

Abstract
Multiprotein complexes are central to our understanding of cellular biology, as they play critical roles in nearly every biological process. Despite many impressive advances associated with structural characterization techniques, large and highly-dynamic protein complexes are too often refractory to analyze by conventional, high-resolution approaches. To fill this gap, ion mobility-mass spectrometry (IM-MS) methods have emerged as a promising approach for characterizing the structures of challenging assemblies due in large part to the ability of these methods to characterize the composition, connectivity, and topology of large, labile complexes. In this Critical Insight, we present a series of bioinformatics studies aimed at assessing the information content of IM-MS datasets for building models of multiprotein structure. Our computational data highlights the limits of current coarse-graining approaches, and compelled us to develop an improved workflow for multiprotein topology modeling, which we benchmark against a subset of the multiprotein complexes within the PDB. This improved workflow has allowed us to ascertain both the minimal experimental restraint sets required for generation of high-confidence multiprotein topologies, and quantify the ambiguity in models where insufficient IM-MS information is available. We conclude by projecting the future of IM-MS in the context of protein quaternary structure assignment, where we predict that a more complete knowledge of the ultimate information content and ambiguity within such models will undoubtedly lead to applications for a broader array of challenging biomolecular assemblies.
Graphical abstract
Introduction
Structural characterization of the multicomponent complexes that form the functional units of the “interactome”, specifically protein complexes, represents a major challenge for structural biology.[1,2] Due to their large size, low copy numbers, and intrinsic heterogeneity and lability, important targets are too often refractory to analysis by traditional techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, or electron microscopy, despite impressive advances in these fields.[3,4] Alternative approaches for characterizing difficult multicomponent structures may result in low-resolution or sparse datasets, such as those generated from small-angle scattering[5] or covalent labeling/crosslinking methodologies.[6] Circumventing the limitations of a single technique, integration of datasets from multiple experiments has been shown to be a potent approach for characterizing multiprotein complexes,[7] as often times these datasets provide complementary information. This workflow, commonly referred to as integrative structural biology, has progressed rapidly due largely to advances in computational techniques that have made it possible to encode different types of experimental datasets as spatial restraints in a single modeling workflow.[8]
Generally, an integrative modeling workflow is an iterative process described by four major steps: 1) the gathering of experimental data, 2) the translation of such data into spatial restraints, 3) the generation of an ensemble of putative structures that satisfy the experimentally-defined restraints, and 4) the characterization of the ensembles generated, where ambiguities are identified and used to refine structural hypotheses. This process may then be iterated as necessary in order to resolve ambiguities to the extent allowed by the experimental restraints utilized.[8] MS-based methods such as chemical crosslinking,[9–11] native-MS,[12,13] and ion mobility-MS[14] have garnered much attention as valuable experiments within such integrative structural biology frameworks. Of these MS-based technologies, ion-mobility-mass spectrometry (IM-MS) is uniquely positioned for the interrogation of multiprotein structure.[15] Unlike solution-phase measurements which may report on the average of an ensemble of proteoforms, conformers, or oligomerization states, IM-MS datasets can be used to discern the relative proportions of these species within mixtures, and interrogate their composition, connectivity, and collision cross sections (CCSs) individually.[16] Due to its unique capabilities in protein structure analysis, IM-MS is often deployed to determine coarse-grained (CG) protein topology models for assemblies that have resisted previous characterization attempts, often in combination with other forms of biophysical data.[17,18]
Figure 1A illustrates the potential information content often derived from native MS datasets. While direct analysis of the masses of intact complexes can often provide unambiguous information about the protein composition and stoichiometry,[19] it is also useful to interrogate solution or gas-phase disassembly products to further elucidate connectivity and structural modularity. To this end, methods for solution[20,21] and gas-phase[22–24] disruption of multiprotein complexes are actively being developed to increase the number of observable sub-complexes, and therefore the overall information content of the experiment. In addition to the composition and connectivity information garnered by MS, IM-MS (Figure 1B) provides 3D structural information on both monomeric and oligomeric protein ions in the form of CCSs.[25,26] Since multiple methods are available for the accurate calculation of CCS values from in silico models,[27–29] it is possible to assign putative structures to the signals observed in the IM-MS experiment.
Figure 1.
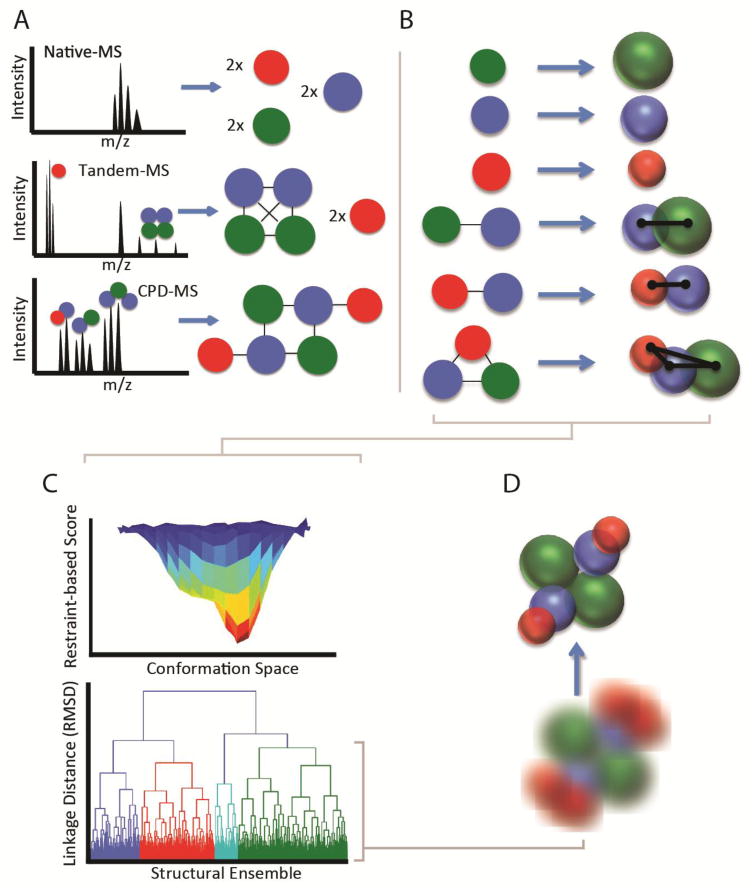
A general workflow for IM-MS-based modeling. A) native-MS, tandem-MS, and solution-phase disruption-MS yield increasing amounts of composition and connectivity information for a multiprotein complex. This information can be encoded with varying levels of ambiguity based on the information available. B) IM-MS data can be included to build a 3D topology mode. Individual subunits or domains can be encoded as spheres with radius derived from their measured CCS, while exact distances between subunits can be derived from CCS measurements of dimeric and trimeric species. C) Optimization of the experimentally-defined scoring function using a Monte Carlo method provides unbiased sampling of potential structures for high-stoichiometry complexes. These structures form an ensemble which is subjected to clustering analysis to mine for predominant structural families D) Structural families detected by clustering can be characterized in aggregate using kernel density functions, mean structures and standard deviations, or individual structures can be identified as representative of the family.
Despite being used to restrain rigorous dynamics experiments for peptides [30] and small proteins [31] for decades, our ability to extract structural information from CCS measurements of large proteins and multiprotein complexes is still evolving. A recent comprehensive analysis of the PDB revealed that the general amount of CCS variance exhibited by proteins increases for high mass and stoichiometry protein complexes, indicating increased CCS information content is available for such assemblies.[28] These observations corroborate earlier experimental results showing that the oligomerization patterns of homomeric protein complexes can be discerned in many cases based on CCS trends as a function of complex stoichiometry.[32]
Methods for extracting topological information for large, heteromeric protein complexes are, however, less developed. Early procedures for optimizing pairwise and trimeric subunit interactions were based on a linear search for conformations, using spherical subunit representations that satisfied experimental CCS restraints.[33] Although the spherical representation of protein subunits possesses obvious limitations when modeling highly aspherical subunits such as multidomain proteins, spheres still represent the primary component in IM-MS based modeling due to their trivial geometric relationship to the CCS parameter, their ease of implementation in computational workflows, and their facile relationship to protein-protein interaction geometries. Subsequently described IM-MS workflows aimed at the generation of protein quaternary structure models (Figure 1C) utilized a Monte Carlo approach for sampling orientations of spheres that satisfied excluded volume, symmetry, connectivity, and CCS restraints in order to yield an ensemble of structures that can be interrogated via hierarchical clustering methodologies.[34,35,33] Such IM-MS derived models have been favorably compared to structures produced using more mature workflows, indicating a promising level of cooperativity between CCS measurements and other biophysical parameters commonly used in protein complex model generation.[34] This general approach has been used to elucidate the topological features of the DNA replisome,[33,36] ribosomal initiation factor complexes,[37] and ATPases,[38] all providing critical structural insights as well as methodological enhancements. More recently, surface induced dissociation (SID) coupled to IM-MS and covalent labeling has been applied to build a complete model of the toyocamycin nitrile hydratase complex[39] by leveraging the sub-complexes produced both through controlled disruption in solution and SID.
Despite these promising examples, many questions remain about the ability to unambiguously assign protein topology using IM-MS datasets (Figure 1D). Most of these questions surround the potential errors introduced when high levels of coarse graining are applied, the interpretation of structural ensembles generated from IM-MS modeling approaches, and the confidence levels associated with IM-MS structures in a general sense.[40] Additionally, questions remain regarding the extent of structural rearrangement apparent in some proteins and complexes in the gas-phase; a topic that has been investigated in detail elsewhere.[35,41] In this Critical Insight, we seek to critically evaluate the information content of IM-MS for protein quaternary structure assignment in cases where we can assume a strong memory of solution-phase structure. Based on many of the challenges described above, we develop a new generalized algorithm for translating IM-MS datasets into structural models and benchmark our new method against many known topologies present in the PDB. We continue by quantifying, for the first time, the ambiguity present in under-restrained models, and suggest approaches for mitigating such effects. We conclude by projecting the future of IM-MS derived models of protein quaternary structure.
Assessing Coarse-Graining Errors in Multiprotein Models Generated from IM-MS Data
In workflows that utilize IM-MS data to restrain models of protein quaternary structure, it is typically assumed that the protein components of the assembly can be accurately represented by spheres defined by either their measured or estimated CCS. Although many reports have demonstrated a strong correlation between experimental CCS measurements and CCS values extracted from solution-phase protein models, the strength of this correlation can depend on the domain structure and globularity of the protein analyte in question.[42,43] Moreover, the magnitude and nature of the errors incorporated into IM-MS multiprotein models through the coarse-graining process are currently unknown. In order to investigate such coarse-graining errors, we extracted a non-redundant set of 191 high-resolution protein complex structures from the 3D complex set database,[44] and developed a method for the rapid generation of CG structures based on these entries where the extent of coarse graining can be treated as a variable. The first step in our protocol involves extracting coordinates and center-of-mass values for each subunit within the protein complex. Next, the CCS values are calculated for each subunit using the projection approximation function within the IMPACT library.[28] To generate the initial CG model at subunit resolution, we placed spheres having radii corresponding to the projected area of the subunits at the center of mass for each subunit in the complex. To evaluate the model, the projected area of the high-resolution structure was compared to that calculated from the CG model.
Our results suggest that a significant number of the protein complexes currently available within the PDB contain subunits that are not accurately represented when subunit-level coarse-graining is applied. As shown in Figure 2A–C, subunit-level coarse-graining very often results in large deviations in CCS compared to the reference. We define CG error as the total percent of atoms found within the high-resolution structure that fit within an average of CG representations determined by our workflow (see Supporting Information for details). We used a 5% deviation in the CCS values obtained for CG models when compared to reference CCSs for the corresponding all-atom reference structure to define a “significant” error threshold in our analysis, as such defects reflect, in our view, both the maximum error that can be introduced into a model before losing significant topology information, as well as the maximum error value carried by experimental restraint information recovered for large protein complexes by IM-MS.[45] Specifically, over 38% of the protein complexes studied here contained significant errors (greater than 5%) when this level of coarse graining was applied. We also note that the error distribution associated with this level of coarse graining is highly asymmetric, containing many structures having CG errors greater than 10%.
Figure 2.
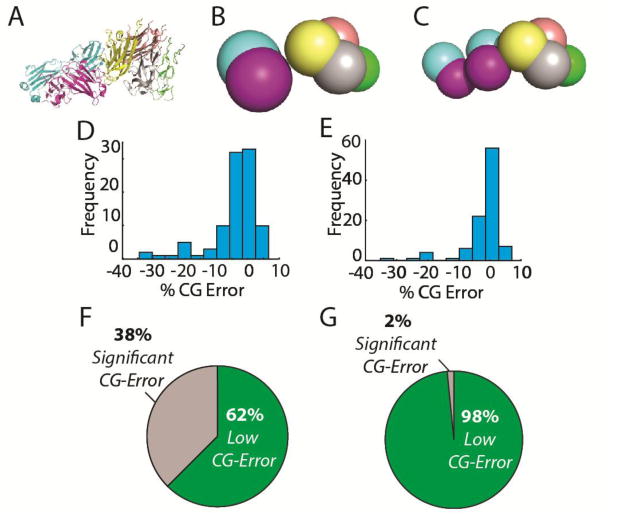
Coarse-graining error for domain and subunit-level representations. A) An example high resolution reference structure PDB ID 4MXW with subunits color coded. B.) A coarsegrained model of 4MXW at the subunit level. C) A coarse-grained model of 4MXW at the domain-level. D) 191 non-redundant protein topologies were coarse grained at the subunit-level. The coarse-graining error distribution for this level of coarse-graining is shown, with bin sizes of approximately 4 Å along the X-axis. E) Subunit-level coarse graining introduced significant CCS errors for 28% of the complexes in our set. F) The coarse-graining error distribution for the same set of protein topologies coarse-grained at the domain level. G) When coarse-grained at the domain level, only 2% of topologies had significant coarse-graining errors introduced.
A more detailed analysis of the structures within the survey reveals that proteins with multiple domains are most susceptible to high CG errors, especially those proteins having domains connected by long linker regions. Interestingly, however, we found no correlation between the CCS/mass ratio of individual subunits and their propensity to introduce error into the model, indicating that the overall packing density of the protein does not play a major role in the CG errors on display in Figures 2D and 2E. Based on this data we hypothesized that coarse-graining at the domain level should eliminate the majority of the errors we observed from our subunit-resolution CG modeling experiments. To investigate this, we implemented a k-means clustering method[46] in SciPy[47] to heuristically detect protein domain structure over a range of thresholds associated with protein and domain mass (See Supporting Information for details). The results associated with these higher-resolution CG structures are shown in Figures 2F and 2G, and reveal a strong relationship between the resolution of the CG structures and the propensity for CG error we record during our analysis. Figure 2G, for example, shows that the fraction of protein complexes with significant errors drops to ∼2% when domain-level CG is applied to the same pool of structures analyzed in Figure 2E. We find that error-prone structures that persist in our analysis are largely those containing extremely long linker regions or aspherical domains that remain inaccurately represented using domain-level coarse-graining.
Benchmarking the Information Content of IM-MS Datasets for Modeling Known Protein Complexes
To generate ensembles of putative structures based on IM-MS-derived data, we developed a program for the interpretation and optimization of diverse MS and/or IM-derived restraint sets. This program, referred to as IMMS_modeler, was built using connectivity and distance restraints from the Integrated Modeling Platform (IMP)[8], some of which were implemented previously.[34] Novel aspects of our approach include: 1) the use of a restraint file for facile input of new data, 2) the ability to use new mathematical definitions within the scoring function, and 3) a new Monte Carlo algorithm that enables a significantly broader sampling restraint space. By default, IMMS_modeler generates ensembles of 1000 structures that satisfy all of the declared restraints. We found this amount of structures to be a representative sample of structural space for most complexes, and have based the following experiments on these ensembles. All CCS calculations were performed offline using the projected area function in the IMPACT library (See Supporting Information for Details).
In order to thoroughly evaluate our method as a general approach for modeling multiprotein complexes, we set out to benchmark IM-MS modeler against known protein complex topologies with varying levels of restraint information. In these experiments, we generated CG models at the resolution of individual protein subunits for a small subset of complex topologies used in the previous experiment. For simplicity, we focused this stage of our analysis only on those protein complexes that did not show significant CG error, as described in the above section (Figure 2). Despite these limitations, the geometric principles described here are transferrable to models created at higher levels of CG resolution.
On the Positive Predictive Power of IM-MS datasets
In order to characterize the information content associated with CCS measurements of intact protein complexes and sub-complexes when used to define inter-protein distances and geometry in the context of a search of potential quaternary structures (which we define as “internal restraints”), we simulated IM-MS datasets for at least five non-redundant complex topologies for protein trimers, tetramers, pentamers, and hexamers (Figure 3). Although some of the complexes used to generate the analysis shown in Figure 3 contain symmetric elements, no symmetry restraints were implemented to avoid bias. All restraint sets contained detailed information regarding the connectivity of the complex, as well as the CCS of the intact assembly, as in our view these restraints are essential for any IM-MS based quaternary structure assignment. In addition to this information, restraint sets contained varying numbers of the “internal restraints” described above, which correspond to the pair-wise distance restraints that are commonly obtained from native IM-MS datasets, are also included in our analyses. [21,32] We note that although 3D systems are completely restrained by a minimum of 3N-6 fixed distance restraints (where N is number of bodies), the restraints used in this report attempt to simulate real IM-MS data. Specifically, restraints mined from IM-MS data contain error, often producing predictive values that are less than those generated through precise distance geometries. For purposes of this analysis, the structures generated using our method were defined as true positives (native-like topologies) if they had RMSD values of less than 5 Å relative to the reference structure; and we defined a positive predictive value (PPV) as the fraction of true positive structures within the ensemble of structures sampled using a given restraint set (see Figure S2 for examples of structures filtered out of these analyses using RMSD).
Figure 3.
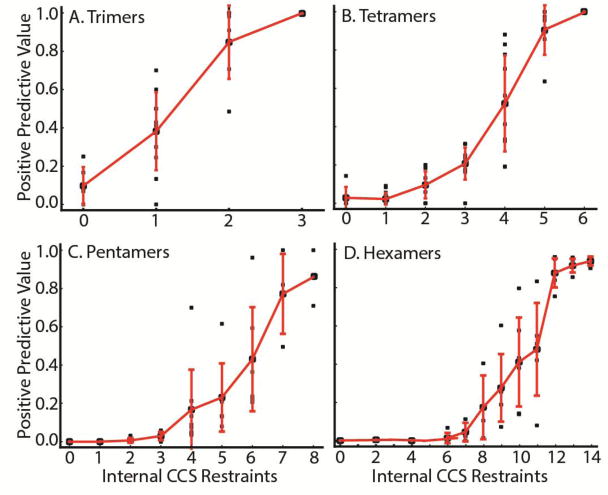
Positive Predictive Values of the IM-MS restraint sets plotted as a function of the number of internal CCS-derived restraints. At least 5 non-redundant topologies from the PDB were considered for each number of subunits, A) Trimers B) Tetramers C) Pentamers and D) Hexamers. Each restraint set was manually curated to ensure the data reflected data that could be reasonably generated through existing IM-MS technologies.
As expected, our results reveal a positive relationship between the number of internal CCS restraints available for a complex and the positive predictive value for a given modeling effort. For trimeric protein complexes (Figure 3A), the ensemble is enriched for true positives with the addition of internal distance restraints between subunits. Here, due to the trivial relationship between the CCS and the angle of subunits within the complex, the model should be fully restrained by the global CCS plus any two IM-derived distance restraints.[33] Notably, there is one outlier structure that seemingly refutes this general conclusion; however, our analysis also suggests that the global CCS restraint becomes less sensitive when large disparities exist in the CCS of each component, allowing us to rationalize all of the results shown. (Figure S3) Higher stoichiometry complexes (Figure 3B–D), exhibit similarly strong increases in PPV in a manner correlated with the number of internal restraints included. We note that the number of restraints necessary to reach a PPV > 0.8, where 80% of the structures identified in the ensemble are within 5 Å of the “true” structure, increases rapidly as the number of subunits increases, further motivating the need to develop new methods and technologies for the comprehensive generation of native-like sub-complexes for IM-MS analysis.[20,23,24]
Characterizing Ambiguity in the Structural Ensembles Defined by IM-MS
Although the PPV is a valuable metric for comparing the information content of multiple restraint sets, interpretation of PPV values for individual datasets can be challenging. This is due to the fact that members of a structural ensemble generated by the IMMS-modeling approach described here are not randomly distributed, and in many cases can be clustered into distinct sub-distributions, or structural families. Pairwise relationships between structures within an ensemble can be described by a pairwise RMSD matrix, which can in turn be interrogated using hierarchical clustering to determine groups of highly related structures. Alternatively, other similarity measures can be implemented to describe structural relationships between models, including the ultrafast similarity score,[48] or distance matrix RMSD,[49] which each may have their own advantages depending on the geometries present in the ensemble. For the computational data described in this Critical Insight, a detailed analysis of the structural ensemble produced from an IM-MS restrained search of protein topology space regularly reveals useful information, in addition to what is provided by the PPV value analysis shown in Figure 3 alone. In the sections below, we discuss the interpretation of hierarchical clustering datasets in the context of such IM-MS restrained models, focusing on our recent efforts to define and quantify the ambiguity and resolution within the IM-MS data.
A hierarchical clustering dendrogram (as shown in Figure 4) illustrates the relationship between all structures within an ensemble. The number of clusters depends on the “cut point” chosen during dendrogram analysis, a value that is typically a user defined parameter. For example, our algorithm automatically defaults to a dendrogram cut point that generates clusters at linkages that exhibit greater than 70% of the maximum RMSD in the entire matrix analyzed. Our ensemble analysis workflow evaluates the in-cluster RMSD as it compares to the average RMSD of the ensemble, as well as the cross-cluster RMSD, revealing distinct structural families that define the identified clusters (Figure 4). It is worth noting that the application of IM-MS restraints often leads to the type of model ambiguity shown in Figure 4 for large hetero-protein targets.[34] Indeed, such ambiguity may, in some cases, represent the native ensemble of protein complex structures associated with function.[50,38] Commonly, however, such uncertainty is due to incomplete structural information and can be resolved through the application of additional restraints [18,51] (see below for examples).
Figure 4.
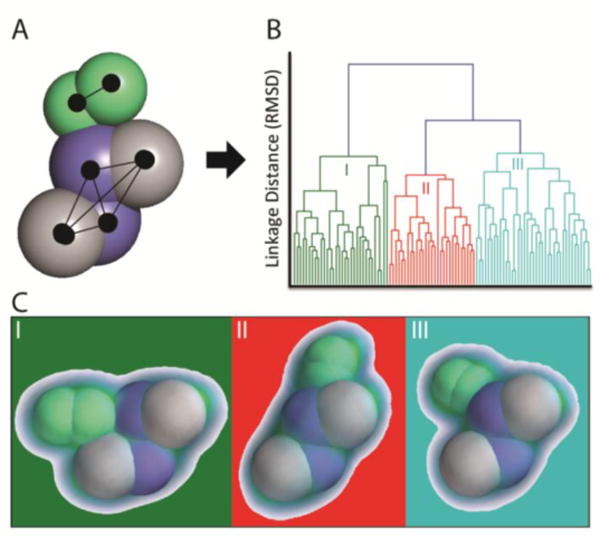
Parsing Structural Ensembles Generated with Ambiguous Restraint Sets. A) A restraint set was generated for 2AFH, a nitrogenase heterotetramer (purple and grey) bound to the dimeric nucleotide switch protein (green). The binding location and pose for the nucleotide switch protein is not defined in the restraint set and a CCS-filtered structural ensemble contains many putative structures. B) Hierarchical clustering of the ensemble reveals three distinct structural families within the ensemble, greatly simplifying the analysis. C) Plotting the kernel density function of each structural family reveals high resolution within all families.
As mentioned above, the in-cluster RMSD can be a valuable metric for quantitatively expressing the ambiguity within a cluster. However, when evaluating biomolecular structures, qualitative and visual expression of ambiguity is often more facile to interpret. In order to fill this gap for IM-MS derived models, we developed a new method for visualizing the ambiguity within a structural family using kernel density functions.[52,47] In this method, the coordinates within a structural family or ensemble are aligned, and each subunit coordinate is uniformly populated with protein density as a sphere corresponding to its collision cross section. Next, the Gaussian kernel function is estimated for this volume of coordinates, and then visualized. For the workflow described here, we utilize the Mayavi Library[53] in Python to visualize the kernel densities. As illustrated in Figure 4C, this kernel density function approach allows for the visualization of structural ambiguity present within an ensemble; information that is likely vital for the detailed interpretation of structural ensembles defined by sparse sets of restraints.
Leveraging Symmetry and Modularity to Resolve Ambiguity within IM-MS Model Ensembles
To further evaluate IM-MS based quaternary structure assignments in a general sense, as well as the newly-developed methods described here, we chose two case studies that illustrate real-world examples of challenging modeling targets. As shown in Figure 3, the number of restraints needed to accurately recapitulate the topology of a multiprotein complex increases linearly, creating challenges for integrative modeling of these complexes. However, in the data shown below, we demonstrate that by leveraging the modularity and symmetry within high-stoichiometry complexes, it is possible to circumvent these limitations.
As an example of a symmetry restraint applied in order to resolve ambiguity within an IM-MS restrained ensemble of protein quaternary structures, we built models of the Large T-antigen (LTag) complex bound to p53. LTag is a hexameric ring structure that binds p53 monomers in a stoichiometric and symmetric fashion around the ring.[54] Assuming a comprehensive protein-protein connectivity dataset from Native MS, we searched for a minimal IM-MS restraint set to recapitulate the known topology of LTag-p53 with C6 symmetry. Our first attempt utilized only connectivity and global CCS information to generate a structural ensemble. (Figure 5A) For this ensemble, we observe three structural families, with relatively little resolution between them. Each family is represented by a very broad distribution of RMSD values relative to the reference structure, indicating that both the accuracy and effective resolution of the structural models created in this search are low. The kernel density function estimated for each structural family also illustrates the poor resolution generated from this restraint set.
Figure 5.
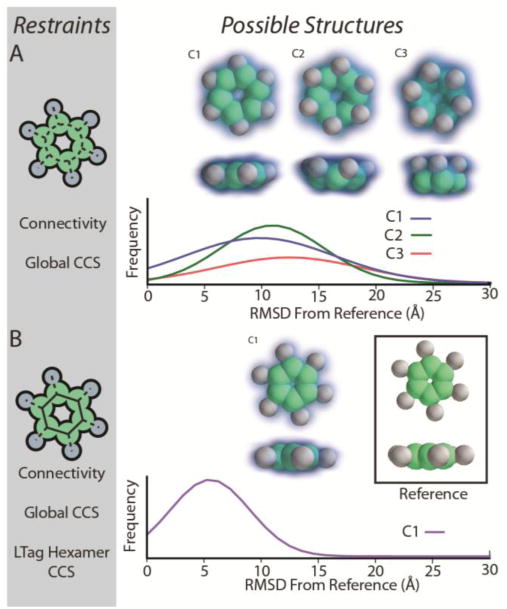
Modeling the topology of hexameric LTag bound to p53 using the symmetry restraint. Two restraint sets (left panels, A and B) were used to generate structural ensembles that were evaluated using hierarchical clustering, kernel density functions, and RMSD distributions. (Right panels).
In order to resolve the above ambiguity, we add restraints associated with the CCS of the LTag hexamer and the overall C6 symmetry of the complex, a likely result given the interface structure known for this assembly.[54] The resulting IM-MS restrained ensemble is homogenous and gives rise exclusively to highly accurate models (Figure 5B). This monomodal ensemble of structures is characterized by a significantly narrower distribution of RMSD values when compared with the distributions observed in Figure 5A, and is centered at an RMSD of 6Å relative to the reference. Such RMSD values are typically achieved by our modeling workflow for structures where additional symmetry restraints can be coupled to the distances mined from IM-MS data.
For our second example, we sought to apply our method to a large, asymmetric protein complex that has been interrogated using MS methods previously.[55] The Actin-Related Protein 2/3 (ARP2/3) complex structure was recently solved by X-ray crystallography (PDB ID 1K8K).[56] In addition, a previous Native MS study identified two modules within the heptameric complex, the trimeric Actin Localization Module (ALM) and the tetrameric Nucleating Module (NM). Extrapolating from the data shown in Figure 3, we predict that the heptameric ARP2/3 requires between 16 and 19 internal CCS restraints to reach a PPV value of 80%. When modeling the ALM and NM individually, we find that even minimal simulated IM-MS restraint sets lead to highly accurate models. We generated high-confidence models for the trimeric ALM using 2 IM-derived distance restraints and a global CCS restraint. In parallel, the correct structure was readily found for the NM using 4 IM-derived distance restraints plus the global CCS restraint. These results agree well with data shown in Figure 3 for trimeric and tetrameric protein complexes.
Next, we attempted to find the minimal IM-MS restraint sets necessary for localization of ALM binding to NM, leading to a precise assignment of ALM-NM topology. We started by attempting to model this complex without providing any information about points of connectivity between ALM and NM, and filtered the resulting ensemble based on global CCS alone. (Figure 6A) The resulting ensemble features two structural families, a larger population family with an RMSD distribution centered on 15 Å from the reference structure, and a less populated cluster with a very broad RMSD distribution centered on 28 Å. Interestingly, although the resolution within both families is poor, the major family appears to correctly localize the general ALM binding site on the NM surface. To reduce the ambiguity in the models, we then added two restraints that enforced connectivity between the p20 subunit of the ALM and the p34 and arp3 subunits of the NM (Figure 6B). This new connectivity information, along with the global CCS restraint gives rise to a new ensemble of potential structures. The new restraint set acts to eliminate the majority of the incorrect structures found in Figure 6A; however, it gives rise to a new, more highly-resolved distribution of structures centered on 25 Å from the reference. Interestingly, we note that the major structural family identified for this restraint set remains essentially unchanged from the one identified in Figure 6A, where the correct localization of ALM is determined, but having a broad RMSD distribution centered on ∼15 Å from the reference structure
Figure 6.
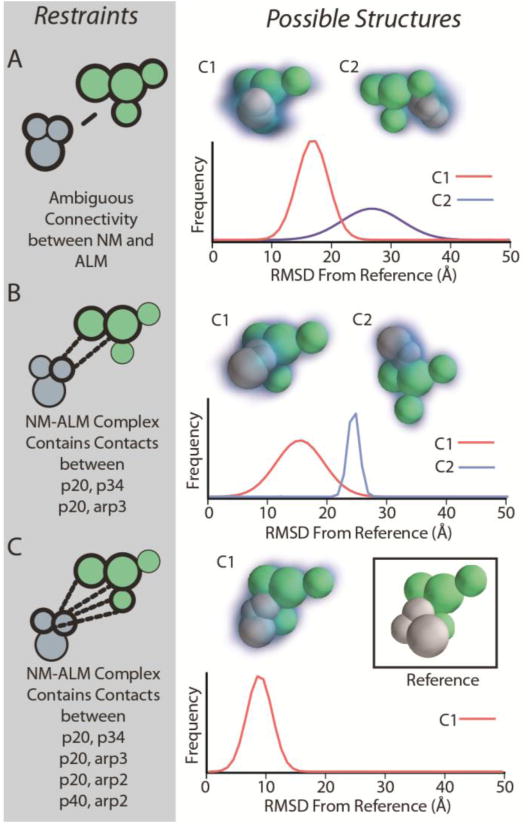
Docking modules within the ARP2/3 complex using connectivity restraints. After encoding the structures of the nucleating module (NM) and the actin localization module (ALM), we tested the global CCS in conjunction with various sets of connectivity restraints (left panels, A, B, and C) for their ability to restraint the docking location and pose of NM on ALM. Structural ensembles were evaluated by hierarchical clustering and the structural families, kernel density functions, and RMSD distributions from the reference are provided.
Finally, we applied a new restraint set with 4 total connectivity restraints linking p20 from the ALM with p34, arp2, and arp3 from the NM; and linking p40 from the ALM with arp2 from the NM. (Figure 6C) These restraints represent the full complement of protein connectivity information accessible through MS methods.[57] When combined with sufficient connectivity information, we find that the global CCS restraint can define not only the location of ALM on the surface of NM, but also the relative orientation of the two sub-complexes. We observe a single, well-resolved family of structures centered around and RMSD value of 9 Å relative to the reference structure. Furthermore, when structures within this family are averaged, the resulting mean structure has an RMSD of only 2 Å from the reference, indicating that in this example, the mean structure is in much closer agreement with the reference than any individual structure in the ensemble. Although the mean structure in this case results in a highly accurate candidate model, we advise caution when averaging structural ensembles generally, as poorly defined structural families may cause the average structure to be distorted, or heavily biased. Combining the connectivity restraints used here with the distance and internal CCS restraints used to build models for each module, we recapitulated the correct topology using only 11 internal restraints, one third fewer internal restraints than that the number of restraints one would predict based on PPV alone (extrapolated from Figure 3).
Conclusions and Future Directions
In this report we explored several questions related to the generation of CG multi-protein topology models restrained using IM-MS data. We outlined a workflow based on integrative modeling principles that allows for facile translation of IM-MS data into ensembles of putative structures for hypothesis refinement or integration with high resolution docking tools. We explored the limits of coarse-grained modeling, and demonstrated that many protein topologies found in the PDB are not amenable to coarse-graining at the subunit-level, mostly due to their intricate domain architectures. However, when sufficient data is available, domain-level coarse-graining results in significant errors in only 2% of cases.
We benchmarked our CG modeling workflow against protein topologies extracted from the PDB, exploring the ambiguity in IM-MS derived structural ensembles as a function of the information content contained in restraint sets. Our results indicated a predictable relationship between the PPV of an ensemble, and the number of internal IM-MS restraints used to generate it. Although the estimated PPV may be used as a benchmark to predict the ambiguity within a CG modeling ensemble, in many cases it underestimates the total possible information content of the IM-MS experiment, as such an analysis does not account for the structural relationships between members of an ensemble. We found that applying hierarchical clustering yields, in many cases, highly-resolved conformational families that can inform future experiments, or be reported as likely structures based on available data. Additionally, we undertook two case studies that showed that highly-symmetric or modular complexes can be modeled with high fidelity using smaller numbers of internal restraints than those predicted by a PPV analysis. In these cases, we observe that in large complexes the information content of the intact CCS is maximal only when one or more substructures are fully defined.
Although the computational results presented in this Critical Insight are encouraging, there are still many challenges ahead in fully harnessing the information content available in IM-MS datasets. Our CG error analysis (Figure 2) clearly motivates the development of domain-level IM-MS models of protein quaternary structure, and a move away from CG at the intact subunit level. The development of IM-MS tools for the generation of such information on protein tertiary structure, such as collision induced unfolding (CIU),[58,59] as well as efforts to integrate IM-MS data with other sources of experimental data sensitive to local protein structure[60,61,51] and computational domain assignment algorithms[62] will, therefore, become increasingly important in future IM-MS protein topology modeling efforts. Similarly, our analysis of ambiguity in IM-MS models of protein quaternary structure strongly points to the need for improved methodologies capable of detecting protein complex connectivity and symmetry. As such, the development of technologies that produce a comprehensive population of protein sub-complexes, either in the gas-phase or in solution, will prove highly valuable.[23,20,24] Finally, the ability of our IMMS-Modeler algorithm to assess, for the first time, the ambiguity present within IM-MS restrained models of protein complex structure will likely lead to a greater ability to integrate such datasets with other forms of structural restraints, derived both from MS and other forms of data. Future iterations of IMMS-Modeler will incorporate the ability to build models based on custom shapes, interface directly with domain-prediction software, and utilize next-generation scoring functions that enable multi-factorial assessments of model fitness. Although not discussed in detail here, it is also clear that increases in CCS precision will drive concomitant increases in the PPV of IM-MS restraints, as decreasing the current ± 3% CCS error value used in the analyses described here will surely reduce the occurrence of spurious structural families within a filtered structural ensemble.[63–65] On the other hand, our data demonstrate that much can be accomplished using current IM-MS capabilities and that the proper application of restraints can be used to build high-confidence models of multi-protein complexes with both full knowledge of their precisions and informed estimates of their accuracies.
Supplementary Material
Figure S1. Coarse-graining CCS error as a function of Subunit Residues/Sphere
Figure S2. Example structures at various levels of RMSD as measured by internal distances for the tetramer 1DXR (left) and hexamer 2BS2 (right). The high resolution and coarse-grained (CG) reference structures are provided, followed by example structures at increasing RMSD. RMSD values of under 5 angstroms represent the cutoff for quality models in this study.
Figure S3. CCS restraints increase in selectivity when subunits are similar in size. In our analysis of the PPVs for trimeric protein complexes from the 3D complex set, we found one outlier in the dataset (PDB ID 2INC) showing extremely low PPV even when restrained by a set of 2 internal CCS restraints and a global CCS restraint. We observed that in this protein complex, one subunit was approximately 75% smaller than the other proteins in the system. In order to determine the impact of this size disparity on the observed PPV, we artificially inflated the CCS of the small subunit until it was in parity with the others. Indeed, as the CCS of the small subunit was increased, the corresponding restraint set yielded a higher PPV.
Acknowledgments
Protein topology modeling efforts in the Ruotolo lab are supported through the National Institute of General Medical Sciences, National Institutes of Health (R01 GM095832). Additionally, we gratefully acknowledge the support of Erik Marklund (Uppsala), Matteo Degiacomi (Oxford), and Justin Benesch (Oxford), who helped us to integrate IMPACT CCS calculations into our computational workflows.
Footnotes
Supplemental Information: All of the software used in this work for modeling and analysis will be made freely available at: https://sites.lsa.umich.edu/ruotolo/software/IMMS_Modeler, Supplemental figures, discussion, and experimental methods can be found in the online Supporting Information document.
References
- 1.Robinson CV, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450(7172):973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- 2.Marsh JA, Teichmann SA. Structure, dynamics, assembly, and evolution of protein complexes. Annu Rev of Biochem. 2015;84:551–575. doi: 10.1146/annurev-biochem-060614-034142. [DOI] [PubMed] [Google Scholar]
- 3.Hansen MR, Graf R, Spiess HW. Solid-State NMR in Macromolecular Systems: Insights on How Molecular Entities Move. Accounts Chem Res. 2013;46(9):1996–2007. doi: 10.1021/ar300338b. [DOI] [PubMed] [Google Scholar]
- 4.Skiniotis G, Southworth DR. Single-particle cryo-electron microscopy of macromolecular complexes. Microscopy. 2016;65(1):9–22. doi: 10.1093/jmicro/dfv366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mertens HDT, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J Struct Biol. 2010;172(1):128–141. doi: 10.1016/j.jsb.2010.06.012. http://dx.doi.org/10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol. 2007;8(8):645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- 7.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007;450(7170):683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 8.Russel D, Lasker K, Webb B, Velázquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, Sali A. Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLoS Biol. 2012;10(1) doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT. Structural Characterization by Cross-linking Reveals the Detailed Architecture of a Coatomer-related Heptameric Module from the Nuclear Pore Complex. Mol Cell Proteomics. 2014;13(11):2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stengel F, Aebersold R, Robinson CV. Joining Forces: Integrating Proteomics and Cross-linking with the Mass Spectrometry of Intact Complexes. Mol Cell Proteomics. 2012;11(3) doi: 10.1074/mcp.R111.014027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 12.Hyung SJ, Ruotolo BT. Integrating mass spectrometry of intact protein complexes into structural proteomics. Proteomics. 2012;12(10):1547–1564. doi: 10.1002/pmic.201100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smits AH, Vermeulen M. Characterizing Proteins and Protein Interactions Using Mass Spectrometry: Challenges and Opportunities. Trends Biotechnol. 34(10):825–834. doi: 10.1016/j.tibtech.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Zhong Y, Hyung SJ, Ruotolo BT. Ion mobility–mass spectrometry for structural proteomics. Expert Rev of Proteomics. 2012;9(1):47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruotolo BT, Benesch JL, Sandercock AM, Hyung SJ, Robinson CV. Ion mobility–mass spectrometry analysis of large protein complexes. Nat Protoc. 2008;3(7):1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 16.Chorev DS, Ben-Nissan G, Sharon M. Exposing the subunit diversity and modularity of protein complexes by structural mass spectrometry approaches. Proteomics. 2015;15(16):2777–2791. doi: 10.1002/pmic.201400517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcoux J, Cianferani S. Towards integrative structural mass spectrometry: Benefits from hybrid approaches. Methods. 2015;89:4–12. doi: 10.1016/j.ymeth.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 18.Politis A, Borysik AJ. Assembling the pieces of macromolecular complexes: Hybrid structural biology approaches. Proteomics. 2015;15(16):2792–2803. doi: 10.1002/pmic.201400507. [DOI] [PubMed] [Google Scholar]
- 19.Snijder J, Heck AJR. Analytical Approaches for Size and Mass Analysis of Large Protein Assemblies. Annu Rev of Anal Chem. 2014;7(1):43–64. doi: 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- 20.Zhong Y, Feng J, Ruotolo BT. Robotically-Assisted Titration Coupled to Ion Mobility-Mass Spectrometry Reveals the Interface Structures and Analysis Parameters Critical for Multiprotein Topology Mapping. Anal Chem. 2013;85(23):11360–11368. doi: 10.1021/ac402276k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsh JA, Hernández H, Hall Z, Ahnert SE, Perica T, Robinson CV, Teichmann SA. Protein Complexes Are under Evolutionary Selection to Assemble via Ordered Pathways. Cell. 2013;153(2) doi: 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benesch JLP. Collisional Activation of Protein Complexes: Picking Up the Pieces. J Am Soc Mass Spectrom. 2009;20(3):341–348. doi: 10.1016/j.jasms.2008.11.014. http://dx.doi.org/10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Zhou MW, Wysocki VH. Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas phase. Accounts Chem Res. 2014;47(4):1010–1018. doi: 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- 24.Samulak BM, Niu S, Andrews PC, Ruotolo BT. Ion Mobility-Mass Spectrometry Analysis of Cross-Linked Intact Multiprotein Complexes: Enhanced Gas-Phase Stabilities and Altered Dissociation Pathways. Anal Chem. 2016;88(10):5290–5298. doi: 10.1021/acs.analchem.6b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uetrecht C, Barbu IM, Shoemaker GK, van Duijn E, Heck AJ. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat Chem. 2011;3(2):126–132. doi: 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- 26.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal Chem. 2010;82(22):9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 27.Bleiholder C, Contreras S, Bowers MT. A novel projection approximation algorithm for the fast and accurate computation of molecular collision cross sections (IV). Application to polypeptides. Int J Mass Spectrom. 2013;354:275–280. doi: 10.1016/j.ijms.2013.06.011. [DOI] [Google Scholar]
- 28.Marklund EG, Degiacomi MT, Robinson CV, Baldwin AJ, Benesch JL. Collision cross sections for structural proteomics. Structure. 2015;23(4):791–799. doi: 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 29.Larriba C, Hogan CJ., Jr Ion mobilities in diatomic gases: measurement versus prediction with non-specular scattering models. J Phys Chem A. 2013;117(19):3887–3901. doi: 10.1021/jp312432z. [DOI] [PubMed] [Google Scholar]
- 30.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH. From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations. J Am Chem Soc. 2013;135(51):19147–19153. doi: 10.1021/ja4114193. [DOI] [PubMed] [Google Scholar]
- 31.Shi HL, Pierson NA, Valentine SJ, Clemmer DE. Conformation Types of Ubiquitin M+8H (8+) Ions from Water:Methanol Solutions: Evidence for the N and A States in Aqueous Solution. J Phys Chem B. 2012;116(10):3344–3352. doi: 10.1021/jp210797x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pukala TL, Ruotolo BT, Zhou M, Politis A, Stefanescu R, Leary JA, Robinson CV. Subunit architecture of multiprotein assemblies determined using restraints from gas-phase measurements. Structure. 2009;17(9):1235–1243. doi: 10.1016/j.str.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Politis A, Park A, Hyung SJ, Barsky D, Ruotolo BT, Robinson CV. Integrating Ion Mobility Mass Spectrometry with Molecular Modelling to Determine the Architecture of Multiprotein Complexes. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall Z, Politis A, Robinson CV. Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure. 2012;20(9):1596–1609. doi: 10.1016/j.str.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Evidence for macromolecular protein rings in the absence of bulk water. Science. 2005;310(5754):1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 36.Politis A, Park A, Hall Z, Ruotolo BT, Robinson CV. Integrative Modelling Coupled with Ion Mobility Mass Spectrometry Reveals Structural Features of the Clamp Loader in Complex with Single-Stranded DNA Binding Protein. J Mol Biol. 2013;425(23) doi: 10.1016/j.jmb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Politis A, Schmidt C, Tjioe E, Sandercock AM, Lasker K, Gordiyenko Y, Russel D, Sali A, Robinson CV. Topological Models of Heteromeric Protein Assemblies from Mass Spectrometry: Application to the Yeast eIF3:eIF5 Complex. Chem & Biol. 2014 doi: 10.1016/j.chembiol.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou M, Politis A, Davies RB, Liko I, Wu KJ, Stewart AG, Stock D, Robinson CV. Ion mobility–mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat Chem. 2014;6(3):208–215. doi: 10.1038/nchem.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song Y, Nelp MT, Bandarian V, Wysocki VH. Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent Sci. 2015;1(9):477–487. doi: 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneidman-Duhovny D, Pellarin R, Sali A. Uncertainty in integrative structural modeling. Curr Opin Struct Biol. 2014;28:96–104. doi: 10.1016/j.sbi.2014.08.001. http://dx.doi.org/10.1016/j.sbi.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han L, Hyung SJ, Mayers JJ, Ruotolo BT. Bound anions differentially stabilize multiprotein complexes in the absence of bulk solvent. J Am Chem Soc. 2011;133(29):11358–11367. doi: 10.1021/ja203527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagel K, Natan E, Hall Z, Fersht AR, Robinson CV. Intrinsically Disordered p53 and Its Complexes Populate Compact Conformations in the Gas Phase. Angew Chem Int Ed. 2013;52(1):361–365. doi: 10.1002/anie.201203047. [DOI] [PubMed] [Google Scholar]
- 43.Pacholarz KJ, Porrini M, Garlish RA, Burnley RJ, Taylor RJ, Henry AJ, Barran PE. Dynamics of Intact Immunoglobulin G Explored by Drift-Tube Ion-Mobility Mass Spectrometry and Molecular Modeling. Angew Chem Int Ed. 2014;53(30):7765–7769. doi: 10.1002/anie.201402863. [DOI] [PubMed] [Google Scholar]
- 44.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D Complex: A Structural Classification of Protein Complexes. PLoS Comput Biol. 2006;2(11):e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ, Robinson CV. Ion mobility–mass spectrometry analysis of large protein complexes. Nat Protoc. 2008;3(7):1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 46.Arthur D, Vassilvitskii S. k-means++: the advantages of careful seeding. Proceedings of the Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms; New Orleans, Louisiana. 2007. [Google Scholar]
- 47.Jones E, Oliphant T, Peterson P. SciPy: Open source scientific tools for Python. 2001 [Google Scholar]
- 48.Ballester PJ, Richards WG. Ultrafast shape recognition for similarity search in molecular databases. Proc R Soc A. 2007;463(2081):1307–1321. doi: 10.1098/rspa.2007.1823. [DOI] [Google Scholar]
- 49.Kloczkowski A, Jernigan RL, Wu Z, Song G, Yang L, Kolinski A, Pokarowski P. Distance Matrix-Based Approach to Protein Structure Prediction. J Struct Funct Genomics. 2009;10(1):67–81. doi: 10.1007/s10969-009-9062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6(4):281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 51.Politis A, Stengel F, Hall Z, Hernandez H, Leitner A, Walzthoeni T, Robinson CV, Aebersold R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat Meth. 2014;11(4):403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss R. Multivariate Density Estimation: theory, practice, and visualization. J Am Stat Assoc. 1994;89:359+. [Google Scholar]
- 53.Ramachandran P, Varoquaux G. Mayavi: 3D Visualization of Scientific Data. Comp in Sci & Engin. 2011;13(2):40–51. doi: 10.1109/mcse.2011.35. [DOI] [Google Scholar]
- 54.Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes & Devel. 2006;20(17):2373–2382. doi: 10.1101/gad.1456306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chorev DS, Moscovitz O, Geiger B, Sharon M. Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat Comm. 2014;5:3758. doi: 10.1038/ncomms4758. [DOI] [PubMed] [Google Scholar]
- 56.Robinson RC, Turbedsky K, Kaiser DA, Marchand JB, Higgs HN, Choe S, Pollard TD. Crystal Structure of Arp2/3 Complex. Science. 2001;294(5547):1679–1684. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 57.Sinz A, Arlt C, Chorev D, Sharon M. Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology. Protein Sci. 2015;24(8):1193–1209. doi: 10.1002/pro.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong Y, Han L, Ruotolo BT. Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angew Chem. 2014;126(35):9363–9366. doi: 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- 59.Eschweiler JD, Martini RM, Ruotolo BT. Chemical Probes and Engineered Constructs Reveal a Detailed Unfolding Mechanism for a Solvent-Free Multidomain Protein. J Am Chem Soc. 2017;139(1):534–540. doi: 10.1021/jacs.6b11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16(12):2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt C, Macpherson JA, Lau AM, Tan KW, Fraternali F, Politis A. Surface Accessibility and Dynamics of Macromolecular Assemblies Probed by Covalent Labeling Mass Spectrometry and Integrative Modeling. Anal Chem. 2017;89(3):1459–1468. doi: 10.1021/acs.analchem.6b02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ansari ES, Eslahchi C, Pezeshk H, Sadeghi M. ProDomAs, protein domain assignment algorithm using center-based clustering and independent dominating set. Proteins: Struct Funct Bioinf. 2014;82(9):1937–1946. doi: 10.1002/prot.24547. [DOI] [PubMed] [Google Scholar]
- 63.Hamid AM, Garimella SVB, Ibrahim YM, Deng LL, Zheng XY, Webb IK, Anderson GA, Prost SA, Norheim RV, Tolmachev AV, Baker ES, Smith RD. Achieving High Resolution Ion Mobility Separations Using Traveling Waves in Compact Multiturn Structures for Lossless Ion Manipulations. Anal Chem. 2016;88(18):8949–8956. doi: 10.1021/acs.analchem.6b01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benigni P, Marin R, Molano-Arevalo JC, Garabedian A, Wolff JJ, Ridgeway ME, Park MA, Fernandez-Lima F. Towards the analysis of high molecular weight proteins and protein complexes using TIMS-MS. Int J Ion Moblity Spectrom. 2016;19(2–3):95–104. doi: 10.1007/s12127-016-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glaskin RS, Ewing MA, Clemmer DE. Ion Trapping for Ion Mobility Spectrometry Measurements in a Cyclical Drift Tube. Anal Chem. 2013;85(15):7003–7008. doi: 10.1021/ac4015066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Coarse-graining CCS error as a function of Subunit Residues/Sphere
Figure S2. Example structures at various levels of RMSD as measured by internal distances for the tetramer 1DXR (left) and hexamer 2BS2 (right). The high resolution and coarse-grained (CG) reference structures are provided, followed by example structures at increasing RMSD. RMSD values of under 5 angstroms represent the cutoff for quality models in this study.
Figure S3. CCS restraints increase in selectivity when subunits are similar in size. In our analysis of the PPVs for trimeric protein complexes from the 3D complex set, we found one outlier in the dataset (PDB ID 2INC) showing extremely low PPV even when restrained by a set of 2 internal CCS restraints and a global CCS restraint. We observed that in this protein complex, one subunit was approximately 75% smaller than the other proteins in the system. In order to determine the impact of this size disparity on the observed PPV, we artificially inflated the CCS of the small subunit until it was in parity with the others. Indeed, as the CCS of the small subunit was increased, the corresponding restraint set yielded a higher PPV.