

Abstract
Purpose of Review
The treatment landscape of multiple myeloma is rapidly changing; however, despite improvement in patients’ survival, it still remains a largely incurable disease. One hallmark of myeloma is substantial immune dysfunction leading to an increased infection rate and the inability of immune surveillance to detect neoplastic cells. Here, we critically analyze clinical approaches to harness the immune system to overcome this defect with a focus on antibody based and adoptive cellular therapies.
Recent Findings
Clinical trials exploring these immunother-apies to treat myeloma are now well underway and show promising results. In relapsed myeloma, monoclonal antibodies directed against plasma cell antigens and immune checkpoints have already shown substantial efficacy. In parallel, trials of adoptive cellular therapy have exciting promise in myeloma, having induced dramatic responses in a handful of early study participants.
Summary
Taken together, immunotherapeutic approaches hold enormous potential in the field of multiple myeloma and in the near future can be combined with or even replace the current standard of care.
Keywords: Checkpoint blockade, PD-1, CD38, CART cell, Cellular therapy, BCMA
Introduction
Multiple myeloma (MM) is a plasma cell neoplasia accounting for 1% of all cancers and 10% of all hematologic malignancies [1]. Unprecedented results have been obtained in the treatment of this disease with the median overall survival more than doubling in the last decade [2, 3]. The driving force of this improvement was an enrichment in our knowledge of MM biology leading to the development of novel drug classes such as proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs), the standard of care in clinical practice. However, MM largely remains an incurable disease characterized by periods of remission followed by relapses with neoplastic cells showing an increasing degree of drug resistance [4]. Therefore, there is still a significant unmet clinical need to develop new, more effective, therapies for MM.
A hallmark of MM biology is the clinically relevant immune dysfunction that can be recognized early in the natural history of the disease [5]. Indeed, even patients with monoclonal gammopathy of undetermined significance (MGUS) showed a ~2-fold risk of bacteremia as compared to age-matched healthy controls [6]. Additional evidence of immune dysfunction comes from suboptimal responses to vaccination against a variety of pathogens in MM patients [7]. Immune defects harnessing B cell [8, 9], T cell [10], dendritic cell [11], and NK cell [12] function can be found in MM patients; many are prompted by an immunosuppressive microenvironment in the bone marrow that promotes tumor survival and decreased immune surveillance [13]. Indeed, the complex network of interactions in the bone marrow microenvironment of patients affected by plasma cell disorders is a key player in the progressive functional impairment of host immune system and already from the MGUS stage, the immune system fails to eradicate malignant cells [14]. However, in this stage, the disease is still controlled by the immune cells, and a balance between plasma cell proliferation and immune effector functions is still present. At the time of disease progression, immune escape by MM cells occurs and is guided by multiple mechanisms. As an example, programmed cell death ligand 1 (PD-L1) expression by malignant plasma cells is upregulated and engages programmed cell death protein 1 (PD1) on activated cytotoxic T cells inhibiting their activity [15].
Each of the immune defects described above represent a unique opportunity to develop immunotherapeutic agents aimed to restore immune function, elicit tumor-specific immune responses, and add new strategies in the therapeutic armamentarium against MM.
Building on the success of immunotherapy in other malignancies, such as melanoma, Hodgkin’s lymphoma, and acute lymphoblastic leukemia, a variety of immunotherapeutic approaches are currently being evaluated in MM.
While therapies for MM such as IMiDs, allogeneic transplant, and vaccine therapy [16–19] broadly fit in the category of immune therapy for MM, three groups of novel immunotherapy strategies have recently generated enormous excitement in the field and will be the focus of this review: monoclonal antibodies targeting MM cell antigens, monoclonal antibodies targeting immune inhibitory molecules (checkpoint blockade therapy), and adoptive cellular therapy.
Monoclonal Antibodies
In many solid and hematologic cancers, monoclonal antibody (mAb)-based therapy has led to a paradigm shift toward the inclusion of immunotherapeutic agents into clinical practice [20]. In MM, similarly, among the last four drugs approved by the food and drug administration (FDA), two of them were mAbs.
The efficacy and the safety of mAbs are based mainly on two factors: the choice of the target antigen and the spectrum of activation of the immune system.
MAbs can either target tumor cells directly or can aim to release the breaks on the immune system. When tumor antigens are targeted, they should be highly expressed on the surface of the malignant cells and ideally not expressed by other essential cell types. By labeling the tumor cells, mAbs can activate many immune effector mechanisms among which the most characterized are complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP) [21]. Additionally, mAbs can act directly on tumor cells, blocking the function of the target molecule or receptor, inducing apoptosis and other intracellular signals, or delivering conjugated toxins [22].
As agents of checkpoint blockade, mAbs target and block inhibitory ligand/receptor interactions between tumor cells or cells in the microenvironment and immune effector cells, thus re-activating these immune effectors to enable the recognition of neo-antigens through their endogenous T cell receptors (TCRs) and subsequent tumor cell lysis.
The main clinical results of the key compounds used in MM are summarized in Table 1.
Table 1.
Clinical data from key studies exploring mAb-based treatment in RRMM patients
| Target | Drug | Combination | No patients evaluable for response |
Median No of prior therapies (range) |
Response rates (%)
|
Frequent toxicities (%, any grade AEs) | Reference | ||
|---|---|---|---|---|---|---|---|---|---|
| ≥PR | VGPR | CR | |||||||
| CD38 | Daratumumab | - | 42a | 4 (2–12)a | 36 a | 5a | 5a | Fatigue 40%, allergic rhinitis 24%, pyrexia 17% | Lokhorst et al. [23] |
| - | 106a | 5 (2–14)a | 29a | 9a | 3a | Fatigue 40%, anemia 33%, nausea 29% | Lonial et al. [24] | ||
| Bortezomib-dexamethasone | 251 | 2 (1–9) | 83 | 40 | 19 | Thrombocytopenia 58%, peripheral sensory neuropathy 47%, diarrhea 32% | Palumbo et al. [25••] | ||
| Lenalidomide-dexamethasone | 286 | 1 (1–11) | 93 | 33 | 43 | Neutropenia 59%, diarrhea 43%, fatigue 35% | Dimopoulos et al. [26••] | ||
| Isatuximab | - | 19b | 6.5 (2–16)c | 32 | 0 | 16 | Fatigue 49%, nausea 34%, pyrexia 29% | Martin et al. [27] | |
| Lenalidomide-dexamethasone | 22b | 4.5 (1–8), 6 (1–10)d | 50 | 23 | 0 | Fatigue 46%, pyrexia 35%, diarrhea 31% | Vij et al. [28] | ||
| Pomalidomide-dexamethasone | 26 | 4(2–11) | 62 | 31 | 4 | Fatigue 62%, diarrhea 35%, dyspnea 31% | Mkhael et al. [29] | ||
| MOR202 | Dexamethasone | 17 | 3(ne) | 29 | 12 | 0 | AE G ≥ 3: lymphopenia 39%, neutropenia 22%, anemia 17%, thrombocytopenia 17% | Raab et al. [30] | |
| Lenalidomide- dexamethasone or pomalidomide-dexamethasone | 18 | 3(ne) | 78 | 6 | 11 | AE G ≥ 3: neutropenia 48%, lymphopenia 39%, pneumonia 17% | Raab et al. [30] | ||
| SLAMF7 | Elotuzumab | - | 34 | 4.5 (2–10) | - | - | - | Chills 32%, pyrexia 18%, flushing 12% | Zonder et al. [31] |
| Bortezomib-dexamethasone | 77 | 1 (1–3) | 66 | 33 | 4 | Infections 67%, diarrhea 44%, constipation 40% | Jakubowiak et al. [32] | ||
| Lenalidomide-dexamethasone | 321 | 2(1–4) | 79 | 28 | 4 | Lymphopenia 99%, anemia 96%, thrombocytopenia 84%, neutropenia 82% | Lonial et al. [33•] | ||
| PD-1 | Nivolumab Pembrolizumab | - | 27 | nr | 0 | 0 | 0 | Skinc (pruritus, rash) 18%, fatiguec 17%, pneumonitisc 11%. | Lesokhin et al. [34] |
| Lenalidomide-dexamethasone | 50 | nr | 50 | 13 | 3 | Thrombocytopenia 28%, neutropenia 24% | Mateos et al. [35] | ||
| pomalidomide-dexamethasone | 45 | 3 (2–6) | 65 | 20 | 9 | AE G ≥ 3: neutropenia 40%, hyperglycemia 25%, upper respiratory tract infections 21% | Badros et al. [36•] | ||
SLAMF7 signaling lymphocytic activation molecule family member 7, PD-1 programmed cell death protein 1, PR partial response, VGPR very good partial response, CR complete response, AE adverse event, G grade, NCT clinicaltrials.gov registry number, nr not reported
Data refers to the 16 mg/kg daratumumab dose
Data refers to patients treated with doses ≥10 mg/kg
Median number across entire study population
Isatuximab 10 and 20 mg/kg cohort, respectively
CD38-Directed mAbs
CD38 is a cyclic ADP-ribose hydrolase glycoprotein located on the cell surface with a variety of functions including the transduction of intracellular proliferative signals and ectoenzymatic activity involved in the catabolism of extracellular nucleotides [37].
CD38 is expressed at low levels on various hematological tissues (i.e., thymocytes, B cells, NK cells, platelets, erythrocytes, and monocytes) and solid tissues (i.e., Purkinje cells and neurofibrillary tangles in the brain, epithelial cells in the prostate, beta-cells in the pancreas) [38]. However, higher expression is present on the surface of neoplastic plasma cells in the majority of MM cases [39].
The binding of anti-CD38 mAbs to healthy tissues has not caused clinically significant on-target/off-tumor toxicity. CD38 targeted MAbs, however, do attach to CD38 molecules expressed on erythrocytes, interfering with blood compatibility testing, thus complicating the safe release of blood products [40]. Patients should undergo red blood cell antigen phe-notyping before starting a CD38 targeted treatment and the transfusion service should be aware of the problem because methods like dithiothreitol incubation of patients’ erythrocytes can remove surface CD38 and neutralize this kind of interference [40].
CD38-targeting mAbs kill MM cells directly inducing apoptosis, modulating ectoenzymatic function, and activating CDC, ADCC, and ADCP on different immune cell types [21]. Different mAbs induce different levels of these effector functions; however, their relative contribution to the clinical efficacy and safety is largely unknown.
Currently, three anti-CD38 mAbs are being clinically developed for the treatment of MM: daratumumab (IgG1-k, fully human), isatuximab (IgG1-k, chimeric), and MOR202 (IgG1-l, fully human).
Daratumumab
Daratumumab was first tested as a single agent in a phase I/II study on heavily pretreated relapsed/refractory MM (RRMM) patients [23]. No maximum tolerated dose (MTD) was reported in the dose escalation cohort (n = 32), and it showed clear single agent activity with dose-related response rates in the expansion phase cohort (n = 72). The overall response rate [ORR, at least partial response (PR)] was 36% in patients receiving the maximum dose tested (16 mg/kg), the only safety warning was represented by a high frequency of infusion-related reactions (71%, all grades); however, these were manageable and rarely severe (1% grade ≥ 3). The results of this trial were confirmed by a second phase II monotherapy trial, testing daratumumab at the 16 mg/kg dose in 106 RRMM patients [24], although median progression-free survival (PFS) was still short in the overall population (3.7 months).
The promising single-agent activity of this drug prompted its evaluation in combination studies with established backbone therapies in lesser pretreated RRMM patients [41, 42].
Early results from a phase III study evaluating bortezomib-dexamethasone ± daratumumab in 498 RRMM patients who were not PI refractory were outstanding [25••]. After a median follow-up of 7.4 months, the hazard ratio for progression or death with daratumumab vs control was 0.39 (p < 0.001) with an ORR of 82.9% in the daratumumab-bortezomib-dexamethasone arm. Twelve-month PFS was 60.7% in the daratumumab cohort versus 26.9% in the control cohort. Regarding safety, higher rates of hematologic adverse events were reported in the daratumumab group (any grade thrombocytopenia 58.8 vs 43.9%, neutropenia 17.7 vs 9.3%, lymphopenia 13.2 vs 3.8%); infusion-related reactions were reported in the 45.3% of patients receiving daratumumab; in ≥95% of cases, they occurred during the first infusion.
In another phase III study evaluating lenalidomide-dexamethasone ± daratumumab in 569 RRMM patients who were not refractory to lenalidomide, early results were equally remarkable [26••]. After a median follow-up of 13.5 months, the hazard ratio for progression or death with daratumumab vs control was 0.37 (p < 0.001) with a 12-month PFS of 83.2 vs 60.1%. ORR in the daratumumab-lenalidomide-dexamethasone group was 92.9 vs 76.4% in the controls. Safety data were consistent with the trials discussed above.
These clinical results led to FDA approval of daratumumab as monotherapy in patients treated with ≥3 prior lines of therapy (including a PI and an IMiD), and in combination with lenalidomide-dexamethasone or bortezomib-dexamethasone in patients who have received at least one prior therapy.
Isatuximab
The main difference between isatuximab and the other CD38-targeting mAbs is its ability to directly induce apoptosis of MM cells without the need to crosslink 2 CD38 molecules and its inhibition of CD38 ectoenzyme activity [43].
Clinically, two phase I/II dose escalation studies are evaluating isatuximab in patients with heavily pretreated RRMM, one as single agent and the other in combination with lenalidomide-dexamethasone. In the first trial [27], isatuximab monotherapy was given at increasing doses ranging from 0.3 to 20 mg/kg in 35 patients. The MTD was not reached, and the ORR was 27%.
In the isatuximab combination study with lenalidomide-dexamethasone [28], the MTD was similarly not reached (maximum tested dose 20 mg/kg), and despite heavy pretreatment (median of 6 prior lines in the 20 mg/kg cohort), the ORR was 50%. The most frequent adverse events were fatigue (46%), pyrexia (35%), and diarrhea (31%). Infusion reaction rates were similar to daratumumab studies.
Recently, at American Society of Clinical Oncology (ASCO) meeting, early data of a dose-escalation phase Ib study evaluating the combination of isatuximab with pomalidomide-dexamethasone in RRMM patient who received ≥2 prior therapies have been presented. In 26 patients who were evaluable for response, ORR was 62%. Of note, 77% of enrolled patients were refractory to prior IMiDs. Fatigue (62%), diarrhea (35%), and dyspnea (31%) were the most frequent adverse events; identification of the MTD is still ongoing [44].
MOR202
Compared to the other anti-CD38 mAbs, MOR202 does not induce CDC, which is likely the predominant immune effect contributing to infusion reactions [30, 45].
In a phase I/II study, MOR202 was evaluated in clinically relevant doses with dexamethasone, pomalidomide-dexameth-asone, and lenalidomide-dexamethasone in 41 RRMM patients [30]. The drug was well tolerated and, as expected, a low incidence of infusion-related reactions was reported (7% all grade). ORR was 29% in the MOR202-dexamethasone cohort and 78% in the MOR202-IMiD-dexamethasone cohort.
SLAMF7-Directed mAbs
Signaling lymphocytic activation molecule family member 7 (SLAMF7; CS1) is highly expressed on plasma cells and MM cells, as well as other lymphocyte subpopulations such as NK, albeit, at lower levels [46]. This molecule plays a role in MM-stromal cell interactions, MM cell growth and survival, and immune response regulation [46].
Elotuzumab (IgGlk, humanized) is a mAb targeting SLAMF7. The anti-myeloma effect of elotuzumab relies on several mechanisms: the inhibition of SLAMF7-dependent MM cell adhesion to bone marrow stromal cells, the induction of ADCC, and the enhancement of NK cell cytotoxicity [47, 48].
The first clinical data obtained in a phase I trial with elotuzumab monotherapy in RRMM were disappointing, with no objective responses observed [31]. However, no MTD was reached and the drug was well tolerated up to the maximum planned dose of 20 mg/kg every 15 days, prompting its evaluation in combination regimens given the strong preclinical rationale.
In a phase II trial of 152 RRMM patients who received 1–3 prior lines of therapy, elotuzumab-bortezomib-dexamethasone compared to bortezomib-dexamethasone produced similar ORRs but longer PFS (9.7 vs 6.9 months in the control arm) without additional toxicity [32].
The most interesting results came from the combination of elotuzumab with lenalidomide-based therapy. Lenalidomide augments ADCC and there is evidence that in vitro pretreatment of effector cells with lenalidomide enhances elotuzumab-induced lysis of MM cells [49]. In a phase III study of 646 RRMM patients, elotuzumab-lenalidomide-dexamethasone compared to lenalidomide-dexamethasone significantly produced a better ORR rate (79 vs 66%) and a prolonged PFS (median 19.4 versus 14.9 months, HR 0.70, p < 0.001) at the cost of a modest incremental increase of adverse events (mainly lymphocytopenia and herpes zoster reactivation) [33•]. This study strongly suggests that lenalidomide and elotuzumab may work synergically in a clinically meaningful fashion and led to FDA approval of this combination in RRMM patients who have received one to three prior therapies [50].
Checkpoint Blockade Therapy
Immune responses in human biology are strictly regulated by inhibitory molecules that function as checkpoints to control the intensity and duration of immune system activation [51]. These immune checkpoint molecules are key regulators of self-tolerance; however, they are exploited by neoplastic cells in general and by MM cells in particular to hamper immune surveillance [52].
The PD-1/PD-L1 axis is one of the best characterized immune checkpoint pathways exploited by MM cells [53–55]. MM cells express PD-L1 which, upon the engagement of PD-1 expressed by T cells and NK cells, decreases their proliferation capacity, cytotoxicity, and cytokine production, making them functionally “exhausted” [56].
The preclinical evidence that blocking PD-1 and/or PD-L1 prevents their interaction and enhances T and NK-cell anti-myeloma cytotoxicity [57], combined with the clinical success of checkpoint inhibitors in other solid [58] and hematologic [59] neoplastic disorders, prompted the evaluation of mAbs blocking the PD1/PDL1 interaction in clinical trials for MM.
Nivolumab (IgGk, fully human) is a mAb targeting PD-1. It was tested as monotherapy in a phase Ib clinical trial enrolling patients with different hematologic malignancies. In a subanalyisis of 27 RRMM patients, no objective responses were reported [34].
Pembrolizumab (IgGk, humanized) is a PD-1 inhibitor tested in combination with IMiDs ad dexamethasone in MM.
Pembrolizumab-lenalidomide-dexamethasone combination was evaluated in a phase 1 dose-escalation phase in 40 RRMM patients who failed more than 2 prior therapies [35]. The MTD for pembrolizumab was 200 mg every 3 weeks and the ORR in the whole population was 50%, with an interesting ORR of 38% in lenalidomide-refractory patients. The most common treatment-related adverse events were thrombocytopenia (28%) and neutropenia (24%). A phase III trial using this combination in transplant ineligible newly diagnosed MM patients is ongoing (NCT02579863).
In a phase II study involving 48 RRMM patients, pembrolizumab was combined with pomalidomide (another IMiD) and dexamethasone [36•]. In this trial, all patients were refractory to lenalidomide and 80% were additionally refractory to a proteasome inhibitor. One of the main risks of checkpoint blockade therapy is to induce autoimmune reactions known as immune-related adverse events, which were represented in this trial as interstitial pneumonitis (13%), hypothyroidism (10%), transaminitis (6%), adrenal insufficiency (4%), and vitiligo (2%). ORR was 56%, with a median duration of response of 8.8 months in responding patients. The ORR was quite similar in patients who were refractory both to PIs and lenalidomide (55%).
The combinations of pembrolizumab plus either lenalidomide (NCT02579863) or pomalidomide (NCT02576977) are currently being tested in phase 3 studies for myeloma. In early July 2017, the FDA placed a clinical hold on these studies, and an arm on another earlier phase study evaluating pembrolizumab plus lenalidomide (NCT02036502). While, at this time, publically available data is scarce, it is possible that increased deaths with the combination may be due to auto-immune toxicities. We eagerly await the availability of more information to better understand the outcomes of these large trials and the promise of these combinations for patients with myeloma. The inhibition of PD-L1 as well as other immune checkpoint molecules are currently being evaluated for MM patients [60]; however, mature clinical data are still lacking.
Other mAbs
Many other approaches are being evaluated in the field. As an example, antibodies against CD 138 and CD56 conjugated to a tubulin-binding agent (antibody-drug conjugates) showed clinical activity as single agents [61, 62] and in combination with lenalidomide-dexamethasone [63, 64]. Targeting bone marrow stroma and cytokines, despite the strong biologic rationale, has not yet produced meaningful results [65].
Adoptive Cellular Therapy
Adoptive cellular therapy, most notably, chimeric antigen receptor (CAR) T cell therapy, has shown dramatic efficacy in relapsed and refractory hematologic malignancies. Multiple groups demonstrated an approximately 80% complete response (CR) rate with CD19 targeted CAR T cell therapy for relapsed or refractory pediatric and adult B cell acute lymphoblastic leukemia (B-ALL) [66–69]. MM may be the next malignancy where adoptive cellular therapy has the potential to change the landscape of management for the relapsed/ refractory population. The results and lessons learned from studies with CAR T cells, as well as TCR modified T cells, and activated myeloma infiltrating lymphocytes (aMILs) are discussed below.
CAR T Cell Therapy
Generation of a CAR T cell therapy product consists of collection of a patient’s own white blood cells, followed by ex vivo stimulation of T lymphocytes and gene transfer and integration (usually via lentivirus or retrovirus) of DNA encoding the CAR. Gene-modified T cells are then further expanded and transfused back to the same patient, usually after a short course of conditioning chemotherapy. The CAR itself is composed of single chain variable fragment (scFv) specific to a cell surface target antigen. This is genetically fused to a spacer, a transmembrane domain, and then, intracellularly, to signaling domains that provide the T cell with activation and co-stimulation signals when cognate antigen is engaged by the scFv. This therapy consists of a “living drug.” To obtain efficacy, it is essential that the cells expand in vivo and persist for some period of time. It is unclear what the minimum requirement for persistence may be. Mechanisms of relapse include T cell centric failures to proliferate and maintain effector function. Alternatively, cancer cell centric mechanisms of relapse include the expansion of an antigen negative clone, so called “antigen escape.”
With this novel therapy comes novel toxicities, most notable are cytokine release syndrome (CRS) and neurologic toxicity. Severe CRS (sCRS) is characterized by hypotension and hypoxia, requiring intensive care unit level support. Neurologic toxicity is characterized by aphasias, obtundation, and sometimes seizures. These severe toxicities were seen to occur in <30% of patients in multiple B-ALL trials, less so in trials for other malignancies, and correlated with high disease burden at the time of treatment [66–69, 70••].
CD19
There has been tremendous efficacy demonstrated with CD19 targeted CAR T cell therapy for B-ALL and other CD19 expressing malignancies. MM, a malignancy of plasma cells (PCs), is generally thought to be CD19 negative; however, there are interesting reasons to test CD19 targeted CAR T cell therapy in MM. Hypotheses include the following: (1) it is possible that CD19 is expressed on MM cells, but at low levels not detected by traditional flow or immunohistochemistry (IHC) assays. The CAR T cell is a uniquely potent therapeutic and may eradicate MM cells even with only a few hundred CD19 molecules on the cell surface. (2) It may be that a less differentiated CD19+ MM precursor cell is necessary for continued MM proliferation. (3) The MM tumor microenvironment may contain B regulatory cells (BREGS) which, analogous to TREGS, may aid MM cells to avoid immune destruction. These hypotheses were evaluated in a trial for MM patients who experienced a short interval relapse after high dose melphalan and autologous stem cell transplantation (MEL-ASCT). On the trial, these patients were again treated with MEL-ASCT and given CD19 targeted CAR T cells in the early post-ASCT period (NCT02135406). The results show that 2/10 patients treated had a “remission inversion” where the remission to the second treatment with high-dose melphalan lasted longer than the first [71•]. The investigators at the University of Pennsylvania (UPenn) who conducted the study have now gone on to focus on using this strategy as consolidation after upfront MEL-ASCT, hopeful that in this earlier setting CD19 targeted CAR T cell therapy may alter the natural history of MM (NCT02794246). Many institutions are now focusing on other MM-specific targets.
Kappa Light Chain
A phase I study of CAR T cell therapy targeting the kappa light chain was conducted by investigators at Baylor. The CAR consisted of a murine scFv specific for kappa and included CD28 and CD3-zeta signaling domains and was cloned into a gamma retroviral plasmid. Cyclophosphamide conditioning chemotherapy was given for patients with ALC > 500/µl. MM patients received 1–2 infusions, with patients receiving between 9.2 × 107 and 1.9 × 108 CAR+ T cells/m2. Stable disease was seen in 4 of 7 MM patients with a durability of 2–17 months. It is possible that the underwhelming responses seen in MM patients on this trial are because light chains are secreted and not maintained on the cell surface of plasma cells [72].
CD138
Investigators at the Chinese PLA General Hospital in Bejing reported on five MM patients treated with a CD138 targeted CAR incorporating 4-1BB and CD3-zeta signaling domains. This was well tolerated with four of five patients treated obtaining stable disease for 3–7 months [73]. While CD138 is clearly highly expressed on MM cells, there is concern that it is also expressed on a variety of essential normal tissue. It is possible that a more efficacious CD138 targeted CAR could bring with it the risk of “on-target/off-tumor” toxicity.
Identifying Novel Targets for CAR T Cell Therapy of MM
Identifying the CD19 equivalent in MM is a non-trivial task as the common markers used to immunophenotypically identify MM cells are all expressed on other potentially essential cell types. CD138 (syndican-1) is expressed on bronchial epithelia, among other tissue types. CD56 (neural crest adhesion molecule-1) is expressed on central nervous system cells [74]. CD38 (cyclic ADP-ribose hydrolase) and signaling lymphocytic activation molecule family-member 7 (SLAMF7), the targets of several mAbs discussed above, are expressed on a variety of other hematopoietic cell types [37, 46].
It is important to keep in mind that a CAR T cell is a significantly more potent warhead than a mAb. In fact, a clinical trial using the same antibody fragment as Herceptin, an antibody well tolerated by hundreds of thousands of breast cancer patients, was fatal in the first two patients treated because of low level antigen expression on lung epithelial cells [75].
BCMA
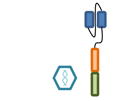
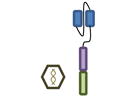
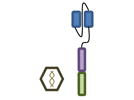
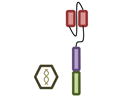
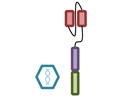
For these reasons, several groups have independently concluded that BCMA (B cell maturation antigen; TNFRSF17) may be an ideal antigen to specifically target MM cells with CAR T cell therapy. BCMA is highly expressed on the MM cells of virtually all patients [76]. As opposed to CD19 where expression is lost as B cells differentiate into PCs, as its name implies, BCMA levels increase during this differentiation process. The designs of the five currently open trials evaluating BCMA targeted CAR T cell therapy are summarized in Table 2.
Table 2.
Design of BCMA targeted CAR T cell vectors and clinical trials for MM
| Institution | NCI | Bluebird Multi-Inst | Nanjing Legend | UPenn | MSK |
|---|---|---|---|---|---|
|
| |||||
| Overview |
|
|
|
|
|
|
| |||||
| scFv derived from | Murine Hybridoma | Murine Hybridoma | Murine Hybridoma | Human Library | Human Library |
|
| |||||
| Co-stimulatory domain | CD28 | 4-1BB | 4-1BB | 4-1BB | 4-1BB |
|
| |||||
| Gene transfer | Retrovirus | Lentivirus | Lentivirus | Lentivirus | Retrovirus |
|
| |||||
| Conditioning | Cyclophosphamide+ Fludarabine | Cyclophosphamide+ Fludarabine | Cyclophosphamide | None (cohort 1)> Cyclophosphamide | Cyclophosphamide+/-Fludarabine |
|
| |||||
| BCMA Ag required | >50% | >50% | “clear expression” | no requirement | >1% |
|
| |||||
| Clinicaltrials.gov Identifier | NCT02215967 | NCT02658929 | NCT03090659 | NCT02546167 | NCT03070327 |
|
| |||||
| Median prior lines | 7 | 7 | 3 | 9 | Not yet reported |
|
| |||||
| Efficacy | CR: 1 | CR: 4 | CR: 15 | CR: 1 | Not yet reported |
| VGPR: 4 | VGPR: 7 | VGPR: 13 | VGPR: 2 | ||
| PR: 3 | PR: 4 | PR: 7 | PR: 1 | ||
| 16 evaluable pts | 18 evaluable pts | 35 evaluable pts | 9 evaluable pts | ||
|
| |||||
| Legend |
|
||||
NCI National Cancer Institute, UPenn University of Pennsylvania, MSK Memorial Sloan Kettering, scFv single chain variable fragment, CD3 ζ T cell receptor zeta-chain
National Cancer Institute (NCI) investigators were the first to publish pre-clinical data using BCMA targeted CAR T cell therapy [77•]. They have continued to conduct a dose escalation trial demonstrating the potential for dramatic responses induced by this treatment modality for MM. Here, at the fourth dose level investigated (9 × 106 CAR+ T cells/kg), they reported on two patients with robust immune responses including dramatic clearance of MM from the marrow which previously contained >80% MM cells and obtained either a stringent CR (lasting 17 weeks) or cleared their bone marrow but had persistent mildly elevated serum markers for a VGPR (ongoing 52+ weeks) [78••, 79]. This dose level, high by B-ALL standards where 1 × 106 CAR+ T cells/kg is commonly used, however, did induce significant toxicities including sCRS and prolonged cytopenias, from which both patients fully recovered. Because of these toxicities and the history of toxicities in B-ALL correlating to disease burden, this trial has limited enrollment to patients with <30% MM cell involvement of their bone marrow and continues to enroll at this dose.
This CAR construct has been licensed by Bluebird Bio and modified for evaluation in a multi-institution trial that is underway. The most recent report of this dose escalation trial revealed that at the second dose level and above (150–900 × 10 total CAR+ T cells), the ORR was 100% with 11 out of 15 patients experiencing robust efficacy with either a VGPR or CR; impressively, all of the responses are ongoing with three patients out approximately 1 year. However, notably, grade 3 toxicity was rarely observed (CRS: grade 3 n = 2; grade 4+ n = 0; neurotoxicity: grade 3/4 n = 0). This includes eight patients treated at these efficacious dose levels with >50% bone marrow involved by MM cells [80]. This trial is continuing to enroll.
Nanjing Legend is conducting a trial in China with exciting early results. Twenty-eight of 35 patients reached a CR or VGPR, with the remaining patients having achieved a PR for an ORR of 100%. Many of the PRs were recently treated and may still convert to deeper responses. Responses seem durable, with five patients treated greater then 12 months ago remaining in CR [81]. There are several unanswered questions, including surrounding the exact specificity of the scFv. Additionally, this trial differs from the others in that the patients were treated at a significantly earlier time in their treatment course, with median prior lines of therapy being 3, compared to 7–9 from the other trials reported.
University of Pennsylvania (UPenn) investigators have reported the clinical trial experience with their BCMA targeted CAR T cell therapy. Notably, unlike the other trials discussed, the first cohort treated (and only cohort reported) did not include any conditioning chemotherapy. This cohort is notable for a single patient with a stringent CR (sCR) ongoing for greater than 12 months. Nine patients have been reported on at a goal 500 × 10 total CAR T cells. In addition to the sCR, two other patients achieved VGPRs. Four of the nine patients reported required anti-IL6 receptor therapy with Tocilizumab for CRS and/or neurotoxicity [82].
Interestingly, trials conducted by NCI and UPenn investigators already reported evidence of antigen escape in MM [78••, 82]. This should not come as a surprise given that it is a common source of relapse in CD19 targeted CAR T cell therapy for B-ALL [67] and highlights the need for additional novel targets for MM.
One difference between these five trial designs is the origin of the scFv’s. The NCI, Blubird, and Nanjing Legend trials use a murine hybridoma derived scFv while the UPenn and Memorial Sloan Kettering (MSK) trials of BCMA targeted CAR T cell therapy utilized a human library screening approach to identify scFv’s. A human library screening approach has two distinct advantages. One library derived scFvs may provide investigators with numerous scFvs to compare efficacy and select a superior lead candidate. Two, as investigators at the Hutchinson Cancer Center noted, second administrations, and potentially persistence, are limited in at least some patients by host anti-murine scFv immune responses [83]. Host anti-CAR immune responses would be expected to be significantly less for human derived CARs. This may be one reason the UPenn trial was able to demonstrate examples of dramatic efficacy even without the use of conditioning chemotherapy, which, to date, was assumed to be essential.
Eligibility based on BCMA expression is another difference in BCMA targeted CAR T cell trial design. BCMA expression is detected by IHC or flow cytometry; the specific assays used by different groups are not available but may be important when comparing patient selection. The NCI and Bluebird use a threshold of 50% BCMA positivity as eligibility for their trials. Investigators at the NCI have reported that this screens out 30% of prospective patients [84]. Nanjing Legend reports requiring “clear BCMA expression,” while MSK uses broader eligibility of “any BCMA positive MM cells”, and UPenn does not screen out for low BCMA expression. Determining if screening for target antigen either enhances efficacy or simply limits the potentially eligible patient population is another advance that may enhance the efficacy of CAR T cell therapy moving forward. Developing and validating the methodology of such companion diagnostics is an often overlooked but important aspect of trial design.
TCR Engineered T Cell Therapy
Similar to CAR T cells, TCR engineered T cell therapy consists of ex vivo genetic manipulation of autologous Tcells and subsequent re-infusion. Advantages over CAR T cells include that intracellular antigens can be targeted as TCRs recognize processed peptide presented in HLA. Limitations include that TCRs, unlike CARs, are HLA restricted, and thus a unique synthetic TCR is required for patients with different HLA types.
TCR engineered T cell therapy has been tried clinically for MM. Synthetic TCRs for clinical use are often affinity enhanced in the lab in the hopes of increasing the potential for efficacy. A MAGE-A3 peptide in HLA-A*01 was initially targeted in one such trial of an affinity enhanced TCR modified T cell product. While extensive off target pre-clinical testing was done, unfortunately, the first two patients treated died of cardiogenic shock as this TCR also recognized a peptide/HLA complex derived from TITIN, a protein expressed by beating cardiomyoctes [85, 86]. This experience is another reminder of the potency of adoptive T cell therapy, and the caution required when translating new synthetic TCR or CAR vectors to the clinic.
A shared NY-ESO/LAGE-1 peptide/HLA-A*0201 complex was the target of another TCR engineered T cell therapy for MM. The clinical trial evaluating this therapy included high-dose melphalan conditioning, ASCT, and then, on day 2 post-ASCT, administration of engineered T cells. This study was well tolerated and showed evidence of long-term persistence of engineered T cells. However, given that for 75% of patients this was their first autologous transplant, it is difficult to make conclusions regarding efficacy [87••].
Therapy with synthetic TCR gene modified T cells for MM continues to move forward on several fronts. Combination therapy with checkpoint blockade, for example, is under evaluation in the clinic with the NY-ESO/LAGE-1 peptide targeted TCR modified T cells in combination with permbrolizumab (NCT03168438). New target intracellular antigens, such as BOB1 in MM [88], are being developed to avoid antigen escape. Importantly, advances in gene editing are allowing for the replacement of the endogenous TCR-alpha gene with the synthetic TCR-alpha and beta cDNA for optimal TCR expression. This leads to increased stem memory and central memory T cell phenotypes over traditional gene modification [89]. Some or all of these advances may eventually lead sTCRT cell therapies to catch up to the clinical results we see with CAR T cell therapy.
Myeloma Infiltrating Lymphocytes
T cells can be re-targeted by gene modification as described above, but evidence from the success of checkpoint blockade indicates that endogenous myeloma infiltrating lymphocytes (MILs) exist that can already recognize neo-antigens on MM cells; however, they are kept in the exhausted or anergic state by the immune suppressive MM microenvironment. Harvesting these MILs and stimulating them ex vivo before re-infusing them may be enough, then, to generate anti-MM immunity. Investigators at Johns Hopkins tested this hypothesis by co-culturing harvested MILs with agonistic CD3/CD28 beads and IL2 in culture to revert this exhausted phenotype and expand MIL numbers before re-infusion. As seen in the study of TCR engineered T cells, activated MILs (aMILs) were given days after high-dose melphalan and ASCT obscuring the contribution of the aMILs to the clinical responses. Importantly, several correlates of functional immunity suggesting an anti-MM response indicating the promise of such therapy [90••].
Conclusions and Future Directions
We are hopeful that advances in immunotherapy, when translated to MM, may lead to a curative approach. Tumor antigen targeted mAbs (e.g., anti-CD38, anti-SLAMF7) are being evaluated earlier in the disease course, and we await results from these trials which may indicate that combination therapy including one of these antibodies for induction therapy of MM may be analogous to the success of Rituximab plus combination chemotherapy in NHLs. In the relapsed setting, we anticipate that targeting the PD1/PDL1 pathway in combination therapy will be a new active regimen that has the potential to change the standard of care for these patients. The PD1/PDL1 pathway, however, is only the tip of the iceberg for modulating immune checkpoints. There are many other antibodies, both antagonistic and agonistic, as well as bispecific antibodies, already in the clinic for targeting additional immune receptor/ligand pairs. It will be exciting to see the results of clinical trials evaluating different combinations from this large class of drugs in MM.
While the trials are in their early stages, adoptive T cell therapy has already shown instances where enormous myeloma burden from heavily pre-treated patients was rapidly eliminated, akin to what was seen in B-ALL. Challenges still exist, including addressing relapse via antigen escape or lack of persistence. To address antigen escape future CAR T cell vectors will be developed targeting additional antigens. CD38 [91, 92], SLAMF7 [93], CD70 [94], CD44v6 [95], Lewis Y [96], CD138 [73], kappa light chain [72], and NKG2D ligands [97, 98] are all being evaluated at various stages of pre-clinical or early phase clinical studies as additional targets for MM. To address lack of persistence, further enhancement to CAR T cell therapy in development includes changes in the production process and vector optimization. Producing a less differentiated final CAR T cell product may increase persistence. One method to accomplish this includes culturing T cells with a small molecule inhibitor to protein kinase B (AKT) during the production process. Pre-clinical evidence shows that this may uncouple cell expansion from differentiation [99]. Bluebird has reported that they plan to investigate this strategy for BCMA targeted CAR T cell therapy in an upcoming clinical trial for MM. Toward this end, another strategy being investigated is immunophenotypic selection of T cells to optimize CD4/CD8 ratio [69, 83] and/or select a central memory phenotype [100]. Additionally, the trend toward more fully human CARs (discussed in detail above), as well as, optimizing conditioning therapy may further enhance persistence [69, 83]. Other options being actively explored include taking advantage of a CART cell’s potential to function as a micropharmacy by developing, so-called “armored” CAR T cells [101]. This is accomplished through the inclusion of a second gene into the CAR T cell vector that may either alter the immunosuppressive tumor microenvironment or directly enhance CAR T cell function. Examples include generating T cells that secrete a pro-inflammatory cytokine [e.g., IL12 [102, 103], express an additional signaling ligand [e.g., CD40L [104], 4-1BBL [105], or suppress immune inhibitory pathways [e.g., dominant negative or knock down of PD-1 [106, 107].
Lastly, combination of mAb therapy with adoptive cellular therapy is a particularly attractive method that may synergize these two immune therapies. We predict that clinical trials in the near future will thoroughly explore this potential avenue.
Acknowledgments
MD and MB have no acknowledgments for this work. ELS is supported by ASH, LLS, SITC, an MSK Technology Development Grant, and the MSK Cancer Center Support Core Grant (P30 CA008748).
Footnotes
Compliance with Ethical Standards
Human and Animal Rights and Informed Consent This article contains no studies with human or animal subjects performed by any of the authors.
Conflict of Interest Mattia D’Agostino declares no potential conflicts of interest.
Mario Boccadoro reports personal fees from Sanofi, Celgene, Amgen, Janssen, Novartis, Abbvie, BMS, personal fees from Celgene, Janssen, Amgen, BMS, Mundipharma, Novartis, Sanofi, outside the submitted work.
Eric L. Smith reports personal fees from Juno Therapeutics. In addition, Dr. Smith has a patent on CAR T cell therapy targeting multiple myeloma specific antigens pending with royalties paid by Juno Therapeutics, and a patent on antibody and bispecifc antibody therapy targeting multiple myeloma specific antigens pending that has not been licensed.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Howlader N, Noone AM, Krapcho M, Garshell J, Neyman N, Altekruse SF, et al. SEER cancer statistics review, 1975–2010 -previous version - SEER cancer statistics review. Bethesda: National Cancer Institute; 2010. [Google Scholar]
- 2.Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–8. doi: 10.1038/leu.2013.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papadas A, Asimakopoulos F. Mechanisms of resistance in multiple myeloma. Berlin Heidelberg: Springer; 2017. pp. 1–38. [DOI] [PubMed] [Google Scholar]
- 5.Tete SM, Bijl M, Sahota SS, Bos NA. Immune defects in the risk of infection and response to vaccination in monoclonal gammopathy of undetermined significance and multiple myeloma Front Immunol. Frontiers Media SA. 2014;5:257. doi: 10.3389/fimmu.2014.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kristinsson SY, Tang M, Pfeiffer RM, Björkholm M, Goldin LR, Blimark C, et al. Monoclonal gammopathy of undetermined significance and risk of infections: a population-based study. Haematologica Ferrata Storti Foundation. 2012;97(6):854–8. doi: 10.3324/haematol.2011.054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robertson JD, Nagesh K, Jowitt SN, Dougal M, Anderson H, Mutton K, et al. Immunogenicity of vaccination against influenza, Streptococcus Pneumoniae and Haemophilus influenzae type B in patients with multiple myeloma. Br J Cancer. 2000;82(7):1261–5. doi: 10.1054/bjoc.1999.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rawstron AC, Davies FE, Owkn RG, English A, Pratt G, Child JA, et al. Br J Haematol. 1. Vol. 100. Blackwell Publishers; 1998. B-lymphocyte suppression in multiple myeloma is a reversible phenomenon specific to normal B-cell progenitors and plasma cell precursors; pp. 176–83. [DOI] [PubMed] [Google Scholar]
- 9.Katzmann JA, Clark R, Kyle RA, Larson DR, Therneau TM, Melton LJ, et al. Suppression of uninvolved immunoglobulins defined by heavy/light chain pair suppression is a risk factor for progression of MGUS. Leukemia. NIH Public. Access. 2013;27(1):208–12. doi: 10.1038/leu.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frassanito MA, Cusmai A, Dammacco F. Deregulated cytokine network and defective Th1 immune response in multiple myeloma. Clin Exp Immunol. 2001 Aug;125(2):190–7. doi: 10.1046/j.1365-2249.2001.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown RD, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-??1 and interleukin-10. Blood. 2001;98(10):2992–8. doi: 10.1182/blood.v98.10.2992. [DOI] [PubMed] [Google Scholar]
- 12.Jurisic V, Srdic T, Konjevic G, Markovic O, Colovic M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med Oncol. 2007;24(3):312–7. doi: 10.1007/s12032-007-0007-y. [DOI] [PubMed] [Google Scholar]
- 13.Pratt G, Goodyear O, Moss P. Immunodeficiency and immuno-therapy in multiple myeloma. Br J Haematol. 2007;138:563–79. doi: 10.1111/j.1365-2141.2007.06705.x. [DOI] [PubMed] [Google Scholar]
- 14.Romano A, Conticello C, Cavalli M, Vetro C, La Fauci A, Parrinello NL, et al. Immunological dysregulation in multiple myeloma microenvironment. [Accessed 10 Jul 2017];Biomed Res Int. 2014 2014:198539. doi: 10.1155/2014/198539. Available from: http://www.hindawi.com/journals/bmri/2014/198539/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yousef S, Marvin J, Steinbach M, Langemo A, Kovacsovics T, Binder M, et al. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J. 2015;5(3):e285. doi: 10.1038/bcj.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu YX, Kortuem KM, Stewart AK. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. [Accessed 12 Apr 2017];Leuk Lymphoma. 2013 54(4):683–7. doi: 10.3109/10428194.2012.728597. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22966948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith E, Devlin SM, Kosuri S, Orlando E, Landau H, Lesokhin AM, et al. CD34-selected allogeneic hematopoietic stem cell transplantation for patients with relapsed, high-risk multiple myeloma. [Accessed 12 Apr 2017];Biol Blood Marrow Transplant. 2016 22(2):258–67. doi: 10.1016/j.bbmt.2015.08.025. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26325439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allegra A, Penna G, Innao V, Greve B, Maisano V, Russo S, et al. Vaccination of multiple myeloma: current strategies and future prospects. [Accessed 12 Apr 2017];Crit Rev Oncol Hematol. 2015 96(2):339–54. doi: 10.1016/j.critrevonc.2015.06.003. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26123319. [DOI] [PubMed] [Google Scholar]
- 19.Festuccia M, Martino M, Ferrando F, Messina G, Moscato T, Fedele R, et al. Allogeneic stem cell transplantation in multiple myeloma: immunotherapy and new drugs. [Accessed 12 Apr 2017];Expert Opin Biol Ther. 2015 15(6):857–72. doi: 10.1517/14712598.2015.1036735. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25865214. [DOI] [PubMed] [Google Scholar]
- 20.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer Nature Publishing Group. 2012;12(4):278–87. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 21.De Donk NWCJV, Moreau P, Plesner T, Palumbo A, Gay F, Laubach JP, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127:681–95. doi: 10.1182/blood-2015-10-646810. [DOI] [PubMed] [Google Scholar]
- 22.Tai Y-T, Anderson KC. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011;2011:1–14. doi: 10.1155/2011/924058. (Figure 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with Daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015 Sep;373(13):1207–19. doi: 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 24.Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Phase II study of daratumumab (DARA) monotherapy in patients with >= 3 lines of prior therapy or double refractory multiple myeloma (MM): 54767414MMY2002 (Sirius) ASCO Meet Abstr. 2015;33(18_suppl):LBA8512. [Google Scholar]
- 25••.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754–66. doi: 10.1056/NEJMoa1606038. Potentially practice-changing data in RRMM patients treated with Daratumumab/Bortezomib-based backbone treatment. [DOI] [PubMed] [Google Scholar]
- 26••.Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, Lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319–31. doi: 10.1056/NEJMoa1607751. Massachusetts Medical Society. Potentially practice-changing data in RRMM patients treated with Daratumumab/Lenalidomide-based backbone treatment. [DOI] [PubMed] [Google Scholar]
- 27.Martin TG, Hsu K, Strickland SA, Glenn MJ, Mikhael J, Charpentier E. A phase I trial of SAR650984, a CD38 monoclonal antibody, in relapsed or refractory multiple myeloma. J Clin Oncol. 2014;32 abstr 8532. [Google Scholar]
- 28.Vij R, Lendvai N, Martin TG, Baz RC, Campana F, Mazuir F, Charpentier E, Benson DM, et al. A phase Ib dose escalation trial of isatuximab (SAR650984, anti-CD38 mAb) plus lenalidomide and dexamethasone (Len/Dex) in relapsed/refractory multiple myeloma (RRMM): Interim results from two new dose cohorts. J Clin Oncol. 2016;34(15_suppl):8009–8009. [Google Scholar]
- 29.Mikhael J, Richardson PG, Usmani Z, Raje N, Bensinger W, Kanagavel D, et al. A phase Ib study of isatuximab in combination with pomalidomide (Pom) and dexamethasone (Dex) in relapsed/ refractory multiple myeloma (RRMM) 2017 ASCO Annual Meeting Abstracts. JCO. 2017;35 (suppl; abstr 8007) [Google Scholar]
- 30.Raab MS, Chatterjee M, Goldschmidt H, Agis H, Blau I, Einsele H, Engelhardt M, Ferstl B, Gramatzki M, öllig CR, Weisel K, Jarutat T, Weinelt D, Endell J, Boxhammer R, Peschel C, et al. A phase I/IIa study of the CD38 antibody MOR202 alone and in combination with pomalidomide or lenalidomide in patients with relapsed or refractory multiple myeloma. Blood. 2016;128:1152. [Google Scholar]
- 31.Zonder JA, Mohrbacher AF, Singhal S, Van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120(3):552–9. doi: 10.1182/blood-2011-06-360552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakubowiak A, Offidani M, Brigitte P, La Rubia JD, Garderet L, Laribi K, et al. Randomized phase 2 study : elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood. 2016;127(23):2833–41. doi: 10.1182/blood-2016-01-694604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–31. doi: 10.1056/NEJMoa1505654. Massachusetts Medical Society. A large phase III randomized trial demonstrating the advantage of Elotuzumab addition to lenalidomide-dexamethasone treatment in RRMM patients. Elotuzumab is not active as single agent in MM, however this study clearly demonstrates the clinical synergy of Elotuzumab and IMiDs. [DOI] [PubMed] [Google Scholar]
- 34.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase ib study. J Clin Oncol. 2016;34(23):2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mateos M-V, Orlowski RZ, Siegel DSD, Reece DE, Moreau P, Ocio EM, Shah JJ, Rodríguez-Otero P, Munshi NC, Avigan D, Ge JY, Marinello PM, et al. Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): Final efficacy and safety analysis. J Clin Oncol. 2016;34(15_suppl):8010–8010. [Google Scholar]
- 36•.Badros AZ, Hyjek E, Ma N, Lesokhin AM, Rapoport AP, Kocoglu MH, Lederer E, Philip S, Lesho P, Johnson A, Dell C, Goloubeva O, Singh B, et al. Pembrolizumab in combination with pomalidomide and dexamethasone for relapsed/refractory multiple myeloma (RRMM) Blood. 2016;128(22) A phase II study on higly preatretad patients with an high percentace of PI and IMiDs double-refractory patients showing benefit from PD-1 directed therapy associated with pomalidomide backbone treatment. [Google Scholar]
- 37.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841–86. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 38.van de Donk NWCJ, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. [Accessed 10 Jul 2017];Immunol Rev. 2016 270(1):95–112. doi: 10.1111/imr.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric Immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol. 2004;121(4):482–8. doi: 10.1309/74R4-TB90-BUWH-27JX. [DOI] [PubMed] [Google Scholar]
- 40.Chapuy CI, Nicholson RT, Aguad MD, Chapuy B, Laubach JP, Richardson PG, et al. Resolving the daratumumab interference with blood compatibility testing. [Accessed 10 Jul 2017];Transfusion. 2015 55(6 Pt 2):1545–54. doi: 10.1111/trf.13069. [DOI] [PubMed] [Google Scholar]
- 41.Plesner T, Arkenau H-T, Lokhorst HM, Gimsing P, Krejcik J, Lemech C, et al. Safety and efficacy of Daratumumab with Lenalidomide and dexamethasone in relapsed or relapsed, refractory multiple myeloma. Blood. 2014;124(21):84. [Google Scholar]
- 42.Mateos MV, Moreau P, Comenzo R, Bladé J, Benboubker L, de la Rubia J, et al. An open-label, multicenter, phase 1b study of daratumumab in combination with oomalidomide-dexamethasone and with backbone regimens in patients with multiple myeloma. 20th Congress of European Hematology Association (EHA); Vienna, Austria. 11–14 June 2015; (abstract P275) [Google Scholar]
- 43.Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574–83. doi: 10.1158/1078-0432.CCR-14-0695. [DOI] [PubMed] [Google Scholar]
- 44.Mikhael J, Richardson PG, Usmani Z, Raje N, Bensinger W, Kanagavel D, et al. A phase Ib study of isatuximab in combination with pomalidomide (Pom) and dexamethasone (Dex) in relapsed/ refractory multiple myeloma (RRMM) 2017 ASCO Annual Meeting Abstracts. JCO. 2017;35 (suppl; abstr 8007) [Google Scholar]
- 45.Tawara T, Hasegawa K, Sugiura Y, Harada K, Mura T, Hayashi S, et al. Complement activation plays a key role in antibody-induced infusion toxicity in monkeys and rats. J Immunol. 2008;180(4):2294–8. doi: 10.4049/jimmunol.180.4.2294. [DOI] [PubMed] [Google Scholar]
- 46.Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol/Hematol. 2013;88:168–77. doi: 10.1016/j.critrevonc.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–84. doi: 10.1158/1078-0432.CCR-07-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841–9. doi: 10.1007/s00262-013-1493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tai Y-T, Dillon M, Song W, Leiba M, Li X-F, Burger P, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. [Accessed 10 Jul 2017];Blood. 2008 112(4):1329–37. doi: 10.1182/blood-2007-08-107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.FDA. FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma. FDA News release. 2015 [Google Scholar]
- 51.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. Nature Publishing Group. 2012;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110(1):296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- 53.Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. Nature Publishing Group. 2012;27(2):464–72. doi: 10.1038/leu.2012.213. [DOI] [PubMed] [Google Scholar]
- 54.Wang L, Wang H, Chen H, Wang W, Chen X-Q, Geng Q-R, et al. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget. 2015;6(38):41228–36. doi: 10.18632/oncotarget.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R, Oyajobi BO, et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015;29(10):2110–3. doi: 10.1038/leu.2015.79. [DOI] [PubMed] [Google Scholar]
- 56.Hallett WHD, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant. 2011;17(8):1133–45. doi: 10.1016/j.bbmt.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 57.Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. 2015;21(20):4617–8. doi: 10.1158/1078-0432.CCR-15-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with Nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2014;372(4):141206100011003. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benson DM. Checkpoint inhibition in myeloma. ASH Educ Progr B. 2016;2016(1):528–33. doi: 10.1182/asheducation-2016.1.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heffner LT, Jagannath S, Zimmerman TM, Lee KP, Rosenblatt J, Lonial S, et al. BT062, an antibody-drug conjugate directed against CD138, given weekly for 3 weeks in each 4 week Cycle : safety and further evidence of clinical activity. Am Soc Hematol Annu Meet Proc. 2012;120(21):653. [Google Scholar]
- 62.Chanan-Khan A, Wolf AJL, Garcia J, Gharibo M, Jagannath S, Manfredi D, et al. Efficacy analysis from phase I study of Lorvotuzumab Mertansine (IMGN901) used as monotherapy in patients with heavily pre-treated CD56-positive multiple myeloma case Description : patient 0226. Blood. 2010 Dec;116 2010. [Google Scholar]
- 63.Kelly KR, Chanan-Khan A, Heffner LT, Somlo G, Siegel DS, Zimmerman T, et al. Indatuximab Ravtansine (BT062) in combination with Lenalidomide and low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma: clinical activity in patients already exposed to Lenalidomide and Bortezomib. Blood. 2014;124(21):4736. [Google Scholar]
- 64.Berdeja JG, Ailawadhi S, Weitman SD, Zildjian S, O’Leary JJ, O’Keeffe J, et al. Phase I study of lorvotuzumab mertansine (LM, IMGN901) in combination with lenalidomide (Len) and dexa-methasone (Dex) in patients with CD56-positive relapsed or relapsed/refractory multiple myeloma (MM) ASCO Meet Abstr. 2011;29(15_suppl):8013. [Google Scholar]
- 65.Kumar SK, Anderson KC. Immune therapies in multiple myeloma. Clin Cancer Res. 2016;22(22):5453–60. doi: 10.1158/1078-0432.CCR-16-0868. [DOI] [PubMed] [Google Scholar]
- 66.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. [Accessed 8 Mar 2015];Sci Transl Med. 2014 6(224):224ra25. doi: 10.1126/scitranslmed.3008226. Available from: http://stm.sciencemag.org/content/6/224/224ra25.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. [Accessed 16 Oct 2014];N Engl J Med. 2014 371(16):1507–17. doi: 10.1056/NEJMoa1407222. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4267531&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. Tcells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. [Accessed 14 Oct 2014];Lancet. 2014 385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. [Accessed 8 Aug 2016];J Clin Invest. 2016 126(6):2123–38. doi: 10.1172/JCI85309. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27111235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70••.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. [Accessed 25 Aug 2016];Sci Transl Med. 2013 5(177):177ra38. doi: 10.1126/scitranslmed.3005930. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23515080. First report of the dramatic efficacy of CD19 targeted CAR T cell therapy in B-ALL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Garfall AL, Maus MV, Hwang W-T, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. [Accessed 10 Jul 2017];N Engl J Med. 2015 373(11):1040–7. doi: 10.1056/NEJMoa1504542. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26352815. Report of CD19 targeted CAR T cell therapy for MM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated K light chains. [Accessed 5 Apr 2017];J Clin Invest. 2016 126(7):2588–96. doi: 10.1172/JCI86000. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27270177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. [Accessed 04 Apr 2017];J Cell Immunother. 2016 2(1):28–35. Available from: www.sciencedirect.com/science/article/pii/S2352177515000023. [Google Scholar]
- 74.Davila ML, Bouhassira DCG, Park JH, Curran KJ, Smith EL, Pegram HJ, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. [Accessed 21 Dec 2015];Int J Hematol. 2014 99(4):361–71. doi: 10.1007/s12185-013-1479-5. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4684946&tool= pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. [Accessed 13 Jul 2015];Mol Ther. 2010 18(4):843–51. doi: 10.1038/mt.2010.24. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2862534&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. [Accessed 5 Apr 2017];Cancer Cell. 2017 31(3):396–410. doi: 10.1016/j.ccell.2017.02.002. Available from: http://www.sciencedirect.com/science/article/pii/S1535610817300168. [DOI] [PubMed] [Google Scholar]
- 77•.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. [Accessed 8 Jun 2015];Clin Cancer Res. 2013 19(8):2048–60. doi: 10.1158/1078-0432.CCR-12-2422. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3630268&tool=pmcentrez&rendertype=abstract. Preclinical evidence of anti-BCMA CAR for treatment of MM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78••.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell-maturation-antigen chimeric antigen receptor cause remissions of multiple myeloma. [Accessed 04 Apr 2017];Blood. 2016 doi: 10.1182/blood-2016-04-711903. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27412889. Initial demonstration of the potential for BCMA targeted CAR T cell therapy to eradiacte large disease burden in patients with MM. [DOI] [PMC free article] [PubMed]
- 79.Kochenderfer JN. Chimeric antigen receptor T cell therapy for multiple myeloma. [Accessed 4 Apr 2017];Blood. 2016 128:SCI-37. doi: 10.1182/blood-2017-06-793869. Available from: http://www.bloodjournal.org/content/128/22/SCI-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berdeja JG, Lin Y, Raje NS, Siegel DSD, Munshi NC, Liedtke M, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: updated results. J Clin Oncol. American Society of Clinical Oncology. 2017;35(15_suppl):3010. doi: 10.1200/JCO.2017.35.15_suppl.3010. [DOI] [Google Scholar]
- 81.Xiaohu FF, Zhao W, Liu J, He A, Chen Y, Cao X, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. American Society of Clinical Oncology. 2017;35(18_suppl):LBA3001. doi: 10.1200/JCO.2017.35.18_suppl.LBA3001. Accessed 10 Jul 2017. [DOI] [Google Scholar]
- 82.Cohen AD, Garfall AL, Stadtmauer EA, Lacey SF, Lancaster E, Vogl DT, et al. B-cell maturation antigen (BCMA)-specific chimeric antigen receptor T cells (CART-BCMA) for multiple myeloma (MM): initial safety and efficacy from a phase I study. [Accessed 4 Apr 2017];Blood. 2016 128:1147. Available from: http://www.bloodjournal.org/content/128/22/1147. [Google Scholar]
- 83.Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkins lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modifiedTcells. [Accessed 4 Apr 2017];Sci Transl Med. 2016 8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ali SA, Shi V, Wang M, Stroncek D, Maric I, Brudno JN, et al. Remissions of multiple myeloma during a first-in-humans clinical trial of T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor. [Accessed 21 Feb 2016];Blood. 2015 126(23):LBA-1. doi: 10.1182/blood-2016-04-711903. American Society of Hematology. Available from: http://www.bloodjournal.org/content/126/23/LBA-1.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. [Accessed 4 Apr 2017];Sci Transl Med. 2013 5(197):197ra103. doi: 10.1126/scitranslmed.3006034. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23926201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. [Accessed 4 Apr 2017];Blood. 2013 122(6):863–71. doi: 10.1182/blood-2013-03-490565. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23770775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87••.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. [Accessed 4 Apr 2017];Nat Med. 2015 21(8):914–21. doi: 10.1038/nm.3910. Available from: http://www.nature.com/doifinder/10.1038/nm.3910. Report of sTCR engineered T cells for the treatment of MM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jahn L, Hombrink P, Hagedoorn RS, Kester MGD, Van Der Steen DM, Rodriguez T, et al. TCR-based therapy for multiple myeloma and other B-cell malignancies targeting intracellular transcription factor BOB1. Blood. 2017;129(10):1284–95. doi: 10.1182/blood-2016-09-737536. [DOI] [PubMed] [Google Scholar]
- 89.Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited central memory and memory stem T cells to treat multiple myeloma without inducing GvHD. [Accessed 10 Jul 2017];Blood. 2017 08:732636. doi: 10.1182/blood-2016-08-732636. Blood-2016. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28637663. [DOI] [PubMed] [Google Scholar]
- 90••.Noonan KA, Huff CA, Davis J, Lemas MV, Fiorino S, Bitzan J, et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. [Accessed 4 Apr 2017];Sci Transl Med. 2015 7(288):288ra78. doi: 10.1126/scitranslmed.aaa7014. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25995224. Clinical evidence of immune response from re-infused aMILs to treat MM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mihara K, Bhattacharyya J, Kitanaka A, Yanagihara K, Kubo T, Takei Y, et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. [Accessed 20 Sep 2015];Leukemia. Macmillan Publishers Limited. 2012 26(2):365–7. doi: 10.1038/leu.2011.205. [DOI] [PubMed] [Google Scholar]
- 92.Drent E, Groen RWJ, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PWHI, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. [Accessed 12 Apr 2017];Haematologica. 2016 101(5):616–25. doi: 10.3324/haematol.2015.137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. [Accessed 20 Sep 2015];Leukemia. 2014 28(4):917–27. doi: 10.1038/leu.2013.279. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3967004&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shaffer DR, Savoldo B, Yi Z, Chow KKH, Kakarla S, Spencer DM, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. [Accessed 5 Apr 2017];Blood. 2011 117:4304–4314. doi: 10.1182/blood-2010-04-278218. Available from: http://www.bloodjournal.org/content/117/16/4304.long?sso-checked=true. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Casucci M, Nicoli di Robilants B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. [Accessed 5 Apr 2017];Blood. 2013 122(20):3461–72. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 96.Peinert S, Prince HM, Guru PM, Kershaw MH, Smyth MJ, Trapani JA, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. [Accessed 5 Apr 2017];Gene Ther. 2010 17(5):678–86. doi: 10.1038/gt.2010.21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20200563. [DOI] [PubMed] [Google Scholar]
- 97.Barber A, Meehan KR, Sentman CL. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. [Accessed 5 Apr 2017];Gene Ther. 2011 18(5):509–16. doi: 10.1038/gt.2010.174. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21209626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barber A, Zhang T, Megli CJ, Wu J, Meehan KR, Sentman CL. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. [Accessed 5 Apr 2017];Exp Hematol. 2008 36(10):1318–28. doi: 10.1016/j.exphem.2008.04.010. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18599182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. [Accessed 5 Apr 2017];J Clin Invest. 2015 126(1):318–34. doi: 10.1172/JCI81217. American Society for Clinical Investigation. Available from: https://www.jci.org/articles/view/81217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. [Accessed 12 Apr 2017];Leukemia. 2015 30(2):492–500. doi: 10.1038/leu.2015.247. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26369987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination im-munotherapy. [Accessed 1 Aug 2016];Nat Rev Clin Oncol. 2016 13(6):394. doi: 10.1038/nrclinonc.2016.65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27118494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. [Accessed 23 Jun 2015];Blood. 2012 119(18):4133–41. doi: 10.1182/blood-2011-12-400044. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3359735&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, Barker JN, et al. IL-12-secreting CD19-targeted cord blood-derived Tcells for the immunotherapy of B-cell acute lymphoblas-tic leukemia. [Accessed 31 Mar 2015];Leukemia. 2015 29(2):415–22. doi: 10.1038/leu.2014.215. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. [Accessed 23 Jun 2015];Mol Ther. 2015 23(4):769–78. doi: 10.1038/mt.2015.4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25582824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T cells. Cancer Cell. 2015;28(4):415–28. doi: 10.1016/j.ccell.2015.09.004. Available from: www.ncbi.nlm.nih.gov/pubmed/26461090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. [Accessed 4 Apr 2017];J Clin Invest. 2016 126(8):3130–44. doi: 10.1172/JCI83092. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27454297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. [Accessed 5 Apr 2017];Clin Cancer Res. 2017 May 1;23(9):2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27815355. [DOI] [PMC free article] [PubMed] [Google Scholar]