we intraperitoneally injected control and Ch25h−/− mice with saline or LPS for 8 hr and assessed Mitotracker staining on peritoneal macrophages. While saline treated mice showed no difference between genotypes, we observed diminished Mitotracker Deep Red and Mitotracker Green staining in peritoneal macrophages derived from Ch25h−/− mice after LPS treatment (Fig. 6I, 6J, and S4J).
Increased mitochondrial cholesterol content results in cytosolic mtDNA accumulation and inflammasome activation
Given our observation that Ch25h−/− macrophages showed increased cellular cholesterol and mitochondrial dysfunction, we asked whether these cells had increased mitochondrial cholesterol content. We used an immunoprecipitation system with anti-Tomm22 coupled beads to isolate mitochondria and mitochondrial-associated membrane (MAM) from either control or 8 hour LPS-stimulated BMDMs. Immunoblots of these fractions showed an expected enrichment in the outer mitochondrial membrane protein Tomm20, depletion of the cytosolic protein β-tubulin, and partial depletion of the ER/MAM protein calreticulin compared to whole cell extracts (Fig. S5A). The incomplete depletion of calreticulin was most likely due to MAM contamination (Hayashi et al., 2007). We saw that Ch25h−/− BMDMs show increased mitochondrial/MAM cholesterol content after LPS stimulation compared to controls (Fig. 7A). Additionally, we found that reconstitution of Ch25h−/− macrophages with wild type but not mutant Ch25h enzyme could decrease mitochondrial/MAM cholesterol levels (Fig 7B). Mitochondrial/MAM cholesterol content was also increased in BMDMs treated with LPS and MCD-Chol (Fig. S5B). These findings together with the evidence of mitochondrial damage and the requirement for AIM2 in cholesterol-dependent inflammasome activation led us to test whether Ch25h-deficiency and cholesterol loading increased cytosolic mtDNA. We stimulated BMDMs with either vehicle or LPS for 8 hr and then extracted cytosolic DNA. qPCR analysis for mtDNA and nuclear DNA (nucDNA) showed that Ch25h−/− BMDMs increased their cytosolic mtDNA, but not nucDNA, after LPS stimulation (Fig. 7C). Additionally, cholesterol treatment of BMDMs was sufficient to drive increased mtDNA but not nucDNA accumulation in the cytosol (Fig. S5C). Consistent with our observation that rapamycin blocks the SREBP2 pathway in Ch25h−/− macrophages, we found that rapamycin could also decrease mitochondrial cholesterol accumulation and cytosolic mtDNA in LPS-treated Ch25h−/− BMDMs (Fig. S5D, S5E). A number of recent studies have demonstrated that cytosolic release of mtDNA can activate the cGAS/STING DNA sensing pathway that triggers the production of type I IFNs (Rongvaux et al., 2014; West et al., 2015; White et al., 2014; Aguirre et al, 2017; Sun et al., 2017). Therefore we asked whether cholesterol treatment of BMDMs would show evidence of type I IFN induction. However, under the LPS stimulation condition there is a large amount of TLR4-mediated IFN production and induction of interferon-stimulated genes (ISGs) (Fig. S5F), and we were unable to detect increased ISG expression with LPS plus MCD-chol stimulation (Supp. Fig. S5F).
Figure 7. Cholesterol buildup in mitochondrial causes cytosolic release of mtDNA.

(A) Quantification of cholesterol in isolated mitochondria from Ch25h+/− and Ch25h−/− BMDMs either untreated or stimulated with LPS for 8 hr. Data from 3 independent experiments (mean+/−SD). (B) Quantification of cholesterol in Ch25h−/− BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut and treated for 8 hr with LPS. Data from three independent experiments (mean+/−SD). (C) mtDNA (Left) or nucDNA (Right) RT-qPCR from cytosolic extracts derived from Ch25h+/− and Ch25h−/− BMDMs treated as above. Data from 4 independent experiments (mean+/−SD). (D) Representation histograms (left) and summary graph (right) of intracellular cyt C staining in Ch25h+/− and Ch25h−/− BMDMs stimulated for 8 hr with LPS and either permeabilized with digitonin or not. Data from two independent experiments (mean+/−SD). (E) mtDNA RT-qPCR of total DNA extractions from BMDMs treated for 7 days with either nil, ethidium bromide (EtBr), or ddC. Data from 3 independent experiments (mean+/−SD). (F) IL-1β ELISA from supernatants of BMDMs depleted of mtDNA as described above, and stimulated with either LPS alone, LPS+MCD-Chol, or LPS+ATP. Data from 3 independent experiments (mean+/−SD). (G) IL-1β bioactivity detected as in Fig. 3 using supernatants from LPS-stimulated BMDMs of the indicated genotypes. Data from three independent experiments (mean+/−SD). (H) IL-1β ELISA of supernatants from BMDMs of the indicated genotypes stimulated with LPS or LPS+ATP. Data from three independent experiments (mean+/−SD). (I) L. monocytogenes CFU from BMDMs of the indicated genotypes infected for 24 hr. Data from three independent experiments (mean+/−SD). (J) IL-1β ELISA of serum from mice of the indicated genotypes injected with LPS (20mg/kg) I.V. for 8 hr. Data from three independent experiments (n = 5 to 8 mice per group, mean+/−SD). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test). See also Figure S5.
The release of mtDNA into the cytosol of Ch25h−/− macrophages would require an increase in mitochondrial membrane permeability. To test this directly, we assessed whether Ch25h−/− macrophages had increased cytochrome C (cyt C), a protein normally sequestered between the mitochondrial inner and outer membrane, in the cytosol (Campos et al., 2006). Intracellular staining for cyt C in non-digitonin treated cells detected both cytosolic and mitochondrial cyt C. This signal was then compared to that for cells that were digitonin-treated, causing cytosolic cyt C to leak out, prior to intracellular staining. We found that LPS treated, Ch25h−/− cells showed a preferential loss of cyt C after digitonin treatment compared to Ch25h+/− cells (Fig. 7D).
To functionally link increased cytosolic mtDNA accumulation with inflammasome activation, we used two approaches to deplete mtDNA from BMDMs. Ethidium bromide (EtBr) is a DNA intercalating agent that has been used in a number of studies to deplete mtDNA (Rongvaux et al., 2014; Shimada et al., 2012; Zhong et al., 2016). 2′-3′-dideoxycytidiine (ddC) is a nucleoside analogue that has selectivity for the mtDNA polymerase (Chen and Cheng, 1989; Rongvaux et al., 2014). We cultured BMDMs for 7 days in the presence of either EtBr or ddC; both conditions resulted in an approximately 66% loss of cellular mtDNA (Fig. 7E). Strikingly, after stimulating these cells with LPS alone, LPS plus MCD-chol, or LPS plus ATP we found that mtDNA depletion selectively reduced IL-1β release in response to cholesterol but not ATP treatment (Fig. 7F). These results suggest that dysregulated cholesterol metabolism results in mitochondrial damage, leading to mtDNA-dependent AIM2 inflammasome activation.
To directly test whether Ch25h-deficiency leads to AIM2-dependent inflammasome activation, we generated Ch25h−/−Aim2−/− mice. BMDMs from these mice and their matched controls were stimulated with LPS and supernatants tested for IL-17A induction in the IL-1β bioassay. Ch25h−/− cells showed increased IL-1β bioactivity as expected (Fig. 7G). However, deletion of AIM2 in the setting of Ch25h-deficiency reduced the IL-1β bioactivity back to Ch25h-sufficient levels (Fig. 7G).
As a second test of whether increased inflammasome activity in Ch25h−/− macrophages required AIM2, we stimulated BMDMs with LPS+ATP, a classic NLRP3 activator, and measured IL-1β by ELISA. We again observed increased IL-1β production in Ch25h−/− macrophages (Fig. 7H). Importantly, deletion of AIM2 in the setting of Ch25h-deficiency resulted in diminished IL-1β production whereas AIM2-deficiency did not alter the response of Ch25h-sufficient cells (Fig. 7H). These results suggest that Ch25h deficiency creates a partial switch in the inflammasome sensor requirement for responses to ATP. We additionally tested the inflammasome response of these cells to FlaTOX, an NLRC4 activator, and poly(dA:dT), an AIM2-activator. With FlaTOX treatment, Ch25h/AIM2 double-deficiency again rescued the overproduction of IL-1β observed in Ch25h−/− macrophages (Fig. S5G), whereas the response to poly(dA:dT) was abrogated in the setting of AIM2-deficiency regardless of Ch25h status, as was expected (Fig. S5H).
We next tested whether the enhanced protection from Listeria infection observed in Ch25h−/− macrophages (Fig. 1C) required AIM2. Listeria engages multiple inflammasome pathways (NLPR3, AIM2, NLRC4) (Wu et al., 2010) and in accord with this knowledge, AIM2 deficiency led to increased Listeria growth (Fig. 7I). Importantly, however, in contrast to the protective effect of Ch25h loss from Aim2+/+ macrophages, Ch25h-deficiency did not lead to a significant protection of Aim2−/− macrophages (Fig. 7I). These findings provide evidence in the context of a bacterial infection that Ch25h restricts AIM2-inflammasome activation.
Caspase-1 activation has been suggested to cause mitochondrial damage under some conditions (Yu et al., 2014). To test whether caspase-1 activity was required for AIM2 inflammasome activation in the setting of Ch25h-deficiency we stimulated BMDMs with LPS and treated with FLICA to irreversibly inhibit caspase-1 and also to allow visualization of inflammasome specks. Consistent with our observations that IL-1β production in response to LPS without ‘signal 2’ is below the limit of detection of ELISA, speck formation was rare in this condition. However, speck formation was increased in Ch25h−/− BMDMs, and this effect was rescued by AIM2 double-deficiency (Fig. S5I). These data suggest that caspase-1 activity is not required for AIM2 inflammasome formation in the setting of Ch25h-deficiency.
Finally, to determine whether AIM2 is required for the overproduction of IL-1β from Ch25h−/− hematopoietic cells in vivo, BM chimeric mice were generated. Following reconstitution the mice were injected intravenously with saline or LPS for 8 hr and then serum was harvested for cytokine analysis. Consistent with our previous results, we saw increased serum IL-1β in LPS-challenged Ch25h−/− BM chimeras (Reboldi et al, 2014) (Fig. 7I). In keeping with our in vitro observations, Ch25h−/−Aim2−/− BM chimeras had diminished serum IL-1β levels that were similar to Ch25h-sufficient levels (Fig. 7I). Collectively, these data suggest that Ch25h deficiency results in altered nucleic acid compartmentalization that triggers inflammation via AIM2.
Discussion
In this study, we identify a metabolic circuit that is crucial for limiting inflammasome activity in activated macrophages. We show that induction of Ch25h by type I IFN plays a key role in restricting the SREBP2 pathway and cholesterol biosynthesis in the context of bacterial infection and LPS exposure. These observations are consistent with a number of recent studies that have demonstrated the ability of type I IFNs to repress cholesterol metabolism (Robertson et al., 2016)(York et al, 2015). However, our study suggests that Ch25h is not required to initiate suppression of sterol biosynthesis, but is instead required to maintain this effect over time. Consistent with work suggesting that mTORC1 signaling can drive SREBP2 activation (Düvel et al., 2010; Porstmann et al., 2008; Wang et al., 2011), we find that the overexpression of cholesterol biosynthesis enzymes in Ch25h−/− cells is rapamycin-sensitive. Both TCR-activated T cells and LPS-activated macrophages switch to aerobic glycolysis and engage mTORC1 signaling due to the mutual requirement of both cell types to massively increase protein synthesis capacity (Powell et al., 2012). However, activated T cells undergo rapid proliferation and thus are constantly using cholesterol to synthesize new cellular membranes, whereas macrophages are largely quiescent and therefore susceptible to mTORC1-driven cholesterol over-accumulation. We speculate that the acute induction of Ch25h in macrophages allows them to selectively utilize the protein synthesis arm of mTORC1 signaling to produce cytokines and other important anti-microbial factors, while preventing cholesterol buildup and cellular pathology.
Inflammasome sensor proteins are positioned in the cytosol and this allows them to distinguish highly pathogenic bacteria from commensals due to the propensity of pathogens to break out of the phagosome. Because mitochondria are derived from endosymbiotic bacteria, they contain a number of potential danger-associated molecular patterns (DAMPs) that could be released into the cytosol to trigger inflammation. Cholesterol levels in mitochondria are about 40-fold lower than in the plasma membrane and these organelles are highly sensitive to cellular cholesterol alterations (Bosch et al., 2011; Marí et al., 2006; Martin et al., 2016). We rationalize that Ch25h upregulation upon extracellular bacterial sensing helps prevent cholesterol-dependent mitochondrial damage and release of endogenous DAMPS into the cytosol. In the absence of this pathway, mitochondrial membrane damage can lead to increases in cytosolic DNA, resulting in engagement of the AIM2 inflammasome and exaggerated IL-1β production.
It is notable that Ch25h-deficiency augments the response to multiple types of inflammasome activators. This could in principle be due to a general antagonistic effect of 25-HC on inflammasome assembly. However, we find that this augmented inflammasome activity is reversed in the setting of AIM2 deletion, suggesting that the broad overproduction of IL-1β is due to ectopic AIM2 activation. This may indicate that the altered metabolic state occurring in the absence of Ch25h makes cells poised to engage AIM2 inflammasomes. With further mitochondrial stress resulting from infection or potentially dietary lipid overload, sufficient mtDNA may become released to engage the AIM2 inflammasome. While our data establish a temporal requirement for Ch25h to restrain IL-1β production by TLR-activated macrophages, Ch25h expression is not preventing IL-1β production. Rather, we suggest that by maintaining the metabolic health of the cell, Ch25h and 25-HC are helping to ensure that macrophages make a beneficial and not pathological amount of IL-1β. While we have mostly tested this circuit in the context of TLR agonists or bacterial infection, Ch25h may also be important in controlling IL-1β production during metabolic inflammation, where it would be critical to keep cytokine concentrations below a pathologic threshold.
Beyond repressing cholesterol biosynthesis through SREBP2, our data suggest that 25-HC can counter mitotoxic effects of cholesterol accumulation, based on the capacity of Ch25h overexpression to block IL-1β release in response to exogenous cholesterol. The LXR family members were intriguing candidates for this effect, since they promote cholesterol efflux through upregulation of ABCA1 and ABCG1, and tentative evidence suggests that 25-HC does have some ligand activity for these nuclear hormone receptors (Cyster et al., 2014). However, Ch25h retained its capacity to blunt cholesterol-dependent effects when expressed in LXRα/β double knockout macrophages. We speculate that this activity of Ch25h may instead be related to the capacity of 25-HC to act as an allosteric activator of ACAT1, an enzyme that esterifies cholesterol to allow storage in lipid droplets (Metherall et al., 1991). However, there is at present no genetic evidence that ACAT1 activity is affected by endogenously produced 25-HC. Future work will be needed to examine effects of 25-HC on cholesterol esterification in activated macrophages.
AIM2 is a well-described sensor of cytosolic DNA that can trigger inflammasome activation (Rathinam and Fitzgerald, 2016). Activation of this sensing pathway is usually described in the context of pathogen-derived nucleic acids that enter the host cell cytosol (Rathinam and Fitzgerald, 2016). However, the AIM2 HIN-200 domain is not sequence-specific, and therefore host DNA represents a potential pool of ligand if the nucleic acid is de-compartmentalized. Cytosolic release of mtDNA can trigger the cGAS-dependent DNA sensing pathway in the context of caspase-3/7- and TFAM-deficiency (Rongvaux et al., 2014; West et al., 2015; White et al., 2014) and in dengue virus infected cells (Aguirre et al, 2017; Sun et al., 2017), but there are few examples of AIM2 activation by mtDNA. A number of studies have suggested that oxidized mtDNA can act as a ligand for NLRP3 (Nakahira et al., 2011; Shimada et al., 2012), which is consistent with our findings that NLRP3 redundantly contributes to cholesterol-dependent IL-1β production. Interestingly, the cited studies had to use AIM2-deficient macrophages to reveal NLRP3-dependent effects in response to transfected mtDNA; but to our knowledge there are no prior examples of endogenous mtDNA release triggering AIM2. It remains an open question as to how exactly cholesterol buildup leads to cytosolic mtDNA release in macrophages. It would be of interest to determine whether this process involves Bax/Bak-mediated membrane permeabilization, as there is conflicting evidence on whether cholesterol promotes or antagonizes Bax oligomerization and pore formation (Christenson et al., 2008; Montero et al., 2010). Cholesterol-induced reduced membrane fluidity and increased oxidative stress might also lead to other forms of mitochondrial membrane damage (Kennedy et al., 2014; Martin et al., 2016). Given the complexities associated with determining the exact nucleic acid sources that trigger cytosolic DNA sensor proteins, and the large size of the nuclear genome, our studies do not exclude the possibility that increased cholesterol content of intracellular membranes causes cytosolic accumulation of nuclear DNA as an additional AIM2 ligand.
Finally, while we believe Ch25h upregulation by type I IFN represents a physiological circuit that prevents mtDNA from engaging AIM2, these observations raise additional questions about the role of cholesterol in driving inflammation in chronic diseases like obesity and metabolic syndrome. A number of studies have implicated inflammatory cytokines like TNF and IL-1β in promoting insulin resistance in high-fat diet (HFD) conditions (de Luca and Olefsky, 2008). Additionally, inflammasomes have been suggested to play a causal role in metabolic inflammation (Stienstra et al., 2011; Vandanmagsar et al., 2011). However, the exact mechanisms underlying lipid-driven inflammation are still unclear. Ceramide accumulation has been argued to engage the NLRP3 inflammasome during HFD (Vandanmagsar et al., 2011), but little is known about the role of cholesterol-driven inflammation in obesity, despite the fact that LDL and HDL levels have been used as risk factors for atherosclerosis for decades. A limited number of reports have noted a connection between cholesterol-dependent mitochondrial dysfunction under high fat conditions and development of metabolic diseases (Martin et al., 2016). While it is recognized that cholesterol levels in cells are tightly controlled and not readily altered by changes in dietary cholesterol, it will be of interest to determine whether Ch25h-deficient mice have exacerbated inflammatory responses to HFD, and whether AIM2 deficiency blunts the inflammatory effects of HFD.
In conclusion, the studies described herein provide mechanistic insight into how regulation of macrophage cholesterol homeostasis during pathogen sensing integrates with inflammasome regulation, and they advance our understanding of how cellular cholesterol can act as an inflammatory driver.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Fixable viability dye eFluor780 | eBioscience | Cat# 65-0865-14 |
| Anti-mouse IL-17A Monoclonal Antibody (eBio17B7), PE | eBioscience | Cat# 12-7177-81 |
| Anti-mouse CD4 Monoclonal Antibody (RM4-5), APC | Tonbo Biosciences | Cat# 20-0042-U100 |
| Anti-mouse TCRβ Monoclonal Antibody (H57-597), Pacific Blue | Biolegend | Cat# 109226 |
| Fixable viability dye eFluor780 | eBioscience | Cat# 65-0865-14 |
| Anti-mouse LDLR Polyclonal Antibody, PE | Sino Biological | Cat# 50305-R032-P-25 |
| Anti-mouse CD4 Monoclonal Antibody (RM4-5), APC | Tonbo Biosciences | Cat# 20-0042-U100 |
| Anti-mouse TCRβ Monoclonal Antibody (H57-597), Pacific Blue | Biolegend | Cat# 109226 |
| Anti-mouse F4/80 Monoclonal Antibody (BM8.1), APC | Tonbo Biosciences | Cat# 20-4801-U100 |
| Anti-Phospho-S6 Ribosomal Protein (Ser240/244) Monoclonal Antibody (D68F8) | Cell Signaling Technologies | Cat# 5364S |
| Anti-Phospho-p70 S6 Kinase (Thr389) Monoclonal Antibody (108D2) | Cell Signaling Technologies | Cat# 9234S |
| Anti-p70 S6 Kinase Monoclonal Antibody (49D7) | Cell Signaling Technologies | Cat# 9234T |
| Anti-mouse Cytochrome C Monoclonal Antibody (6H2.4), Alexa488 | Biolegend | Cat# 612308 |
| Anti-mouse NLRP3/NALP3 Monoclonal Antibody (Cryo-2) | Adipogen | Cat# AG-20B-0014-C100 |
| Anti-mouse Asc Polyclonal Antibody (AL177) | Adipogen | Cat# AG-25B-0006-C100 |
| Anti-mouse Caspase-1 Polyclonal Antibody (M-20) | Santa Cruz Biotechnology | Cat# sc-514 |
| Anti-mouse Tomm20 Polyclonal Antibody | One World Lab | Cat# C16678 |
| Anti-mouse α-Tubulin Monoclonal Antibody (DM1A), Biotin | eBioscience | Cat# 13-4502-80 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes | A gift from D. Portnoy | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant murine IL-β | Peprotech | Cat# 211-11B |
| MitoSOX Red Mitochondrial Superoxide Indicator | ThermoFisher Scientific | Cat# M36008 |
| MitoTracker Deep Red FM | ThermoFisher Scientific | Cat# M22426 |
| MitoTracker Green FM | ThermoFisher Scientific | Cat# M7514 |
| Tetramethylrhodamine, Methyl Ester, Perchlorate (TMRM) | ThermoFisher Scientific | Cat# T668 |
| MitoTEMPO | Millipore Sigma | Cat# SML0737 |
| DAPI, FluoroPure grade | ThermoFisher Scientific | Cat# D21490 |
| ATP | InvivoGen | Cat# TLRL-ATP |
| Nigericin sodium salt | Millipore Sigma | Cat# N7143 |
| Poly(deoxyadenylic-thymidylic)(dA:dT) acid sodium salt | Millipore Sigma | Cat# P0883-10UN |
| FlaTOX | von Moltke et al., 2012 (a gift from R. Vance) | N/A |
| Imject Alum Adjuvant | ThermoFisher Scientific | Cat# 77161 |
| Lipopolysaccharide from E. coli O111:B4 | Millipore Sigma | Cat# L4391 |
| 25-hydroxycholesterol | Avanti Polar Lipids | Cat# 700019P |
| Cholesterol-Water Soluble | Millipore Sigma | Cat# C4951 |
| Methyl-β-cyclodextrin | Millipore Sigma | Cat# C4555 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| Cytochalasin D | Millipore Sigma | Cat# C8273 |
| Filipin III from Streptomyces filipinensis | Millipore Sigma | Cat# F4767 |
| Digitonin | Millipore Sigma | Cat# D141 |
| Ethidium bromide | Millipore Sigma | Cat# E1510 |
| 2′,3′-Dideoxycytidine (ddC) | Millipore Sigma | Cat# D5782 |
| GW3965 hydrochloride | Millipore Sigma | Cat# G6295 |
| Lipoprotein, High Density | Lee Biosolutions | Cat# 361-10-0.01 |
| Brain Heart Infusion Agar, Deep Fill, 15×100mm plate R | Hardy Diagnostics | Cat # W15 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mitochondria Stress Test Kit | Seahorse Biosciences/Agilent | Cat# 103015-100 |
| MACS Mitochondrial Isolation Kit | Miltenyi Biotec | Cat# 130-096-946 |
| Amplex Red Cholesterol Assay Kit | ThermoFisher Scientific | Cat# A12216 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human Embryonic Kidney Cells (HEK293T) | ATCC | Cat# CRL-3216 |
| Platinum-E (Plat-E) Retroviral Packaging Cell Line | A gift some S. Schwab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | Stock No: 000664 |
| Mouse: B6-Ly5.1/Cr Strain ID: B6.SJL- PtprcaPepcb/BoyCrCrl | NCI at Charles River | Stock No: 564 |
| Mouse: Ch25h−/− Strain ID: B6.129S6- Ch25htm1Rus/J | Jackson Laboratories | Stock No: 016263 |
| Mouse: Aim2−/− Strain ID: B6.129P2- Aim2Gt(CSG445)Byg/J | Jackson Laboratories | Stock No: 013144 |
| Mouse: Asc−/− Strain ID: B6.129-Pycardtm1Vmd | Genentech | N/A |
| Mouse: Nlrp3−/− Strain ID: B6.129S6-Nlrp3tm1Bhk/J | Jackson Laboratories | Stock No: 021302 |
| Mouse: Casp1/11−/− Strain ID: B6N.129S2- Casp1tm1Flv/J | Jackson Laboratories | Stock No: 016621 |
| Mouse: Nr1h3−/− Strain ID: 129-Nr1h3tm1Djm/J | Jackson Laboratories | Stock No: 013762 |
| Mouse: Nr1h2−/− Strain ID: B6.129S-Nr1h2tm1Djm/J | Jackson Laboratories | Stock No: 014635 |
| Mouse: Nlrc4−/− Strain ID: B6N-Nlrc4tm1Vmd | Genentech | N/A |
| Oligonucleotides | ||
| RT-PCR primers for Ch25h Fwd: 5′-GCGACGCTACAAGATCCA-3′ Rev: 5′-CACGAACACCAGGTGCTG-3′ |
This paper | N/A |
| RT-PCR primers for Hmgcs1 Fwd: 5′-GGAAGCCTTTGGGGACGTTA-3′ Rev: 5′-ACACTCCAACCCTCTTCCCT-3′ |
This paper | N/A |
| RT-PCR primers for Lss Fwd: 5′-GGGCCCTGAATGGAGTAACC-3′ Rev: 5′-GTCCTCGATGTGCAAGCCC-3′ |
This paper | N/A |
| RT-PCR primers for Dhcr24 Fwd: 5′-GGACATCATCCCTTTCGGCA-3′ Rev: 5′-TGGGGTAGACGTGGATGTCA-3′ |
This paper | N/A |
| RT-PCR primers for Sqle Fwd: 5′-AGTGAACAAACGAGGCGTCC-3′ Rev: 5′-AGCAAGCTTTTCGGAGCTGA-3′ |
This paper | N/A |
| RT-PCR primers for Tert nuclear DNA | West et al., 2015 | N/A |
| RT-PCR primers for mtDNA D loop | West et al., 2015 | N/A |
| Recombinant DNA | ||
| MSCV-IRES-Thy1.1 | Addgene | Plasmid ID: 17442 |
| Software and Algorithms | ||
| FlowJo v10 | FlowJo, LLC | N/A |
| Prism 6 | GraphPad Software | N/A |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jason G. Cyster (Jason.cyster@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
CD45.1 congenic C57BL/6 (B6) male and female mice, 6–12 weeks old were from National Cancer Institute. Ch25h−/− mice were backcrossed onto C57BL/6J for 10 generations. Nr1h2−/−, Nr1h3−/−, Nlrc4−/−, Aim2−/−, Casp11−/−, Casp1/11−/−, Asc−/− and Ifnar1−/− mice have all been previously described. Animals were housed in a specific pathogen-free environment in the Laboratory Animal Research Center at UCSF and all experiments conformed to the ethical principles and guidelines approved by the UCSF Institutional and Animal Care and Use Committee.
Cells
Bone marrow cells were grown in DMEM with 10% FBS and 10% M-CSF media in bacterial petri dishes for 6 days, then plated at 1×106 cells/ml and left to adhere overnight. BMDMs were stimulated with 100ng/ml LPS (E. coli 0111:B4; Sigma) for the indicated times. For ELISA detection of IL-1β in response to MCD-Chol (Sigma), BMDMs were primed with 100ng/ml LPS for 4 hr, and then MCD-Chol (100ug/ml unless indicated) was added for the final 4 hr of culture. For other inflammasome activation experiments, ATP (Sigma) was added for the final 45 minutes of culture at a concentration of 5mM, alum (400mg/ml; Pierce) and poly(dA:dT) (1ug/ml; Invivogen) was added for the final 4 hr of culture, and FlaTox (1:100 dilution) was added for the final 45 minutes of culture. For two sets of experiments, rapamycin (20nM) or cytochalasin D (2uM; Sigma) was also added for the final 4 hr of culture.
METHOD DETAILS
Bone marrow chimeras
CD45.1 congenic mice were lethally irradiated with 1,100 rad in split doses 3 hr apart, and reconstituted with 1–3 × 106 BM cells per mouse from wild type, Aim2−/−, Nlrp3−/−, or Casp1/11−/−, Ch25h+/−, Ch25h−/−, Ch25h+/−Aim2−/−, or Ch25h−/−Aim2−/− mice. Mice were analyzed 8–10 weeks later.
BMDM transduction
BM cells were cultured in DMEM with 10% FBS and 10% M-CSF conditioned medium for 2 days, then spinfected for 2 hr on days 2 and 3 with retroviruses expressing Ch25h, Ch25hmut, Hmgcr, Dhcr24, or control and an IRES-thy1.1 cassette as a reporter. Cells were then plated at 1×106 cells/ml and left to adhere overnight on day 6. Cells were always cultured in 10% M-CSF conditioned medium.
Listeria monocytogenes infection of BMDMs
L. monocytogenes single colonies were incubated in brain heart infusion (BHI) broth overnight at 30°C without shaking. Cultures were then diluted 1:10 in BHI broth and incubated at 37°C with shaking. When cultures were in log-phase growth, bacteria were added to BMDMs seeded on TC-treated plates at a multiplicity of infection (MOI) of 10 to 1. After 30 minutes infected cells were washed, and media containing gentamicin (50ug/ml) was added. For CFU determination, cells were lysed in cold ddH2O and lysates were streaked onto BHI agar plates.
Flow cytometry
Cells were stained with Abs to CD4 (RM4-5), TCRβ (H57-597), IL-17A (17B7), CD11b (M1/70), or Ly6G (1A8). For mitochondrial studies, cells were treated as indicated and then stained with 30 minutes with Mitotracker Deep Red (100nM), Mitotracker Green (100nM), TMRM (100nM) or MitoSOX (2.5 uM) at 37°C. All mitochondrial staining reagents were from Life Technologies. For intracellular cytochrome C staining, cells were stimulated as indicated and then treated on ice with either control or digitonin (25 ug/ml) for 10 minutes. Cells were then treated with BM Cytofix Buffer and Perm/Wash reagent (BD Biosciences), and stained with anti-cytochrome C (6H2.B4). Antibodies were purchased from BD Biosciences or eBiosciences.
IL-1β T cell bioassay
Peripheral lymph nodes (LNs) and spleens from wild type mice were harvested and mashed through a 70 micron strainer. CD4+ T cells were enriched by positive selection using CD4+ MACS beads (Miltenyi). T cells (5 × 105) were then cultured for 4 days in 24-well plates with plate-bound anti-CD3 (2 ug/mL), soluble anti-CD28 (1 ug/mL), and recombinant TGF-β (2 ng/mL) and the indicated BMDM supernatants. On day 4, T cells were stimulated with PMA/iononmycin and brefeldin A (BD Biosciences) for 4 hr. Cells were stained with fixable viability dye (eFluor780; eBiosciences) to exclude dead cells, surface stained with Abs to CD4 (RM4-5) and TCRβ (H57-597), treated with BM Cytofix Buffer and Perm/Wash reagent (BD Biosciences), and stained with anti-IL-17A (17B7). Antibodies were purchased from BD Biosciences or eBiosciences.
Cytokine enzyme-linked immunosorbent assays
Supernatants from cultured BMDMs and serum from LPS-injected mice were collected from the indicated conditions and times points. Unconjugated anti-IL-1β (B122; Biolegend) (2ug/ml) was coated onto 96-well high binding plates and then plates were washed and blocked for 1 hour with PBS containing 5% FBS. Supernatants were added to the plates O/N at 4°C. Plates were washed again and biotinylated anti-IL-1β (polyclonal; eBiosciences) (1ug/ml) was added for 1–2 hr at RT. Biotinylated detection antibodies were then labeled by streptavidin-conjugated horseradish peroxidase (HRP) and visualized by addition of Substrate Reagent Pack (R&D). Color development was stopped by addition of 2N H2SO4. Recombinant IL-1β (eBiosciences) was used as a standard. Absorbance at 450 nm was measured on a tunable microplate reader (VersaMax, Molecular Devices). Cytokine supernatant concentrations were calculated by extrapolating absorbance values from standard curves where known concentrations were plotted against absorbance using SoftMax Pro 5 software.
In vivo peritonitis
For neutrophil recruitment into the peritoneum, mice were injected I.P. with saline containing either LPS (10ug), LPS(10ug)+MCD-Chol(100ug), or LPS+Alum(400ug). 8–10 hr after injection, mice were sacrificed, peritoneal cavities were lavaged using 5 ml of PBS with 1% FBS and 1mM HEPES, and peritoneal cells were analyzed using FACS. For mitochondrial cholesterol measurements and Mitotracker staining on peritoneal macrophages, mice were injected I.P. with saline or LPS (2mg/kg) for 8 hr.
Quantification of sterols
BMDMs were treated as indicated, sterols were extracted with organic solvents, and quantitated using a SCIEX API 5000 mass spectrometer coupled to a Shimadzu LC-20XR high performance liquid chromatograph as previously described(McDonald et al., 2012).
RNA isolation and Real-time RT-PCR
Total RNA was isolated from 5×105 BMDMs treated as indicated with the Trizol reagent (Life Technology) following the manufacturer’s protocol. Real-time PCR was performed using SYBR Green PCR Mix (Roche) and an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA). Hprt levels were used as internal controls. Primer sequences are available upon request.
Seahorse analysis of mitochondrial respiration
For real-time analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), BMDMs were analyzed with an XF-24 Extracellular Flux Analyzer (Seahorse Bioscience). 2×105 BMDMs were plated in Seahorse 24-well plates and the Seahorse XF Cell Mitochondria Stress assay was performed per manufacturer’s instructions. Oligomycin was added at 1uM, FCCP at 1uM, and Rotenone/Antimycin A at 0.5uM each (Seahorse Biosciences).
293T inflammasome reconstitution
1.5×105 293T cells were seeded in 24-well plates O/N in DMEM with 10% FBS and antibiotics. The next day, cells were switched to antibiotic-free DMEM with 10% FBS and transfected using Lipofectamine 2000 (Life Technologies) with the indicated plasmids (all 50ng). 24 hr later, cells were stimulated with nigericin (15uM; Sigma) for 45 minutes. Supernatants were then collected, and cells were lysed in RIPA buffer (25mM Tris-HCl pH 7.5, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100). Western blots were performed with rabbit polyconal anti-caspase-1 p10 (M-20, Santa Cruz), mouse monoclonal anti-NLRP3 (Cryo-2, Adipogen), rabbit polyclonal anti-ASC (AL177, Adipogen), and goat anti-IL-1β (polyclonal; R&D).
Cytosolic and total DNA extraction
2×106 BMDMs per well seeded in 6-well plates were stimulated as indicated, washed with ice cold PBS, and scraped off the plates. Cells were then pelleted in a table top centrifuge by spinning at 800g for 5 minutes at 4°C. Cell pellets were resuspended in 500ul of cytosolic extraction buffer (150mM NaCl, 50mM HEPES pH 7.4, 25 ug/ml digitonin) and rotated for 10 minutes at 4°C. Cells were then centrifuged at 800g for 3 minutes. Supernatants were collected and centrifuged at 800g for 3 minutes twice more, followed by a 5 minute centrifugation at 10,000g. DNA was then extracted from the cell pellets and cytosolic fraction using a QIAamp DNA Micro Kit (Qiagen). DNA was quantified using qPCR with mtDNA (D loop) and nucDNA (Tert) primers; cytosolic DNA signals were normalized to cell pellet DNA signals. Primer sequence are available upon request.
Mitochondrial DNA depletion
BMDMs were grown as described above. On day 6, media was replaced with DMEM containing 10% FBS and 10% M-CSF conditioned media containing either ethidium bromide (100ng/ml) or 2′,3′-dideoxycytidine (40ug/ml). After 5–7 days, BMDMs were detached and mitochondrial DNA depletion was assessed by total DNA extraction followed by qPCR for mtDNA and nucDNA as described above.
Amplex Red Cholesterol Quantification
For total cholesterol content, BMDMs were seeded in 6-well plates at 2×106 cells per well and stimulated as described. Peritoneal macrophages were sorted using a FACS Aria II (BD Biosciences). Cells were then lysed in 500 ul RIPA buffer. Cholesterol quantification was then performed using the Amplex Red Cholesterol Assay (Life Technologies) per manufacturer’s instructions and readout on a Qubit 3.0 Fluorometer (Life Technologies). For mitochondrial cholesterol content, BMDMs were seeded in 10cm dishes at 1×107 cells per dish and treated as indicated. Mitochondria were then isolated using anti-TOM22 MACS beads with the Mouse Mitochondria Isolation Kit (Miltenyi Biotec) per manufacturer’s instructions. Isolated mitochondria were then lysed in 200ul RIPA buffer. Amplex Red fluorescent concentrations were normalized based on total protein content using the BCA Protein Assay Kit (Pierce).
Confocal microscopy
BMDMs (1×105) were seeded on round glass coverslips in 24-well plates overnight and then stimulated with LPS for 8 hr along with 150X FLICA reagent (ImmunoChemistry Technologies). Cells were then fixed with 1% PFA for 20min at 37°C and washed 3x with 1X PBS and stained with DAPI. Images were then taken on a Leica SP5 laser-scanning confocal microscope using a 63X oil immersion lense.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed in Graphpad Prism version 6, as defined throughout the text and figure legends.
DATA AND SOFTWARE AVAILABLITY
The data presented in this manuscript are tabulated in the main paper and in the STAR Methods. The primer sequences used for quantitative RT-PCR are available upon request.
Supplementary Material
(A) RT-qPCR analysis of Ch25h mRNA in Ch25h+/−, Ch25h−/−, or Ifnar1−/− BMDMs infected with L. monocytogenes (MOI = 10:1) for 0, 4, or 8 hr. Data are pooled from 3 independent experiments (mean+/−SD). (B) RT-qPCR analysis of Hmgcs1 and Sqle mRNA in Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. Data are pooled from 3 independent experiments (mean+/−SD). (C) Quantification of cholesterol by Amplex Red fluorescence in sorted peritoneal macrophages from mice injected intraperitoneally with saline or LPS for 8 hr (n=3 mice per group, mean+/−SD). (D) Immunoblotting for pS6 (S240/244) and β-tubulin in Ch25h+/− and Ch25h−/− BMDMs treated for 4 hr with LPS followed by 4 hr of LPS alone, LPS+Rapa (20nM), or LPS+Rapa (100nM). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test).
(A) RT-qPCR analysis of Sqle mRNA in Ch25h−/− BMDMs transduced with MSCV-vector, MSCV-Ch25h, or MSCV-Ch25hmut (mean+/-SD). (B) IL-1β ELISA from supernatants LXRα/β double-deficient BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut (mean+/−SD).
(A) Immunoblot for IL-1β on cell pellets and trichloroacetic acid (TCA)-precipitated supernatants from Ch25h+/− and Ch25h−/− BMDMs treated with LPS alone, LPS+ATP, or LPS+ATP+MitoTEMPO. Data are representative of two independent experiments. (B) RT-qPCR analysis of Ldlr (Left), Hmgcr (Middle), or Fasn (Right) from 293T cells transfected with either MSCV-thy1.1, MSCV-Srebp1a, or MSCV-Srebp2 (mean+/−SD). (C) IL-1β ELISA of supernatants from 293T cells co-transfected with NLRP3, ASC, Caspase-1, pro-IL-1β, and either vector, SREBP1a, or SREBP2 (all 50ng) and 24 hr later treated with 15uM nigercin for 45 min. Data are pooled from 3 independent experiments (mean+/-SD). (D) Immunoblotting for NLRP3, ASC, Caspase-1, pro-IL-1β, and IL-1β p17 on cell lysates from 293T cells prepared and treated as in C. *P<0.05, **P<0.01, ***P < 0.005 (one way ANOVA with bonferroni test).
(A, B) MFI of Mitotracker Deep Red (A) and Mitotracker Green (B) staining on Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. (C, D) MFI of Mitotracker Deep Red (C) and Mitotracker Green (D) staining on Asc+/+ or Asc−/− BMDMs treated for 4 hr with LPS, followed by 4 hr of MCD-Chol (10ug/ml). Data are pooled from 3 independent experiments (mean+/−SD). (E, F) MFI of Mitotracker Deep Red (E) or Mitotracker Green (F) staining on BMDMs transduced with MSCV-vector, -Hmgcr, or -Dhcr24 and stimulated with LPS for 8 hr. Data are pooled from two independent experiments (mean+/−SD). (G) MFI of Mitotracker Deep Red staining on Ch25h+/− and Ch25h−/− BMDMs stimulated for 8 hr with LPS, LPS+HDL (100ug/ml), or LPS+GW3965 (2.5uM). Data are pooled from two independent experiments (mean+/−SD). (H) MFI of TMRM staining on Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. (I) Seahorse analysis of oxygen consumption rate (OCR) from BMDMs treated for 4 hr with LPS followed by 4 hr of MCD-Chol. Data are representative of 3 independent experiments (mean+/−SD). (J) Representative histograms of Mitotracker Deep Red (Top) and Mitotracker Green (Bottom) staining of Ch25h+/− and Ch25h−/− peritoneal macrophages 8 hr after intraperitoneal saline or LPS injection. Data are representative of three independent experiments.
(A) Immunoblot for Tomm20 (top), β-tubulin (middle), and calreticulin (bottom) on whole cell extracts (WCE) or mitochondria immunoprecipitated using anti-Tomm22 MACS beads. (B) Quantification of cholesterol by Amplex Red fluorescence in mitochondria isolated with anti-Tomm22 MACS beads from wild type BMDMs stimulated with LPS alone or 4 hr of LPS followed by 4hr of MCD-Chol. (C) mtDNA (left) or nucDNA (right) RT-qPCR of total DNA from cytosolic extracts derived from BMDMs treated with either LPS alone, or 4 hr of LPS followed by 4 hr of MCD-Chol. Data are pooled from 4 independent experiments (mean+/−SD). (D) Quantification of cholesterol by Amplex Red fluorescence in mitochondria isolated with anti-Tomm22 MACS beads from Ch25h−/− BMDMs stimulated with LPS alone or 4 hr of LPS followed by 4 hr of rapamycin (20nM). (E) mtDNA RT-qPCR of total DNA from cytosolic extracts derived from BMDMs treated with either LPS alone, or 4 hr of LPS followed by 4 hr of rapamycin (20nM). (F) RT-qPCR analysis of Rsad (Left) and Ch25h (Right) transcripts in BMDMs treated for 4 hr with either nil, LPS, MCD-Chol or LPS+MCD-Chol (mean+/−SD). (G, H) IL-1β ELISA from supernatants of BMDMs of the indicated genotypes stimulated with either LPS alone, LPS+FlaTOX (G) or LPS+poly(dA:dT) (H). Data are pooled from three independent experiments (mean+/−SD). (I) Representative images (left) and quantification (right) of confocal microscopy on BMDMs of the indicated genotypes incubated for 8hr with LPS and FLICA and subsequently stained with DAPI. Data are pooled from 4 technical replicates of two independent experiments (mean+/−SD). At least 600 cells were counted for each replicate. *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test).
Highlights.
Ch25h upregulation in activated macrophages restricts cholesterol biosynthesis
High cholesterol synthesis leads to AIM2-dependent inflammasome activation
Elevated cholesterol causes impaired mitochondrial metabolism and mtDNA release
25-HC maintains mitochondrial integrity and prevents AIM2 inflammasome activation
Acknowledgments
We thank J. An and Y. Xu for expert technical assistance, A. Ma for Casp1/11−/−, Asc−/−, and Nlrp3−/− bone marrow, T. DeFranco for Nlrc4−/− bone marrow, K. Fitzgerald for Nlrp3−/−Aim2−/− bone marrow, R. Vance for FlaTox, C. Paillart for help with Seahorse setup, and J. Cox, R. Locksley, A. Reboldi, E. Wolf, and J. Wu for helpful discussions. Confocal imaging was supported by UCSF DRC Center grant P30 DK063720. EVD is supported by NIH F30 grant F30AI120527 and the UCSF Medical Scientist Training Program (MSTP) and JGC is an Investigator of the Howard Hughes Medical Institute. This work was supported in part by NIH grants AI040098 (to JGC) and HL20948 (to JGM and DWR).
Footnotes
Author contributions
J.G.C. and E.V.D. conceived and designed the study. J.G.C. and E.V.D. designed and oversaw all experiments. E.V.D. performed the experiments. J.G.M. and D.W.R. performed the mass spectrometry experiments. J.G.C. and E.V.D. analyzed the data. J.G.C. and E.V.D. wrote the manuscript. J.G.M. and D.W.R. reviewed and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, Fredericks AC, Tripathi S, Zhu T, Pintado-Silva J, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nature Microbiology. 2017;2:17037. doi: 10.1038/nmicrobiol.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, et al. The Transcription Factor STAT-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity. 2013;38:106–118. doi: 10.1016/j.immuni.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Marí M, Herms A, Fernández A, Fajardo A, Kassan A, Giralt A, Colell A, Balgoma D, Barbero E, et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr Biol. 2011;21:681–686. doi: 10.1016/j.cub.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CBL, Paim BA, Cosso RG, Castilho RF, Rottenberg H, Vercesi AE. Method for monitoring of mitochondrial cytochrome C release during cell death: Immunodetection of Cytochrome C by flow cytometry after selective permeabilization of the plasma membrane. Cytometry. 2006;69:515–523. doi: 10.1002/cyto.a.20273. [DOI] [PubMed] [Google Scholar]
- Chen CH, Cheng YC. Delayed cytotoxicity and selective loss of mitochondrial DNA in cells treated with the anti-human immunodeficiency virus compound 2”,3-“dideoxycytidine. J Biol Chem. 1989;264:11934–11937. [PubMed] [Google Scholar]
- Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nature Reviews Immunology. 2014;14:731–743. doi: 10.1038/nri3755. [DOI] [PubMed] [Google Scholar]
- de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Letters. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Mangan MS, Latz E. Crystal Formation in Inflammation. Ann Rev Immuno. 2016;34:173–202. doi: 10.1146/annurev-immunol-041015-055539. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, Farlik M, Decker T, Pasquier Du RA, Romero P, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell. 131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BE, Madreiter CT, Vishnu N, Malli R, Graier WF, Karten B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J Biol Chem. 2014;289:16278–16289. doi: 10.1074/jbc.M114.559914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity. 2013;38:92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Martin LA, Kennedy BE, Karten B. Mitochondrial cholesterol: mechanisms of import and effects on mitochondrial function. J Bioenerg Biomembr. 2016;48:137–151. doi: 10.1007/s10863-014-9592-6. [DOI] [PubMed] [Google Scholar]
- McDonald JG, Russell DW. Editorial: 25-Hydroxycholesterol: a new life in immunology. Journal of Leukocyte Biology. 2010;88:1071–1072. doi: 10.1189/jlb.0710418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherall JE, Ridgway ND, Dawson PA, Goldstein JL, Brown MS. A 25-hydroxycholesterol-resistant cell line deficient in acyl-CoA: cholesterol acyltransferase. J Biol Chem. 1991;266:12734–12740. [PubMed] [Google Scholar]
- Montero J, Marí M, Colell A, Morales A, Basañez G, García-Ruiz C, Fernandez-Checa JC. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim Biophys Acta. 2010;1797:1217–1224. doi: 10.1016/j.bbabio.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell. 2016;165:792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. 25-Hydroxycholesterol suppresses interleukin-1–driven inflammation downstream of type I interferon. Science. 2014;345:679–684. doi: 10.1126/science.1254790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KA, Hsieh WY, Forster T, Blanc M, Lu H, Crick PJ, Yutuc E, Watterson S, Martin K, Griffiths SJ, et al. An Interferon Regulated MicroRNA Provides Broad Cell-Intrinsic Antiviral Immunity through Multihit Host-Directed Targeting of the Sterol Pathway. PLOS Biology. 2016;14:e1002364. doi: 10.1371/journal.pbio.1002364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LE, Shridar M, Smith P, Murrell-Lagnado RD. Plasma membrane cholesterol as a regulator of human and rodent P2X7 receptor activation and sensitization. J Biol Chem. 2014;289:31983–31994. doi: 10.1074/jbc.M114.574699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CCD, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USa. 2011;108:15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Sundstrom KB, Chew JJ, Bist P, Gan ES, Tan HC, Goh KC, Chawla T, Tang CK, Ooi EE. Dengue virus activates cGAS through release of mitochondrial DNA. Scientific Reports. 2017;7:3594. doi: 10.1038/s41598-017-03932-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunology. 2010;5:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USa. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;475:122–122. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) RT-qPCR analysis of Ch25h mRNA in Ch25h+/−, Ch25h−/−, or Ifnar1−/− BMDMs infected with L. monocytogenes (MOI = 10:1) for 0, 4, or 8 hr. Data are pooled from 3 independent experiments (mean+/−SD). (B) RT-qPCR analysis of Hmgcs1 and Sqle mRNA in Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. Data are pooled from 3 independent experiments (mean+/−SD). (C) Quantification of cholesterol by Amplex Red fluorescence in sorted peritoneal macrophages from mice injected intraperitoneally with saline or LPS for 8 hr (n=3 mice per group, mean+/−SD). (D) Immunoblotting for pS6 (S240/244) and β-tubulin in Ch25h+/− and Ch25h−/− BMDMs treated for 4 hr with LPS followed by 4 hr of LPS alone, LPS+Rapa (20nM), or LPS+Rapa (100nM). *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test).
(A) RT-qPCR analysis of Sqle mRNA in Ch25h−/− BMDMs transduced with MSCV-vector, MSCV-Ch25h, or MSCV-Ch25hmut (mean+/-SD). (B) IL-1β ELISA from supernatants LXRα/β double-deficient BMDMs transduced with MSCV-vector, −Ch25h, or −Ch25hmut (mean+/−SD).
(A) Immunoblot for IL-1β on cell pellets and trichloroacetic acid (TCA)-precipitated supernatants from Ch25h+/− and Ch25h−/− BMDMs treated with LPS alone, LPS+ATP, or LPS+ATP+MitoTEMPO. Data are representative of two independent experiments. (B) RT-qPCR analysis of Ldlr (Left), Hmgcr (Middle), or Fasn (Right) from 293T cells transfected with either MSCV-thy1.1, MSCV-Srebp1a, or MSCV-Srebp2 (mean+/−SD). (C) IL-1β ELISA of supernatants from 293T cells co-transfected with NLRP3, ASC, Caspase-1, pro-IL-1β, and either vector, SREBP1a, or SREBP2 (all 50ng) and 24 hr later treated with 15uM nigercin for 45 min. Data are pooled from 3 independent experiments (mean+/-SD). (D) Immunoblotting for NLRP3, ASC, Caspase-1, pro-IL-1β, and IL-1β p17 on cell lysates from 293T cells prepared and treated as in C. *P<0.05, **P<0.01, ***P < 0.005 (one way ANOVA with bonferroni test).
(A, B) MFI of Mitotracker Deep Red (A) and Mitotracker Green (B) staining on Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. (C, D) MFI of Mitotracker Deep Red (C) and Mitotracker Green (D) staining on Asc+/+ or Asc−/− BMDMs treated for 4 hr with LPS, followed by 4 hr of MCD-Chol (10ug/ml). Data are pooled from 3 independent experiments (mean+/−SD). (E, F) MFI of Mitotracker Deep Red (E) or Mitotracker Green (F) staining on BMDMs transduced with MSCV-vector, -Hmgcr, or -Dhcr24 and stimulated with LPS for 8 hr. Data are pooled from two independent experiments (mean+/−SD). (G) MFI of Mitotracker Deep Red staining on Ch25h+/− and Ch25h−/− BMDMs stimulated for 8 hr with LPS, LPS+HDL (100ug/ml), or LPS+GW3965 (2.5uM). Data are pooled from two independent experiments (mean+/−SD). (H) MFI of TMRM staining on Ch25h+/− and Ch25h−/− BMDMs infected with L. monocytogenes for 8 hr. (I) Seahorse analysis of oxygen consumption rate (OCR) from BMDMs treated for 4 hr with LPS followed by 4 hr of MCD-Chol. Data are representative of 3 independent experiments (mean+/−SD). (J) Representative histograms of Mitotracker Deep Red (Top) and Mitotracker Green (Bottom) staining of Ch25h+/− and Ch25h−/− peritoneal macrophages 8 hr after intraperitoneal saline or LPS injection. Data are representative of three independent experiments.
(A) Immunoblot for Tomm20 (top), β-tubulin (middle), and calreticulin (bottom) on whole cell extracts (WCE) or mitochondria immunoprecipitated using anti-Tomm22 MACS beads. (B) Quantification of cholesterol by Amplex Red fluorescence in mitochondria isolated with anti-Tomm22 MACS beads from wild type BMDMs stimulated with LPS alone or 4 hr of LPS followed by 4hr of MCD-Chol. (C) mtDNA (left) or nucDNA (right) RT-qPCR of total DNA from cytosolic extracts derived from BMDMs treated with either LPS alone, or 4 hr of LPS followed by 4 hr of MCD-Chol. Data are pooled from 4 independent experiments (mean+/−SD). (D) Quantification of cholesterol by Amplex Red fluorescence in mitochondria isolated with anti-Tomm22 MACS beads from Ch25h−/− BMDMs stimulated with LPS alone or 4 hr of LPS followed by 4 hr of rapamycin (20nM). (E) mtDNA RT-qPCR of total DNA from cytosolic extracts derived from BMDMs treated with either LPS alone, or 4 hr of LPS followed by 4 hr of rapamycin (20nM). (F) RT-qPCR analysis of Rsad (Left) and Ch25h (Right) transcripts in BMDMs treated for 4 hr with either nil, LPS, MCD-Chol or LPS+MCD-Chol (mean+/−SD). (G, H) IL-1β ELISA from supernatants of BMDMs of the indicated genotypes stimulated with either LPS alone, LPS+FlaTOX (G) or LPS+poly(dA:dT) (H). Data are pooled from three independent experiments (mean+/−SD). (I) Representative images (left) and quantification (right) of confocal microscopy on BMDMs of the indicated genotypes incubated for 8hr with LPS and FLICA and subsequently stained with DAPI. Data are pooled from 4 technical replicates of two independent experiments (mean+/−SD). At least 600 cells were counted for each replicate. *P<0.05, **P<0.01, ***P < 0.005 (unpaired Students t test or one way ANOVA with bonferroni test).