

Abstract
Neuron-secreted factors induce astrocytic expression of the glutamate transporter, GLT-1 (EAAT2). In addition to their elaborate anatomic relationships with neurons, astrocytes also have processes that extend to and envelop the vasculature. Although previous studies have demonstrated that brain endothelia contribute to astrocyte differentiation and maturation, the effects of brain endothelia on astrocytic expression of GLT-1 have not been examined. In the present study, we tested the hypothesis that endothelia induce expression of GLT-1 by co-culturing astrocytes from mice that utilize non-coding elements of the GLT-1 gene to control expression reporter proteins with the mouse endothelial cell line, bEND.3. We found that endothelia increased steady state levels of reporter and GLT-1 mRNA/protein. Co-culturing with primary rat brain endothelia also increases reporter protein, GLT-1 protein, and GLT-1-mediated glutamate uptake. The Janus kinase/signal transducer and activator of transcription 3, bone morphogenic protein/transforming growth factor β, and nitric oxide pathways have been implicated in endothelia-to-astrocyte signaling; we provide multiple lines of evidence that none of these pathways mediate the effects of endothelia on astrocytic GLT-1 expression. Using transwells with a semi-permeable membrane, we demonstrate that the effects of the bEND.3 cell line are dependent upon contact. Notch has also been implicated in endothelia-astrocyte signaling in vitro and in vivo. The first step of Notch signaling requires cleavage of Notch intracellular domain (NICD) by γ-secretase. We demonstrate that the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) blocks endothelia-induced increases in GLT-1. We show that the levels of NICD are higher in nuclei of astrocytes co-cultured with endothelia, an effect also blocked by DAPT. Finally, infection of co-cultures with shRNA directed against recombination signal binding protein for immunoglobulin kappa J (RBPJ), a Notch effector, also reduces endothelia-dependent increases in eGFP and GLT-1. Together, these studies support a novel role for Notch in endothelia-dependent induction of GLT-1 expression.
Keywords: Astrocytes, GLT-1, blood-brain barrier, endothelial cells, Notch, transcription
Graphical Abstract
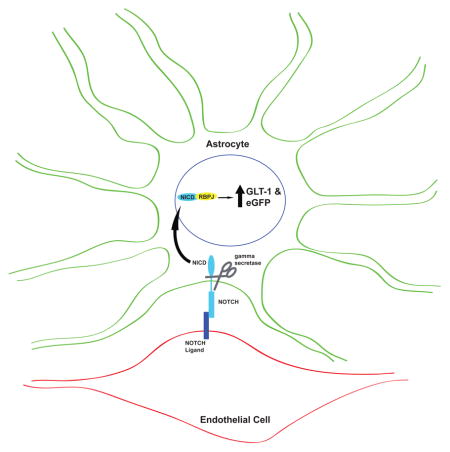
Signaling from endothelial cells has been shown to affect astrocyte specification and maturation. We show that co-culturing astrocytes with endothelial cells increases expression of the glial glutamate transporter GLT-1 as well as expression of a transcriptional reporter in a contact-dependent mechanism. This increase is dependent on Notch signaling, as inhibition of the Notch pathway by treatment with γ-secretase inhibitor or knock-down of RBPJ prevents the endothelia-induced increase in GLT-1.
Introduction
Glutamate, the main excitatory neurotransmitter in the nervous system, executes its functions by activation of ionotropic and metabotropic glutamate receptors (Zhou & Danbolt 2014). Clearance of extracellular glutamate is crucial, as over-activation of glutamate receptors induces neuronal death by a process known as ‘excitotoxicity’ (Roberts & Davies 1987; Frandsen et al. 1989). Glutamate clearance is carried out by a family of five Na+-dependent transporters: glutamate transporter 1 (GLT-1, also called EAAT2), glutamate/aspartate transporter (GLAST, also known as EAAT1), excitatory amino acid carrier 1 (EAAC1, also called EAAT3), and excitatory amino acid transporters 4 and 5 (EAAT4 and EAAT5) (for reviews, see Danbolt 2001; Vandenberg & Ryan 2013; Martinez-Lozada et al. 2016). GLT-1 contributes up to 90% of total glutamate clearance in the forebrain (for reviews, see Robinson 1998; Danbolt 2001).
Decreased expression of GLT-1 is observed in animal models and in post-mortem samples from patients with many different neurologic disorders, including amyotrophic lateral sclerosis, Alzheimer’s disease, Huntington’s disease, and epilepsy (for reviews, see Sheldon & Robinson 2007; Fontana 2015; Miladinovic et al. 2015). This led to the suggestion that drugs targeting GLT-1 up-regulation might have a therapeutic benefit (Dunlop 2006; Sheldon & Robinson 2007; Fontana 2015; Takahashi et al. 2015). In fact, Rothstein and colleagues identified ceftriaxone in a screen of molecules that increase GLT-1 expression and demonstrated that it delays the onset of neurologic impairment in an animal model of amyotrophic lateral sclerosis (ALS) (Rothstein et al. 2005). Ceftriaxone has subsequently been tested in several other animal models of neurologic disease and injury, including hypoxia-ischemia and stroke (Chu et al. 2007; Lipski et al. 2007; Thone-Reineke et al. 2008; Lai et al. 2011; Inui et al. 2013; Hu et al. 2015), multiple sclerosis (Melzer et al. 2008), seizure (Jelenkovic et al. 2008), and alcohol abuse (Alhaddad et al. 2014; for review, see Fontana 2015). Additionally, the pyridazine-based compound LDN/OSU-0212320 was identified in a high-throughput screen of EAAT2 translational activators (Colton et al. 2010). This small molecule delays motor function decline in a mouse model of ALS and prevents excitotoxic neuronal death after pilocarpine-induced status epilepticus (Kong et al. 2014). These reports clearly demonstrate that understanding the endogenous mechanisms that control GLT-1 expression is essential for appropriately targeting GLT-1 regulation.
GLT-1 is enriched in astrocytes, where its expression increases dramatically during synapse formation in both rodents and in humans (Rothstein et al. 1994; Danbolt 1994; Chaudhry et al. 1995; Furuta et al. 1997; Regan et al. 2007). Almost twenty years ago three different groups demonstrated that co-culturing astrocytes with neurons increases GLT-1 expression in astrocytes (Gegelashvili et al. 1997; Schlag et al. 1998; Swanson et al. 1997). This effect of neurons is mimicked by transferring media from cortical neuronal cultures (Gegelashvili et al. 1997) or by separating neurons and astrocytes in a transwell configuration (Schlag et al. 1998), suggesting that secreted factors contribute to this effect. Although membranes isolated from neurons were not sufficient to induce expression of GLT-1 in original studies (Schlag et al. 1998), more recently Yang et al., demonstrated that neuron membrane fractions also induce expression of GLT-1 in astrocytes (Yang et al. 2009).
In addition to having processes that surround synapses, astrocytes have specialized endfeet that contact blood vessels (for reviews, see Bushong et al. 2004; Abbott et al. 2006; Cheslow & Alvarez 2016). While astrocyte differentiation coincides with and induces synaptogenesis (Ullian et al. 2001), astrocyte migration and development also correlate with angiogenesis temporally and spatially (Caley & Maxwell 1970; Marin-Padilla 1995; Zerlin & Goldman 1997). It has been shown that astrocytes affect blood vessel growth and blood brain barrier formation and stability (Igarashi et al. 1999; Lee et al. 2003; Gerhardt et al. 2004; Ma et al. 2013 for reviews, see Abbott et al. 2006; Alvarez et al. 2013; Obermeier et al. 2013; Cheslow & Alvarez 2016), and several groups have demonstrated that brain endothelial cells induce the expression of several astrocytic proteins (Covacu et al. 2006; Imura et al. 2008; Mi et al. 2001; Zerlin & Goldman 1997), but none of these studies have examined the effects of endothelia on expression of GLT-1.
In the present study, we provide the first demonstration that endothelia increase astrocytic expression of GLT-1. We show that this effect is dependent upon contact and is blocked by an inhibitor of γ-secretase as well as knock-down of the transcription factor RBPJ.
Materials and Methods
Animals
A colony of transgenic 8.3 tdTomato /BAC eGFP GLT-1 (“dual reporter”) mice, which are homozygous for both traits, were maintained at the animal facility of the Children’s Hospital of Philadelphia (Regan et al. 2007; Yang et al. 2011b). For studies involving astrocytes or endothelial cells prepared from rats, the Sprague-Dawley strain (RRID:RGD_734476, Charles River Laboratories, Wilmington, MA) was used as described below. All studies were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia and followed the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Animals were housed in standard controlled temperature, humidity, and light, and had ad libitum access to food and water.
Primary enriched astrocyte cultures
Mice of both sexes and 1–3 days of age were used to prepare astrocyte-enriched cultures as previously described (Li et al. 2006; Ghosh et al. 2016). Briefly, mouse brain cortices were dissociated by trypsin and trituration into a single-cell suspension and plated at a density of approximately 2.5 × 105 cells/mL in 75 cm2 flasks. The media was changed every 3–4 days. After 7–10 days (when cells were ~90% confluent), A2B5 hydridoma supernatant (1:50 dilution; from the laboratory of Dr. Judy Grinspan, Children’s Hospital of Philadelphia) and Low Tox-M rabbit complement (Cederlane, Burlington, Canada) were used to eliminate A2B5-positive oligodendrocyte precursors that express GLT-1 (Zelenaia et al. 2000). After 2–3 days of recovery, the astrocytes were split into 6- or 12-well culture plates for experiments. The wells were randomly assigned to different groups.
Culture of the bEND.3 endothelioma cell line
The mouse brain endothelioma cell line bEND.3 was purchased from American Type Culture Collection (cat. # CRL-2299, RRID:CVCL_0170; Manassas, VA). These cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L D-glucose (Thermo Fisher Gibco, Waltham, MA) and supplemented with 10% defined heat-inactivated fetal bovine serum (FBS) (GE Healthcare Hyclone, Marlborough, MA), 4 mM L-glutamine, and 1 mM sodium pyruvate at 37°C and 5% CO2. Cells were never used past passage 30 as per the vendor’s recommendation to ensure similar expression of endothelial markers across experiments. Two lots of bEND.3 cells were used during the course of these studies with no differences in effect noted.
Primary enriched rat brain endothelial cells (RBEC)
Primary endothelial cells were prepared from gray matter blood vessels from adolescent 3 week-old rats (both sexes) as previously described, with modifications (Takata et al. 2013; Welser-Alves et al. 2014). Rats were anesthetized with isofluorane and euthanized by rapid decapitation. The brains were harvested into ice-cold Hank’s buffered saline solution and the brainstem, cerebellum, and white matter were removed. The meninges were also removed and the remaining tissue was finely chopped with a sterile razor blade and incubated in 1 mg/mL collagenase and 100 μg/mL DNase (Roche, Indianapolis, IN) at 37°C for 1.5 hours, with occasional agitation. The tissue was triturated until a homogenous suspension was obtained and then centrifuged at 600 × g for 10 minutes. The enzyme-containing media was removed and replaced with 25% bovine serum albumin (BSA; Sigma, St. Louis, MO) in DMEM (Thermo Fisher Gibco), resuspended, and spun at 1475 × g for 10 minutes. The myelin-containing layer on top and the BSA were removed, the cells were washed in media, and then plated on culture dishes coated with 5 μg/cm2 (75 μg/mL) collagen (Corning, Corning, NY) and 1 μg/cm2 (15 μg/mL) fibronectin (Corning) in media containing 4 μg/mL puromycin (Thermo Fisher Gibco) to select for endothelial cells. This media was replaced with media without puromycin after 3 days. The cells were allowed to grow to confluence (~10–14 days) and then split onto 6- or 12-well plates for further studies. Media for RBEC was DMEM/F12 (Thermo Fisher Gibco) supplemented with 10% heat-inactivated FBS (GE Healthcare Hyclone), 1% penicillin/streptomycin (Thermo Fisher Gibco), and 30 mg of endothelial cell growth supplement from bovine neural tissue (Sigma).
Co-culture of astrocytes and endothelial cells
In experiments in which astrocytes and endothelial cells were cultured together directly, endothelia [either bEND.3 cells (ATCC) or primary rat brain endothelial cells] were split 1:3 (from a just-confluent plate) onto 6- or 12-well plates 2–3 days prior to the astrocytes being split (1:1.5, from a fully-confluent plate) directly on top of the endothelia. In transwell (Corning) experiments, bEND.3 cells were either split onto the top or onto the underside of polyester transwell inserts (4.67 cm2 of cell growth area, 0.4 μm pore size) and allowed to adhere for 2 hours, at which time the inserts were placed in 6-well plates. Astrocytes were split onto either the bottom of the 6-well plate (9.5 cm2 of cell growth area) or onto the upper side of the transwell inserts (see Figure 5a). Co-cultures were maintained for 10–14 days, with complete media changes every 3–4 days, after which the cultures were shaken in radioimmunoprecipitation assay (RIPA) buffer for 1.5 hours and scraped to harvest the protein for Western blot analysis.
Figure 5. Effects of astrocytes from dual reporter mice and bEND.3 co-cultured in direct contact or separated on eGFP protein levels.

Cortical astrocytes and bEND.3 were cultured in transwells as schematized in (a) for 10–14 days and then harvested for Western blot analysis of eGFP protein levels (b). Top: Representative blot and Bottom: summary of quantification, normalized to β-actin. Data are the mean ± SEM of three independent experiments. *** p ≤ 0.001 for indicated comparison.
Western blot analysis
Cell lysis and collection of protein were performed as described previously (Li et al. 2006; Ghosh et al. 2011; Ghosh et al. 2016). For Western blot analysis, 15–20 μg of protein were loaded in each lane and resolved on 10% or 12% SDS-polyacrylamide gels by electrophoresis. The protein was transferred onto polyvinylidine fluoride membranes (transblot apparatus, Bio-Rad Laboratories, Hercules, CA) and blocked for 30 minutes with 5% non-fat dry milk blocking buffer. Membranes were then probed with mouse anti-eGFP (clone N86/8, UC Davis/NIH NeuroMab Facility Cat# 75-414 RRID:AB_2532048; 1:1,000 dilution), rabbit anti-GLT-1 (J.D. Rothstein, Johns Hopkins University; Maryland; USA Cat# GLT-1a RRID:AB_2314565; C-terminal-directed; 1:5,000 dilution) (Rothstein et al. 1994), mouse anti-GLAST (clone ACSA-1, Miltenyi Biotec Cat# 130-095-822 RRID:AB_10829302; 1:200 dilution; Bergisch Gladbach, Germany), mouse anti-STAT3 (Cell Signaling Technology Cat# 9139 RRID:AB_331757; 1:1000 dilution, Danvers, MA), rabbit anti-pSTAT3 (Cell Signaling Technology Cat# 9145 RRID:AB_2491009; 1:1,000 dilution), rabbit anti-RBPJ (Cell Signaling Technology, Cat# 5313, RRID: AB_2665555; 1:1,000 dilution), mouse anti-β-actin (Cell Signaling Technology Cat# 3700, RRID:AB_2242334; 1:10,000 dilution), or a combination of these antibodies in 1% milk blocking buffer. After washing three times in 1% blocking buffer, the membranes were probed with secondary antibodies (either anti-mouse or anti-rabbit fluorescently conjugated antibodies, 1:10,000 dilution, LI-COR Biosystems, Lincoln, NE) for 45 minutes in 1% blocking buffer. The membranes were washed three more times and visualized using a LI-COR Odyssey Infrared Imaging System. All data generated from Western blots were normalized to total protein. As β-actin immunoreactivity was quantified and analyzed for all experiments and found to be unchanged in all cases (data not shown), data were also normalized to β-actin immunoreactivity to control for possible uneven transfer. In most cases, GLT-1 and GLAST were probed on the same immunoblots, and thus their corresponding actin immunoblots are the same and are duplicated in Figures 1b–e, 3b–c, and 6b–c.
Figure 1. Effects of co-culturing cortical astrocytes prepared from dual reporter mice or wild-type rat with bEND.3 cells on glutamate transporter protein and mRNA levels.

Astrocytes from dual reporter mice (a–c) or wild-type rat (d–e) were cultured alone or directly on top of an intact monolayer of bEND.3 cells for 10–14 days, and then were harvested for Western blot analysis of eGFP (a), GLT-1 (b) & (d), and GLAST (c) & (e) protein levels. Top: Representative blots and Bottom: summary of quantification, normalized to β-actin. The β-actin blots are identical in panels (b) and (c) and in panels (d) and (e), as GLT-1 and GLAST are quantified from the same immunoblot. Data are the mean ± SEM of six (dual reporter mice) or three (wild-type rat) independent experiments. mRNA was isolated from co-cultures of astrocytes from dual reporter mice with bEND.3 cells or from astrocytes alone and reverse transcribed. qPCR was performed for eGFP (f) and GLT-1 (g). Data were plotted on a standard curve and normalized to GAPDH. Data are the mean ± SEM of four independent experiments. * p ≤ 0.05, ** p ≤ 0.01, **** p ≤ 0.0001 for indicated comparisons.
Figure 3. Effects of co-culturing with bEND.3 cells or primary endothelial cells from rat forebrain on GLT-1 and GLAST expression and the effect of co-culture with RBEC on DHK-sensitive Na+-dependent uptake in cortical astrocytes from dual reporter mice.

Astrocytes were cultured directly on top of an intact monolayer of either bEND.3 cells or RBEC for 10–14 days and then harvested for Western blot analysis of eGFP (a), GLT-1 (b), and GLAST (c) protein levels. Top: Representative blots and Bottom: summary of quantification, normalized to β-actin. The β-actin blots are identical in panels (b) and (c), as GLT-1 and GLAST are quantified from the same immunoblot. Data are the mean ± SEM of four independent experiments. ** p ≤ 0.01 for indicated comparison. (d) Glutamate uptake (0.5 μM) was measured after 12–13 days in culture in the absence or presence of 300 μM dihydrokainic acid (DHK). (e) Data from (d) were analyzed to only show DHK-sensitive uptake. Data are the mean + SEM of four independent experiments. * p < 0.05 for indicated comparisons.
Figure 6. Effect of DAPT on eGFP, GLT-1, and GLAST protein levels and on Notch activation.

Cortical astrocytes from dual reporter mice were cultured alone or directly on top of an intact monolayer of bEND.3 cells and treated with 10μM DAPT for 10 days, and then harvested for Western blot analysis of eGFP (a), GLT-1 (b), and GLAST (c) protein levels. Top: Representative blots and Bottom: summary of quantification, normalized to β-actin. The β-actin blots are identical in panels (b) and (c), as GLT-1 and GLAST are quantified in the same immunoblot. Data are the mean ± SEM of three independent experiments. **** p ≤ 0.0001, * p ≤ 0.05 for indicated comparisons. (d) Expression of NICD was examined in cortical astrocytes from dual reporter mice, in astrocytes co-cultured with bEND.3 cells for 24 hours, and in co-cultures treated with 10μM DAPT. An anti-GFAP antibody was used as a marker of astrocytes, an anti-CD31 antibody was used as a marker of endothelia, and DAPI staining was used to identify nuclei. The magnification of the first two columns are the same (40x with 3x optical zoom, scale bar 25μm) and the last column is an 8x optical zoom of the region outlined by the white box in the middle column (scale bar 10μm). Representative of 3 independent experiments.
Immunocytochemistry
Primary dual reporter astrocytes or bEND.3 cells were plated as described above onto sterile coverslips coated with poly-D-lysine (50μg/mL) or with a mixture of collagen (5μg/cm2; Corning) and fibronectin (1 μg/cm2; Corning). Where indicated, DAPT (10μM) was also added to the culture media. Cultures were maintained for either 10–14 days (GLT-1 staining, Fig. 2) or for 24 hours (NICD staining, Fig. 6d). The cells were washed once in 1X PBS and then fixed with either ice-cold methanol or with 4% paraformaldehyde in PBS (10 min). After three rinses in washing solution (0.4% triton X-100 in 1X PBS, 5 min each), cells were treated with blocking solution (5% goat serum, in washing solution) for one hour at 23°C. Cells were incubated overnight at 4°C with primary antibodies diluted in blocking solution, either a mix of rabbit anti-GLT-1 (J.D. Rothstein, Johns Hopkins University; Maryland; USA Cat# GLT-1a RRID:AB_2314565; C-terminal-directed; 1:250 dilution) (Rothstein et al. 1994) and rat anti-CD31 (BD Biosciences Cat# 550274, RRID:AB_393571; Clone MEC 13.3; 1:200 dilution; San Jose, CA) or a mix of mouse anti-GFAP (Cell Signaling Technology Cat# 3670, RRID:AB_561049; 1:500) with rabbit anti-activated Notch1 (Abcam Cat# ab8925, RRID:AB_306863; 1:500; Cambridge, MA). We confirmed that this antibody recognizes NICD by transiently transfecting human embryonic kidney (HEK)-293T cells (ATCC Cat# CRL-11268, RRID:CVCL_1926) with 3XFlagNICD1 (a gift from Raphael Kopan, addgene plasmid #20183) (Ong et al. 2006) using a Ca2+ phosphate transfection kit (Clontech, Mountain View, CA). With the Abcam antibody, we observed a clear increase in NICD staining in transfected cells compared with untransfected cells. After rinsing three times in washing solution, coverslips were incubated in blocking solution containing goat anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific Cat# A-11034, RRID:AB_2576217) and donkey anti-rat Alexa Fluor 594 (Thermo Fisher Scientific Cat# A-21209, RRID:AB_2535795) or goat anti-mouse Alexa Fluor 633 (Thermo Fisher Scientific Cat# A-21052, RRID:AB_2535719) conjugates (1:250 dilution) for one hour at room temperature. Nuclei were counterstained using a mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories Inc, Burlingame, CA). Control incubations without primary antibodies were included in each experiment to confirm the absence of non-specific signal due to the secondary antibodies as well as to confirm quenching of endogenous GFP fluorescence. Pictures were taken with a DMi8 Leica confocal microscope equipped with a 40X objective (Leica Microsystems, Wetzler, Germany) using the 405, 488, 594 and 633nm laser lines. For some images an additional 2x, 3x, or 8x optical zoom was used. The imaging settings (laser power, gain, etc.) were the same between conditions in each experiment, and all post-imaging processing was done in parallel and was identical for each condition. The images were taken using sequential mode to avoid cross-contamination of fluorophores.
Figure 2. Immunofluorescence in co-cultures of cortical astrocytes from dual reporter mice with bEND.3 cells.

Expression of GLT-1 was examined in cortical astrocytes from dual reporter mice, in bEND.3 cells, and in astrocytes co-cultured with bEND.3 cells. An anti-CD31 antibody was used as a marker of endothelial cells. The magnification is the same in the first three columns (scale bar 30μm). The fourth column is a 2X optical zoom of the yellow-outlined portion of the third column (scale bar 15μm). Nuclei were counterstained with DAPI. No staining was observed when the primary antibodies were omitted. Data are representative of three independent experiments.
Isolation of mRNA and Quantitative PCR
Astrocytes and bEND.3 cells were cultured as described above, and mRNA was harvested after 14 days using the RNeasy Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Equal amounts of isolated mRNA were reverse transcribed into cDNA using a high capacity RNA-to-DNA kit (Applied Biosystems, Foster City, CA), and one tenth of this reaction was used per quantitative polymerase chain reaction (qPCR). Quantitative PCR was performed using Taqman primer-probe mixes (Applied Biosystems) and a Stratagene Mx3005 qPCR system was used for detection.
Measurement of Na+-dependent transport activity
Co-cultures of astrocytes from dual reporter mice and rat primary brain endothelia (RBEC) were plated as described above and transport assays were performed as previously described (Dowd & Robinson 1996; Ghosh et al. 2011). Briefly, astrocytes or co-cultures of astrocytes and RBEC were placed in a 37°C water bath and rinsed twice with either 1 mL of sodium- or choline-containing buffer then incubated with 0.5μM [3H]-Glutamate for 5 min in the absence or presence of 300μM dihydrokainic acid (DHK; Tocris Bioscience, Bristol, UK), a GLT-1 selective inhibitor (Arriza et al. 1994; for review, see Robinson & Dowd 1997). Assays were stopped with the addition of 1 mL 4°C choline-containing buffer and subsequently solubilized in 1 mL 0.1M sodium hydroxide. The suspension was analyzed for radioactivity and protein concentration. Na+-dependent transport was calculated as the difference in radioactivity accumulated in the presence or absence of sodium and normalized to the amount of protein per well. As little or no DHK-sensitive uptake was observed in the absence of RBEC, DHK-sensitive uptake was normalized to the amount observed in the presence of RBEC.
Pharmacological treatments
Astrocytes and/or bEND.3 cells were cultured as described above. Three days after astrocytes were re-plated onto bEND.3 cells, leukemia inhibitory factor (LIF, 0.1μg/mL; Sigma), human recombinant bone morphogenic protein 2 (BMP-2, 40ng/mL dilution, PeproTech, Rocky Hill, NJ), human recombinant Noggin (Fc chimera, 250ng/mL dilution, R&D Systems, Minneapolis, MN), N5-[imino(nitroamino)methyl]-L-ornithine, methyl ester, monohydrochloride (L-NAME, 100μM, Cayman Chemical, Ann Arbor, MI), or N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT, 10μM, Santa Cruz Biotechnology, Dallas, TX) were randomly added to different wells during the normal media change. Subsequent media changes were of 25% of the media, with the drugs at the above concentrations. Treatments continued for 10 days, after which the cells were lysed and the protein harvested for analysis.
Lentivirus production and cell transduction
Lentiviruses were prepared as previously described (Ghosh et al., 2011). Briefly, HEK-293T cells were grown in poly-D-lysine (500μg/mL) coated 15 cm plates and transfected (Ca2+ phosphate transfection kit, Clontech) with pCMVΔr8.2 (15μg, a packing plasmid), JS-86 (5μg, an envelope plasmid), and the transfer plasmid (20μg, pLKO.1) containing either an shRNA directed against recombination signal binding protein for immunoglobulin kappa J region (RBPJ) (Sigma, CCGGCTGTATCACAACTCCACAAATCTCGAGATTTGTGGAGTTGTGATACAGTTTTTG) or a scrambled shRNA control (Sigma, CCGGCAACAAGATGAAGAGCACCAACTC- GAGTTGGTGCTCTTCATCTTGTTGTTTTT) that has no known mammalian gene targets. After 16h, the cells were rinsed with fresh media. 24 and 48h later the conditioned media containing the lentivirus particles were harvested. Media from three plates were filtered through 0.45μm filters and concentrated by centrifugation at 26,000rpm in a SW28 rotor (Beckman Coulter, Brea, CA) for 2h at 4°C. The pellet was suspended in ice-cold medium (500μL) and aliquots were maintained at −80°C. The concentration of lentiviral particles was determined using the HIV-p24 Elisa assay kit (Perkin Elmer, Waltham, MA). For transduction of dual reporter astrocytes or co-cultures of astrocytes with bEND.3 cells, lentiviruses were thawed on ice, mixed with media and added to cultures 3–5 days after plating astrocytes on top of endothelia (400 ng p24/mL). The media was replaced with fresh media after 24h. As previously reported (Ghosh et al., 2011) we do not observe toxicity after infection. Cell lysates were harvested 11–13 days after infection for Western blot analysis.
Statistical analyses
As indicated above, all cultures were randomly assigned to different groups. Data analyses were not conducted in a blinded fashion, but essentially every experiment was conducted by more than one of the authors. All independent experiments were performed on different days. Sample sizes were based on earlier studies in which we or others performed similar analyses and were able to identify statistically significant results (Li et al. 2006; Yang et al. 2009; Ghosh et al. 2011; Ghosh et al. 2016). All statistics were performed using Graphpad Prism software (La Jolla, CA). Analyses were by one-way analysis of variance (ANOVA) with a Bonferroni post-test for multiple comparisons or by paired t-test.
Results
In previous studies, we have used astrocytes derived from mice in which 192 kb of sequence containing the entire mouse GLT-1 gene (with 45 kb upstream and 24 kb downstream) is used to control expression of eGFP (Yang et al. 2009; Ghosh et al. 2011; Yang et al. 2011b; Ghosh et al. 2016). In these mice, there is complete overlap between eGFP and GLT-1, and the levels of eGFP correlate in astrocytes from different regions of the central nervous system expressing different levels of GLT-1 protein (Regan et al. 2007). More recently, mice in which 8.3 kb of 5′ non-coding region of the human EAAT2 gene is used to control expression of tdTomato have been generated (Yang et al. 2011b). In mice homozygous for both of these traits (termed ‘dual reporter’ mice), tdTomato is only found in cells that express eGFP but not all eGFP expressing cells express tdTomato (Yang et al. 2011b). The signals that differentially activate these two reporters have not been identified, but our laboratory uses these dual reporter mice in many of our analyses of the mechanisms that regulate GLT-1 transcription.
Endothelial cells have been implicated in astrocyte specification and maturation (Mi et al. 2001; Imura et al. 2008; Sakimoto et al. 2012), but the effects on GLT-1 expression have not been reported. To determine if endothelial cells affect GLT-1 expression in astrocytes, we prepared astrocytes from the dual reporter mice, eliminated A2B5+ cells that express GLT-1 (Zelenaia et al. 2000), and then sub-cultured these astrocytes onto an intact monolayer of a mouse brain endothelioma cell line (bEND.3). In our earlier studies, we found that neuronal induction of GLT-1 expression in astrocytes requires at least 7 days (Schlag et al. 1998). Every 2–3 days, we used microscopy to monitor expression of fluorescent protein. We consistently observed a delayed increase in eGFP with a peak around 7–10 days (data not shown, reproduced by M.C.L., Z. M.-L., and E.N.K.). Although occasional cells expressed tdTomato, this was not a consistent finding. We quantified the effects of bEND.3 cells on eGFP, GLT-1, and GLAST protein levels by Western blot after 10 days. Co-cultures of bEND.3 cells and astrocytes expressed 25-fold higher levels of eGFP than that observed in astrocytes alone (Fig. 1a). Although the effects on GLT-1 protein levels was lower, co-cultures of bEND.3 cells and astrocytes expressed 4-fold higher levels of GLT-1 than that observed in astrocytes alone (Fig. 1b). We observe no detectable GLT-1 protein in the bEND.3 cells under these conditions. The levels of GLAST protein were comparable in lysates from astrocytes and co-cultures of astrocytes with bEND.3 cells (Fig. 1c). Glutamate transporters are known to form homomultimers that are evident in immunoblots (Haugeto et al. 1996). When quantifying the levels of glutamate transporter protein in lysates by Western blot, we measure the levels of the lower molecular weight monomer and the higher molecular weight multimer and report the sum of the two, as in no case were the monomers and multimers differentially regulated. It should be noted that the levels of total protein in cell lysate are consistently ~2-fold higher in co-cultures than in monocultures of either cell. This indicates that the increases in eGFP or GLT-1 protein calculated in figures 1a and b are underreported because equal amounts of total protein were loaded in each lane. This also suggests that expression of GLAST may increase in the presence of bEND.3 cells.
When we and others first demonstrated an effect of neurons on astrocytic expression of GLT-1 and in a few subsequent follow up studies, we used rat astrocytes and we reported that these cultured astrocytes express essentially no detectable GLT-1 (Gegelashvili et al. 1997; Swanson et al. 1997; Schlag et al. 1998; Zelenaia et al. 2000; Li et al. 2006). As one can see from Fig 1b, GLT-1 protein is clearly detectable in astrocyte cultures prepared from mice. To determine if there might be some uncontrolled experimental variable that has changed over the years, we compared the expression of GLT-1 and GLAST protein in rat astrocyte cultures to those observed in co-cultures with bEND.3 cells. As we previously reported, we detect essentially no GLT-1 protein in rat astrocyte cultures (Fig. 1d). The levels of GLT-1 protein are about 6-fold higher in co-cultures. There is no difference in the levels of GLAST protein (Fig. 1e).
The fact that eGFP levels are higher in co-cultures is consistent with increased transcription. To further test this hypothesis, we measured the levels of eGFP and GLT-1 mRNA levels in astrocytes from dual reporter mice and co-cultures of mouse astrocytes with bEND.3 cells using reverse transcriptase quantitative PCR. eGFP and GLT-1 mRNA levels were ~8-fold higher and 1.4-fold higher, respectively, when astrocytes were co-cultured with bEND.3 cells (Fig. 1f & g).
We examined the expression patterns of GLT-1 protein by immunofluorescence in order to confirm that the increase in GLT-1 was occurring in astrocytes specifically, as a few groups have observed GLT-1 expression in endothelial cells (Shawahna et al. 2011; Cohen-Kashi-Malina et al. 2012; Lecointre et al. 2014). We compared GLT-1 expression in astrocytes and endothelia using CD31 as an endothelial cell surface marker (Legros et al. 2009; Yang et al. 2011a; Chen et al. 2013; Liu et al. 2013; Welser-Alves et al. 2014; Zehendner et al. 2014). As expected, cultures of mouse astrocytes contained few weakly positive GLT-1-expressing cells and CD31 immunoreactivity was not detected (Fig. 2 top row). bEND.3 cells expressed low levels of GLT-1 immunoreactivity that in optical cross-sections appeared to be cytoplasmic and did not co-localize with the CD31 (Fig. 2 middle row), suggesting that this pool of GLT-1 immunoreactivity is intracellular. To avoid issues of the limited resolution of microscopy in the Z-plane, we examined the expression of GLT-1 and CD31 in non-confluent co-cultures to allow identification of GLT-1-expressing cells that are not on top of bEND.3 cells. Under these conditions, we observe clear increases in the levels of GLT-1 immunoreactivity in non-CD31-positive cells and this increased expression was consistently restricted to cells that were in contact with CD31-positive cells (Fig. 2 bottom row). This suggests that contact between endothelia and astrocytes may be required for induction of GLT-1 expression in astrocytes.
bEND.3 cells are an immortalized mouse brain endothelial cell line and have the advantage of being a homogeneous population and allows one to reduce the numbers of animals used for experiments. However, we wished to replicate our findings using endothelial cells prepared from brain blood vessels. We used forebrains from adolescent rats as a source of these endothelial cells. By bright field microscopy, the morphology of these isolated cells was similar to that of the bEND.3 cell line (not shown). By Western blot, co-cultures of dual reporter astrocytes and these rat brain endothelial cells (RBEC) increased both eGFP protein ~70-fold (Fig. 3a) and GLT-1 protein ~4.5-fold (Fig. 3b). In contrast, GLAST protein levels are significantly lower (Fig. 3c) in co-cultures of astrocytes with RBEC.
GLT-1 is subject not only to transcriptional regulation, but also to control at the mRNA level, by post-translational modifications, and by trafficking to and from the plasma membrane. These latter two levels of control are especially important to the functionality of GLT-1 as a transporter (Foran et al. 2014; for review, Robinson 1998). To investigate the functionality of the GLT-1 protein induced by endothelial cells, we measured the amount Na+-dependent glutamate uptake that is sensitive to the selective GLT-1 inhibitor dihydrokainate (DHK). We found no DHK-sensitive uptake in co-cultures of astrocytes and bEND.3 cells (data not shown, n=4). As this was an unexpected result, we performed biotinylation experiments to tag membrane proteins and probed for GLT-1 protein on the surface of the cells (Davis et al. 1998; Gonzalez et al. 2007). We found that essentially 100% of GLT-1 is on the cell surface when either astrocytes from dual reporter mice (n = 1) or rats (n = 1) are co-cultured with bEND.3 cells. We considered the possibility that the contribution of GLAST to total uptake may obscure GLT-1 mediated uptake. As co-cultures of RBECs and astrocytes contain lower levels of GLAST (Fig. 3c), we tested for DHK-sensitive glutamate uptake in co-cultures of astrocytes and RBEC. We do observe significantly more DHK-sensitive uptake in co-cultures of astrocytes and RBECs than in astrocytes alone, but the levels are low (Fig. 3d and e). It is possible that GLT-1 function requires an accessory protein and/or post-translational mechanism that is not induced/activated by endothelia or GLT-1 function is chronically inhibited in these co-cultures. While this would be interesting, this has not been explored.
Among the known signaling pathways previously shown to affect astrocyte specification, maturation, or function are LIF/STAT3, BMP/TGF-β, and nitric oxide (Bonni et al. 1997; Mi et al. 2001; Nakashima et al. 2001; Fan et al. 2005; Covacu et al. 2006; Imura et al. 2008; Brix et al. 2012; Urayama et al. 2013; Balderas et al. 2014; Hong & Song 2014; Stipursky et al. 2014; Raju et al. 2015). Therefore, we investigated the potential involvement of these pathways in astrocytic GLT-1 induction by endothelial cells. Treatment with leukemia inhibitory factor and activation of the STAT3 pathway has been previously shown to drive astrocyte specification and to increase astrocytic markers in culture (Bonni et al. 1997; Mi et al. 2001; Fan et al. 2005; Imura et al. 2008 Urayama et al. 2013; Hong & Song 2014). In our experiments, treatment with LIF (0.1μg/mL) did not increase eGFP or GLT-1 protein levels (Fig. 4a). As a positive control for pathway activation, we investigated STAT3 phosphorylation at tyrosine 705, which increased upon treatment with LIF (Fig. 4b). Furthermore, although co-culture with bEND.3 cells did increase STAT3 protein levels (~2-fold, p<0.0001), there was no significant increase in pSTAT3, whether the levels were normalized to total STAT3 protein or to β-actin only (Fig. 4b). The BMP2/TGF-β pathway also regulates gliogenesis (Nakashima et al. 2001; Imura et al. 2008; Stipursky et al. 2014). Treatment of astrocytes with bone morphogenic protein 2 (BMP-2, 40ng/mL) increased eGFP and GLT-1 protein slightly (though not significantly), an effect which was blocked by treatment with the BMP inhibitor Noggin (250ng/mL) (Fig. 4c). However, Noggin did not block the increase in eGFP/GLT-1 induced by bEND.3 (Fig. 4c). Nitric oxide diverts neural precursors to a glial fate (Covacu et al. 2006), enhances astrocytic glycolysis, (Brix et al. 2012), and regulates glutamate uptake and metabolism (Balderas et al. 2014; Raju et al. 2015). Treatment with the nitric oxide synthase inhibitor L-NG-Nitroarginine methyl ester (L-NAME, 100μM) to block nitric oxide signaling did not block endothelia-dependent eGFP/GLT-1 induction (Fig. 4d). Together, these studies provide evidence that the induction of GLT-1 by endothelial cells is not dependent on LIF/STAT3, BMP/TGF-β, and nitric oxide.
Figure 4. Effect of LIF, BMP2, or L-NAME treatment on eGFP and GLT-1 protein levels.

Cortical astrocytes from dual reporter mice were either co-cultured with bEND.3 cells or treated with leukemia inhibitory factor (LIF), and then harvested for Western blot analysis of eGFP (Top: representative blot, Bottom: white bars) and GLT-1 (Bottom: black bars) (a) or STAT3 and pSTAT3, where pSTAT3 is normalized to total STAT3 levels (b). Data are the mean ± SEM of four independent experiments. Other astrocytes were treated with bone morphogenic factor 2 (BMP-2), Noggin, or both, with or without co-culture with bEND.3 cells as indicated (c), or L-NAME, with or without co-culture with bEND.3 cells as indicated (d) and harvested for Western blot analysis. Blots were probed for eGFP and GLT-1. Top: Representative eGFP blots and Bottom: summary of quantification of both eGFP (white bars) and GLT-1 (black bars), normalized to β-actin. Data are the mean ± SEM of three independent experiments. * p ≤0.05, **** p ≤ 0.0001 for indicated comparisons.
It has been observed by other groups and in our own laboratory that GLT-1 expression can be induced by neuron-conditioned media (Gegelashvili et al. 1997; Swanson et al. 1997; Schlag et al. 1998; Perego et al. 2000) and neurons separated from astrocytes by a transwell membrane are still able to induce GLT-1 (Schlag et al. 1998), suggesting a mechanism that is not dependent on contact. The results of our immunofluorescence studies show that the increase in GLT-1 protein observed in co-cultures with bEND.3 cells is restricted to astrocytes adjacent to endothelial cells (Fig. 2, bottom row). We used transwell inserts, a common blood brain barrier model system (Nakagawa et al. 2009; Cohen-Kashi-Malina et al. 2012; Takata et al. 2013), to investigate whether induction of GLT-1 by endothelial cells requires contact. For these studies, astrocytes prepared from dual reporter mice were either in direct contact with bEND.3 cells (on either side of the transwell membrane), separated from the bEND.3 cells (with bEND.3 on the membrane and astrocytes on the bottom of the culture dish), or on the bottom of the culture dish without any other cell type (Fig. 5a). Increases in eGFP and GLT-1 protein levels are observed only in the condition in which astrocytes and bEND.3 cells are cultured in direct contact (Fig. 5b), indicating that the induction of GLT-1 by endothelial cells is contact-dependent and is not due to a diffusible signal.
In contrast to the signaling mechanisms discussed previously, Notch signaling occurs through cell contact, and several studies have implicated Notch signaling in astrocytic differentiation, proliferation, and activation (for reviews, see Wang & Barres 2000; Gaiano & Fishell 2002; Cau & Blader 2009). Notch signaling is dependent upon the cleavage of the Notch intracellular domain by γ-secretase, which can only occur once the EGF-like repeat-containing extracellular domain binds to a ligand on an adjacent cell. Endothelial cells and astrocytes express both the Notch receptor and Notch ligands (Daneman et al. 2010; Zhu et al. 2011). Therefore, we investigated the potential involvement of Notch signaling by treating co-cultures of dual reporter astrocytes and bEND.3 cells with 10μM N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), an inhibitor of γ-secretase. Treatment with DAPT eliminated eGFP, GLT-1, and GLAST protein expression in co-cultures of astrocytes and bEND.3 cells, but had no effect when astrocytes were cultured by themselves (Fig. 6a–c). When astrocytes and endothelial cells are stained with an antibody specific for cleaved Notch1, there are more NICD puncta in the nuclei of astrocytes cultured with bEND.3 cells than are seen in nuclei of astrocytes alone (Fig. 6d, see white arrows in far right column). This effect is attenuated by treatment with 10μM DAPT (Fig. 6d, bottom row). These results show that co-culturing with endothelia increases nuclear NICD and suggest that the Notch signaling pathway is necessary for endothelia-dependent induction of GLT-1.
As DAPT is a γ-secretase inhibitor, and γ-secretase has additional targets beyond Notch (Haapasalo & Kovacs 2011), we used shRNA directed against RBPJ, a protein that interacts with NICD in the nucleus and affects transcription of Notch target genes (for reviews, see Artavanis-Tsakonas et al. 1999; Ables et al. 2011), to decrease Notch signaling at a point downstream in the pathway. Viral infection with shRNA against RBPJ decreased RBPJ expression (Fig. 7a) and also attenuated endothelia-dependent induction of eGFP/GLT-1 (Fig. 7b&c). It should be noted that the scrambled shRNA control also decreased RBPJ, eGFP, and GLT-1 protein expression, though there was also a significant difference between treatment with shRBPJ and the scrambled control in RBPJ (in both astrocytes alone and in co-cultures) and GLT-1 (in co-cultures). Since the scrambled shRNA has no known mammalian targets, we can only conclude that viral infection itself has a small effect on RBPJ expression in astrocytes. These results provide additional evidence that Notch signaling is responsible for the induction of GLT-1 in astrocytes when they are co-cultured with endothelia.
Figure 7. Effects of shRBPJ on eGFP and GLT-1 protein levels.

Cortical astrocytes from dual reporter mice were cultured alone or on top of an intact monolayer of bEND.3 cells and infected with an shRNA directed against RBPJ (shRBPJ), a scrambled shRNA control (shScr), or no virus 3–5 days after re-plating. Cell lysates were harvested for Western blot 11–13 days after transduction for analysis of (a) RBPJ, (b) eGFP, and (c) GLT-1 protein levels. Top: Representative blots and Bottom: summary of quantification, normalized to β-actin. Data are the mean ± SEM of six independent experiments. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001 for indicated comparisons.
Discussion
In the current study, we demonstrate that co-culturing astrocytes with brain endothelial cells increases the expression of GLT-1 protein and mRNA, as well as expression of a fluorescent reporter of GLT-1 transcription that is only expressed in astrocytes. This induction of GLT-1 is dependent on cell contact. Furthermore, treatment with the γ-secretase inhibitor DAPT and knock-down of the Notch effector RBPJ attenuate this induction, suggesting that Notch signaling is necessary for the upregulation of GLT-1 by endothelial cells. Although endothelial cells have been implicated in astrocyte specification and maturation (Mi et al. 2001; Imura et al. 2008; Sakimoto et al. 2012), this is the first report of endothelial cells affecting GLT-1 expression in astrocytes. These observations suggest that endothelial cells play a role in astrocytic function, and this has implications in development of the central nervous system, protection against injury, and response to changes in blood flow and metabolism.
Astrocytes play an important role in the development and maintenance of the blood brain barrier (BBB). The vasculature of the brain is unique, having more complex tight junctions than those of the peripheral vasculature that increase the transendothelial electrical resistance 50–500-fold (for review, see Abbott et al. 2006). Though mature astrocytes are not present in the cortex until after birth (Sauvageot & Stiles 2002), radial glia contact nascent blood vessels and there is evidence that signaling from radial glia is involved in blood vessel growth and stabilization (Zerlin & Goldman 1997; Gerhardt et al. 2004; Ma et al. 2013; for reviews, see Alvarez et al. 2013; Obermeier et al. 2013; Cheslow & Alvarez 2016). Postnatally, mature astrocytes are polarized, with their endfeet in contact with the blood vessels (for reviews, see Bushong et al. 2004 Abbott et al. 2006; Cheslow & Alvarez 2016), and they induce key components of BBB functionality in endothelial cells that are not observed in isolation (Igarashi et al. 1999; Lee et al. 2003 for review, see Abbott et al. 2006). Several studies have suggested that endothelial cells play a role in astrocyte development as well (Mi et al. 2001; Imura et al. 2008; Sakimoto et al. 2012). Endothelial signals have been shown to change astrocyte morphology and increase expression of astrocytic intermediate filaments in vivo (Zerlin & Goldman 1997), as well as induce accumulation of aquaporin 4 in endfeet in vitro (Camassa et al. 2015). Endothelial cells contribute to astrocyte differentiation, maturation, and function through several important pathways: LIF/STAT3 (Bonni et al. 1997; Mi et al. 2001; Fan et al. 2005; Urayama et al. 2013; Hong & Song 2014), BMP/TGFβ (Nakashima et al. 1999; Nakashima et al. 2001; Stipursky et al. 2014), and NO (Covacu et al. 2006; Brix et al. 2012). However, despite the apparent importance of these signaling mechanisms to astrocyte specification and maturation, we found that none of them significantly induce GLT-1 expression in vitro, and their inhibition does not affect endothelia-dependent induction.
GLT-1 is known to be the predominant glutamate transporter in cortical astrocytes (for reviews, see Robinson 1998; Danbolt 2001) and its expression increases postnatally concurrently with synapse formation (Rothstein et al. 1994; Danbolt 1994; Chaudhry et al. 1995; Furuta et al. 1997; Regan et al. 2007); therefore, GLT-1 can be thought of as a marker of a functionally mature astrocyte. Our data show that the upregulation of GLT-1 by endothelial cells is contact-dependent, in contrast with the induction of other astroglial markers by LIF or BMP2. Notch signaling has been suggested to select and maintain astrocyte identity in the central nervous system (for reviews, see Wang & Barres 2000; Gaiano & Fishell 2002; Cau & Blader 2009). Although activation of STAT3 signaling is apparently necessary for astrocyte differentiation, it has also been demonstrated that neural precursors are unresponsive to gliogenic cues in vivo until late gestation in the mouse (for review, see Temple 2001). It is possible that this might explain the lack of effect seen with LIF treatment in our studies. Notch signaling inhibits cell differentiation in general, and neurogenesis in particular, pointing to a permissive effect of Notch on gliogenesis (Dorsky et al. 1995; Nye et al. 1994; Bao & Cepko 1997; Mizutani & Saito 2005; for review, see Louvi & Artavanis-Tsakonas 2006). However, more recent studies have shown that Notch also directly drives glial development by inducing demethylation of the STAT3 binding sites in astrocytic gene promoters such as gfap and s100β (Deneen et al. 2006; Namihira et al. 2009; Urayama et al. 2013). In this way, Notch signaling can also instruct neural precursor cells to become astrocytes. Namihira et al. show that neurons induce GFAP expression that is blocked by the γ-secretase inhibitor DAPT (Namihira et al. 2009). While the current manuscript was under revision, Hasel et al. showed that astrocytes co-cultured with neurons show evidence of Notch pathway activation, as well as increases in many astrocytic genes (including GLT-1) that are blocked by treatment with DAPT (Hasel et al. 2017). However, endothelial cells also express Notch ligands (Daneman et al. 2010; Zhu et al. 2011) and are in a position to contact neural precursors/radial glia during this time.
These observations have important developmental and disease implications. Following acute traumatic brain injury in mice, Bardehle and colleagues observed heterogeneous juxtavascular proliferation of astrocytes (Bardehle et al. 2013), indicating something different about the astrocytes that are next to blood vessels. This difference could be a signal from the endothelial cells. Data from Magnusson and colleagues lead us to believe that this signal is likely a Notch ligand. Following transient ischemia, Notch signaling is decreased in astrocytes, astrocytic markers are down-regulated, and neurogenic markers are upregulated, an effect that can be mimicked by abolishing Notch signaling through genetic deletion of RBPJ (Magnusson et al. 2014). Our data show that inhibition of the Notch pathway attenuates the induction of GLT-1 by endothelial cells. In fact, when astrocytes are co-cultured with bEND.3 cells and treated with DAPT, GLT-1 protein levels fall below control GLT-1 levels, which does not occur when astrocytes alone are treated with DAPT. We have also observed this same phenomenon with the other glial glutamate transporter GLAST (Fig. 6c, n=4) and GFAP (n=2, data not shown). We speculate that, in addition to inducing GLT-1 expression, endothelial cells might also be providing a constant signal (Notch) that, when disrupted, could lead to a reversion to a precursor state.
As yet it is unclear why some astrocytic gene promoters are inactive in primary cell culture. Our lab and others have found other signals that could contribute to GLT-1 expression in vivo, including nuclear factor-κB (NF-κB) (Zelenaia et al. 2000; Rodriguez-Kern et al. 2003; Sitcheran et al. 2005; Lee et al. 2008; Tai et al. 2008; Ghosh et al. 2011), and Pax6 (Ghosh et al. 2016). NF-κB-dependent mechanisms have been linked to upregulation of GLT-1 by epidermal growth factor (Zelenaia et al. 2000; Sitcheran et al. 2005), β-amyloid and brain-derived neurotrophic factor (Rodriguez-Kern et al. 2003), the β-lactam antibiotic ceftriaxone (Lee et al. 2008), and neurons (Ghosh et al. 2011). Pax6 has been recently shown to contribute to neuron-dependent GLT-1 induction in astrocytes, as short hairpin RNAs directed against Pax6 block the increase in GLT-1 expression that occurs when astrocytes are co-cultured with neurons (Ghosh et al. 2016). Given these findings, we cannot rule out the possibility that one or both of these mechanisms also play a role in endothelia-dependent GLT-1 induction; these will be investigated in future studies.
GLT-1 is an integral part of the maintenance of glutamate homeostasis in the brain. GLT-1 is responsible for the majority of glutamate uptake in brain (for reviews, see Danbolt 2001; Robinson 1998), and inhibition, knock-down, or genetic deletion of GLT-1 leads to excitotoxicity, spontaneous seizures, and premature death in mice (Rothstein et al. 1996; Tanaka et al. 1997). Astrocytes in contact with endothelial cells of the blood brain barrier are in a prime position to sense and respond to changes in blood flow and oxygen. It has been repeatedly shown that blood flow is increased in response to increased neuronal activity (for reviews, see Raichle & Mintun 2006; Hillman 2014), and that astrocytes mediate this response (for reviews, see Koehler et al. 2009; Attwell et al. 2010; Figley & Stroman 2011; Petzold & Murthy 2011; Howarth 2014; Filosa et al. 2016), possibly through the activity of glutamate transporters (Voutsinos-Porche et al. 2003; Gurden et al. 2006; Petzold et al. 2008; Schummers et al. 2008; for review, see Robinson & Jackson 2016). These observations serve to demonstrate that the endothelia-dependent induction of GLT-1 is likely functionally relevant. Down-regulation of GLT-1 is associated with a variety of disorders such as amyotrophic lateral sclerosis (ALS) (Rothstein et al. 1995; Medina et al. 1996; Fray et al. 1998), Huntington’s disease (Arzberger et al. 1997; Lievens et al. 2001; Shin et al. 2005), and epilepsy (Tanaka et al. 1997; Ingram et al. 2001; Wong et al. 2003; Morita et al. 2005), among others (for review, see Sheldon & Robinson 2007). Cerebral ischemia leads to a decrease in GLT-1 expression that is hypothesized to contribute to the delayed neuronal death seen in the hippocampus after an ischemic insult or oxygen glucose deprivation (Raghavendra Rao et al. 2000; Mitani & Tanaka 2003; Ouyang et al. 2007; for review, see Ouyang et al. 2014). Disruption of the BBB reduces GLT-1 expression, reduces glutamate uptake, and results in spontaneous epileptic seizures in vivo (David et al. 2009). On the other hand, increasing GLT-1 has been shown to reduce damage due to excitotoxicity (Jelenkovic et al. 2008; Beurrier et al. 2010; Kong et al. 2012). Therefore, further elucidation of the mechanisms behind the upregulation of GLT-1 by endothelial cells would be beneficial to the study of a variety of disease and disorders of the nervous system.
In conclusion, this study presents the first evidence that the interaction between endothelial cells and astrocytes alters astrocytic GLT-1 expression, suggesting an important role of endothelial cells in astrocytic function that was previously unknown. GLT-1 is of vital importance to glutamate homeostasis in the brain, and signals that affect the expression of this transporter are therefore of considerable interest. We show that Notch signaling, widely studied for its developmental roles both in the nervous system and beyond, is necessary for endothelial induction of GLT-1. This offers further evidence that this contact-dependent pathway may be involved in the maintenance of astrocytic identity, and explains the observation by David and colleagues that disruption of astrocyte-endothelial interactions results in perturbations of glutamate homeostasis (David et al. 2009). Future studies will determine if Notch signaling is sufficient to recapitulate the induction of GLT-1 by endothelial cells, or if a confluence of signaling events are required for GLT-1 expression.
Acknowledgments
This work was supported by NIH grant R01 NS092067. In addition, the Preclinical Models (Judy Grinspan) and Neuroimaging and Neurocircuitry (Hajime Takano/Douglas Coulter) Cores of the Institutional Intellectual and Developmental Research Center at CHOP/Penn (U54 HD086984) provided the A2B5 hybridoma supernatant and assistance with imaging, respectively. The authors would also like to thank Joshua Jackson for suggesting that endothelia might provide a signal to affect GLT-1. We would like to acknowledge advice and suggestions from Joshua Jackson and John O’Donnell over the course of these experiments. The authors have no conflicts of interest to declare.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BBB
blood brain barrier
- BMP2
bone morphogenic protein 2
- DAPT
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester
- DHK
dihydrokainate
- EAAC1
excitatory amino acid carrier 1
- EAAT
excitatory amino acid transporter
- GFAP
glial fibrillary acidic protein
- eGFP
enhanced green fluorescent protein
- GLAST
glutamate-aspartate transporter
- GLT-1
glutamate transporter 1
- LIF
leukemia inhibitory factor
- L-NAME
N5-[imino(nitroamino)methyl]-L-ornithine methyl ester monohydrochloride
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- Pax6
paired homeobox protein 6
- pSTAT3
phospho- signal transducer and activator of transcription 3
- RBEC
rat brain endothelial cells
- RBPJ
recombination signal binding protein for immunoglobulin kappa J region
- STAT3
signal transducer and activator of transcription 3
Bibliography
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhaddad H, Das SC, Sari Y. Effects of ceftriaxone on ethanol intake: a possible role for xCT and GLT-1 isoforms modulation of glutamate levels in P rats. Psychopharmacology (Berl) 2014;231:4049–4057. doi: 10.1007/s00213-014-3545-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease--an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderas A, Guillem AM, Martinez-Lozada Z, Hernandez-Kelly LC, Aguilera J, Ortega A. GLAST/EAAT1 regulation in cultured Bergmann glia cells: role of the NO/cGMP signaling pathway. Neurochem Int. 2014;73:139–145. doi: 10.1016/j.neuint.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Bao ZZ, Cepko CL. The expression and function of Notch pathway genes in the developing rat eye. J Neurosci. 1997;17:1425–1434. doi: 10.1523/JNEUROSCI.17-04-01425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci. 2013;16:580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Faideau M, Bennouar KE, Escartin C, Kerkerian-Le Goff L, Bonvento G, Gubellini P. Ciliary neurotrophic factor protects striatal neurons against excitotoxicity by enhancing glial glutamate uptake. PLoS One. 2010;5:e8550. doi: 10.1371/journal.pone.0008550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Brix B, Mesters JR, Pellerin L, Johren O. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J Neurosci. 2012;32:9727–9735. doi: 10.1523/JNEUROSCI.0879-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci. 2004;22:73–86. doi: 10.1016/j.ijdevneu.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Caley DW, Maxwell DS. Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J Comp Neurol. 1970;138:31–47. doi: 10.1002/cne.901380104. [DOI] [PubMed] [Google Scholar]
- Camassa LM, Lunde LK, Hoddevik EH, Stensland M, Boldt HB, De Souza GA, Ottersen OP, Amiry-Moghaddam M. Mechanisms underlying AQP4 accumulation in astrocyte endfeet. Glia. 2015 doi: 10.1002/glia.22878. [DOI] [PubMed] [Google Scholar]
- Cau E, Blader P. Notch activity in the nervous system: to switch or not switch? Neural Dev. 2009;4:36. doi: 10.1186/1749-8104-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, Strickland S. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202:381–395. doi: 10.1083/jcb.201212032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheslow L, Alvarez JI. Glial-endothelial crosstalk regulates blood-brain barrier function. Curr Opin Pharmacol. 2016;26:39–46. doi: 10.1016/j.coph.2015.09.010. [DOI] [PubMed] [Google Scholar]
- Chu K, Lee ST, Sinn DI, et al. Pharmacological Induction of Ischemic Tolerance by Glutamate Transporter-1 (EAAT2) Upregulation. Stroke. 2007;38:177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- Cohen-Kashi-Malina K, Cooper I, Teichberg VI. Mechanisms of glutamate efflux at the blood-brain barrier: involvement of glial cells. J Cereb Blood Flow Metab. 2012;32:177–189. doi: 10.1038/jcbfm.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CK, Kong Q, Lai L, Zhu MX, Seyb KI, Cuny GD, Xian J, Glicksman MA, Lin CL. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15:653–662. doi: 10.1177/1087057110370998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covacu R, Danilov AI, Rasmussen BS, et al. Nitric oxide exposure diverts neural stem cell fate from neurogenesis towards astrogliogenesis. Stem cells. 2006;24:2792–2800. doi: 10.1634/stemcells.2005-0640. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. The high affinity uptake system for excitatory amino acids in the brain. Progress in neurobiology. 1994;44:377–396. doi: 10.1016/0301-0082(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Progress in neurobiology. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52:953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Rapaport DH, Harris WA. Xotch inhibits cell differentiation in the Xenopus retina. Neuron. 1995;14:487–496. doi: 10.1016/0896-6273(95)90305-4. [DOI] [PubMed] [Google Scholar]
- Dowd LA, Robinson MB. Rapid stimulation of EAAC1-mediated Na+-dependent L-glutamate transport activity in C6 glioma cells by phorbol ester. J Neurochem. 1996;67:508–516. doi: 10.1046/j.1471-4159.1996.67020508.x. [DOI] [PubMed] [Google Scholar]
- Dunlop J. Glutamate-based therapeutic approaches: targeting the glutamate transport system. Curr Opin Pharmacol. 2006;6:103–107. doi: 10.1016/j.coph.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Fan G, Martinowich K, Chin MH, et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- Figley CR, Stroman PW. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur J Neurosci. 2011;33:577–588. doi: 10.1111/j.1460-9568.2010.07584.x. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Morrison HW, Iddings JA, Du W, Kim KJ. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience. 2016;323:96–109. doi: 10.1016/j.neuroscience.2015.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana AC. Current approaches to enhance glutamate transporter function and expression. J Neurochem. 2015;134:982–1007. doi: 10.1111/jnc.13200. [DOI] [PubMed] [Google Scholar]
- Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D. Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia. 2014;62:1241–1253. doi: 10.1002/glia.22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen A, Drejer J, Schousboe A. Direct evidence that excitotoxicity in cultured neurons is mediated via N-methyl-D-aspartate (NMDA) as well as non-NMDA receptors. J Neurochem. 1989;53:297–299. doi: 10.1111/j.1471-4159.1989.tb07327.x. [DOI] [PubMed] [Google Scholar]
- Fray AE, Ince PG, Banner SJ, Milton ID, Usher PA, Cookson MR, Shaw PJ. The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci. 1998;10:2481–2489. doi: 10.1046/j.1460-9568.1998.00273.x. [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471–490. doi: 10.1146/annurev.neuro.25.030702.130823. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Ruhrberg C, Abramsson A, Fujisawa H, Shima D, Betsholtz C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn. 2004;231:503–509. doi: 10.1002/dvdy.20148. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Lane M, Krizman E, Sattler R, Rothstein JD, Robinson MB. The transcription factor Pax6 contributes to the induction of GLT-1 expression in astrocytes through an interaction with a distal enhancer element. J Neurochem. 2016;136:262–275. doi: 10.1111/jnc.13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Yang Y, Rothstein JD, Robinson MB. Nuclear factor-kappaB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J Neurosci. 2011;31:9159–9169. doi: 10.1523/JNEUROSCI.0302-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MI, Susarla BT, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007;103:1917–1931. doi: 10.1111/j.1471-4159.2007.04881.x. [DOI] [PubMed] [Google Scholar]
- Gurden H, Uchida N, Mainen ZF. Sensory-evoked intrinsic optical signals in the olfactory bulb are coupled to glutamate release and uptake. Neuron. 2006;52:335–345. doi: 10.1016/j.neuron.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasel P, Dando O, Jiwaji Z, et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun. 2017;8:15132. doi: 10.1038/ncomms15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Hillman EM. Coupling mechanism and significance of the BOLD signal: a status report. Annu Rev Neurosci. 2014;37:161–181. doi: 10.1146/annurev-neuro-071013-014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Song MR. STAT3 but not STAT1 is required for astrocyte differentiation. PLoS One. 2014;9:e86851. doi: 10.1371/journal.pone.0086851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci. 2014;8:103. doi: 10.3389/fnins.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YY, Xu J, Zhang M, Wang D, Li L, Li WB. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem. 2015;132:194–205. doi: 10.1111/jnc.12958. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Utsumi H, Chiba H, et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- Imura T, Tane K, Toyoda N, Fushiki S. Endothelial cell-derived bone morphogenetic proteins regulate glial differentiation of cortical progenitors. Eur J Neurosci. 2008;27:1596–1606. doi: 10.1111/j.1460-9568.2008.06134.x. [DOI] [PubMed] [Google Scholar]
- Ingram EM, Wiseman JW, Tessler S, Emson PC. Reduction of glial glutamate transporters in the parietal cortex and hippocampus of the EL mouse. J Neurochem. 2001;79:564–575. doi: 10.1046/j.1471-4159.2001.00612.x. [DOI] [PubMed] [Google Scholar]
- Inui T, Alessandri B, Heimann A, Nishimura F, Frauenknecht K, Sommer C, Kempski O. Neuroprotective effect of ceftriaxone on the penumbra in a rat venous ischemia model. Neuroscience. 2013;242:1–10. doi: 10.1016/j.neuroscience.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Jelenkovic AV, Jovanovic MD, Stanimirovic DD, Bokonjic DD, Ocic GG, Boskovic BS. Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions. Experimental biology and medicine. 2008;233:1389–1394. doi: 10.3181/0803-RM-83. [DOI] [PubMed] [Google Scholar]
- Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–169. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Kong Q, Chang LC, Takahashi K, et al. Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest. 2014;124:1255–1267. doi: 10.1172/JCI66163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis. 2012;47:145–154. doi: 10.1016/j.nbd.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai PC, Huang YT, Wu CC, Lai CJ, Wang PJ, Chiu TH. Ceftriaxone attenuates hypoxic-ischemic brain injury in neonatal rats. J Biomed Sci. 2011;18:69. doi: 10.1186/1423-0127-18-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointre M, Hauchecorne M, Chaussivert A, Marret S, Leroux P, Jegou S, Leroux-Nicollet I, Gonzalez BJ, Henry VJ. The efficiency of glutamate uptake differs between neonatal and adult cortical microvascular endothelial cells. J Cereb Blood Flow Metab. 2014;34:764–767. doi: 10.1038/jcbfm.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Legros H, Launay S, Roussel BD, et al. Newborn- and adult-derived brain microvascular endothelial cells show age-related differences in phenotype and glutamate-evoked protease release. J Cereb Blood Flow Metab. 2009;29:1146–1158. doi: 10.1038/jcbfm.2009.39. [DOI] [PubMed] [Google Scholar]
- Li LB, Toan SV, Zelenaia O, Watson DJ, Wolfe JH, Rothstein JD, Robinson MB. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem. 2006;97:759–771. doi: 10.1111/j.1471-4159.2006.03743.x. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Lipski J, Wan CK, Bai JZ, Pi R, Li D, Donnelly D. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience. 2007;146:617–629. doi: 10.1016/j.neuroscience.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xue Q, Tang Q, et al. A simple method for isolating and culturing the rat brain microvascular endothelial cells. Microvasc Res. 2013;90:199–205. doi: 10.1016/j.mvr.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 2013;11:e1001469. doi: 10.1371/journal.pbio.1001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O, Kokaia Z, Frisen J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science. 2014;346:237–241. doi: 10.1126/science.346.6206.237. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Prenatal development of fibrous (white matter), protoplasmic (gray matter), and layer I astrocytes in the human cerebral cortex: a Golgi study. J Comp Neurol. 1995;357:554–572. doi: 10.1002/cne.903570407. [DOI] [PubMed] [Google Scholar]
- Martinez-Lozada Z, Guillem AM, Robinson MB. Transcriptional Regulation of Glutamate Transporters: From Extracellular Signals to Transcription Factors. Advances in pharmacology. 2016;76:103–145. doi: 10.1016/bs.apha.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina L, Figueredo-Cardenas G, Rothstein JD, Reiner A. Differential abundance of glutamate transporter subtypes in amyotrophic lateral sclerosis (ALS)-vulnerable versus ALS-resistant brain stem motor cell groups. Exp Neurol. 1996;142:287–295. doi: 10.1006/exnr.1996.0198. [DOI] [PubMed] [Google Scholar]
- Melzer N, Meuth SG, Torres-Salazar D, et al. A beta-lactam antibiotic dampens excitotoxic inflammatory CNS damage in a mouse model of multiple sclerosis. PLoS One. 2008;3:e3149. doi: 10.1371/journal.pone.0003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Haeberle H, Barres BA. Induction of astrocyte differentiation by endothelial cells. J Neurosci. 2001;21:1538–1547. doi: 10.1523/JNEUROSCI.21-05-01538.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miladinovic T, Nashed MG, Singh G. Overview of Glutamatergic Dysregulation in Central Pathologies. Biomolecules. 2015;5:3112–3141. doi: 10.3390/biom5043112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani A, Tanaka K. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci. 2003;23:7176–7182. doi: 10.1523/JNEUROSCI.23-18-07176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Saito T. Progenitors resume generating neurons after temporary inhibition of neurogenesis by Notch activation in the mammalian cerebral cortex. Development. 2005;132:1295–1304. doi: 10.1242/dev.01693. [DOI] [PubMed] [Google Scholar]
- Morita T, Takahashi M, Takeuchi T, Hikasa Y, Ikeda S, Sawada M, Sato K, Shibahara T, Shimada A. Changes in extracellular neurotransmitters in the cerebrum of familial idiopathic epileptic shetland sheepdogs using an intracerebral microdialysis technique and immunohistochemical study for glutamate metabolism. J Vet Med Sci. 2005;67:1119–1126. doi: 10.1292/jvms.67.1119. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int. 2009;54:253–263. doi: 10.1016/j.neuint.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Takizawa T, Ochiai W, et al. BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A. 2001;98:5868–5873. doi: 10.1073/pnas.101109698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K, Yanagisawa M, Arakawa H, Taga T. Astrocyte differentiation mediated by LIF in cooperation with BMP2. FEBS Lett. 1999;457:43–46. doi: 10.1016/s0014-5793(99)00997-7. [DOI] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Nye JS, Kopan R, Axel R. An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development. 1994;120:2421–2430. doi: 10.1242/dev.120.9.2421. [DOI] [PubMed] [Google Scholar]
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Cheng HT, Chang LW, Ohtsuka T, Kageyama R, Stormo GD, Kopan R. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J Biol Chem. 2006;281:5106–5119. doi: 10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu L, Liu S, Giffard RG. Role of Astrocytes in Delayed Neuronal Death: GLT-1 and its Novel Regulation by MicroRNAs. Adv Neurobiol. 2014;11:171–188. doi: 10.1007/978-3-319-08894-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego C, Vanoni C, Bossi M, Massari S, Basudev H, Longhi R, Pietrini G. The GLT-1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem. 2000;75:1076–1084. doi: 10.1046/j.1471-4159.2000.0751076.x. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Albeanu DF, Sato TF, Murthy VN. Coupling of neural activity to blood flow in olfactory glomeruli is mediated by astrocytic pathways. Neuron. 2008;58:897–910. doi: 10.1016/j.neuron.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Rao AM, Dogan A, Bowen KK, Hatcher J, Rothstein JD, Dempsey RJ. Glial glutamate transporter GLT-1 down-regulation precedes delayed neuronal death in gerbil hippocampus following transient global cerebral ischemia. Neurochem Int. 2000;36:531–537. doi: 10.1016/s0197-0186(99)00153-9. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Raju K, Doulias PT, Evans P, et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal. 2015;8:ra68. doi: 10.1126/scisignal.aaa4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, Davies SW. Excitatory receptors and their role in excitotoxicity. Biochem Soc Trans. 1987;15:218–219. doi: 10.1042/bst0150218. [DOI] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Dowd LA. Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Advances in pharmacology. 1997;37:69–115. doi: 10.1016/s1054-3589(08)60948-5. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Jackson JG. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem Int. 2016 doi: 10.1016/j.neuint.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Kern A, Gegelashvili M, Schousboe A, Zhang J, Sung L, Gegelashvili G. Beta-amyloid and brain-derived neurotrophic factor, BDNF, up-regulate the expression of glutamate transporter GLT-1/EAAT2 via different signaling pathways utilizing transcription factor NF-kappaB. Neurochem Int. 2003;43:363–370. doi: 10.1016/s0197-0186(03)00023-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Sakimoto S, Kidoya H, Naito H, Kamei M, Sakaguchi H, Goda N, Fukamizu A, Nishida K, Takakura N. A role for endothelial cells in promoting the maturation of astrocytes through the apelin/APJ system in mice. Development. 2012;139:1327–1335. doi: 10.1242/dev.072330. [DOI] [PubMed] [Google Scholar]
- Sauvageot CM, Stiles CD. Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol. 2002;12:244–249. doi: 10.1016/s0959-4388(02)00322-7. [DOI] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Molecular pharmacology. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–1643. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Shawahna R, Uchida Y, Decleves X, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8:1332–1341. doi: 10.1021/mp200129p. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitcheran R, Gupta P, Fisher PB, Baldwin AS. Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. The EMBO journal. 2005;24:510–520. doi: 10.1038/sj.emboj.7600555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipursky J, Francis D, Dezonne RS, Bergamo de Araujo AP, Souza L, Moraes CA, Alcantara Gomes FC. TGF-beta1 promotes cerebral cortex radial glia-astrocyte differentiation in vivo. Front Cell Neurosci. 2014;8:393. doi: 10.3389/fncel.2014.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YH, Tsai RY, Wang YH, Cherng CH, Tao PL, Liu TM, Wong CS. Amitriptyline induces nuclear transcription factor-kappaB-dependent glutamate transporter upregulation in chronic morphine-infused rats. Neuroscience. 2008;153:823–831. doi: 10.1016/j.neuroscience.2008.02.055. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Foster JB, Lin CL. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. 2015;72:3489–3506. doi: 10.1007/s00018-015-1937-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata F, Dohgu S, Yamauchi A, et al. In vitro blood-brain barrier models using brain capillary endothelial cells isolated from neonatal and adult rats retain age-related barrier properties. PLoS One. 2013;8:e55166. doi: 10.1371/journal.pone.0055166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- Thone-Reineke C, Neumann C, Namsolleck P, et al. The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J Hypertens. 2008;26:2426–2435. doi: 10.1097/HJH.0b013e328313e403. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Urayama S, Semi K, Sanosaka T, Hori Y, Namihira M, Kohyama J, Takizawa T, Nakashima K. Chromatin accessibility at a STAT3 target site is altered prior to astrocyte differentiation. Cell Struct Funct. 2013;38:55–66. doi: 10.1247/csf.12034. [DOI] [PubMed] [Google Scholar]
- Vandenberg RJ, Ryan RM. Mechanisms of glutamate transport. Physiol Rev. 2013;93:1621–1657. doi: 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- Voutsinos-Porche B, Bonvento G, Tanaka K, Steiner P, Welker E, Chatton JY, Magistretti PJ, Pellerin L. Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron. 2003;37:275–286. doi: 10.1016/s0896-6273(02)01170-4. [DOI] [PubMed] [Google Scholar]
- Wang S, Barres BA. Up a notch: instructing gliogenesis. Neuron. 2000;27:197–200. doi: 10.1016/s0896-6273(00)00028-3. [DOI] [PubMed] [Google Scholar]
- Welser-Alves JV, Boroujerdi A, Milner R. Isolation and culture of primary mouse brain endothelial cells. Methods Mol Biol. 2014;1135:345–356. doi: 10.1007/978-1-4939-0320-7_28. [DOI] [PubMed] [Google Scholar]
- Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol. 2003;54:251–256. doi: 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dai M, Wilson TM, et al. Na+/K+-ATPase alpha1 identified as an abundant protein in the blood-labyrinth barrier that plays an essential role in the barrier integrity. PLoS One. 2011a;6:e16547. doi: 10.1371/journal.pone.0016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, et al. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011b;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehendner CM, White R, Hedrich J, Luhmann HJ. A neurovascular blood-brain barrier in vitro model. Methods Mol Biol. 2014;1135:403–413. doi: 10.1007/978-1-4939-0320-7_33. [DOI] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Molecular pharmacology. 2000;57:667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]
- Zerlin M, Goldman JE. Interactions between glial progenitors and blood vessels during early postnatal corticogenesis: blood vessel contact represents an early stage of astrocyte differentiation. J Comp Neurol. 1997;387:537–546. doi: 10.1002/(sici)1096-9861(19971103)387:4<537::aid-cne5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna) 2014;121:799–817. doi: 10.1007/s00702-014-1180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu TS, Costello MA, Talsma CE, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061–6072. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]