

Abstract
This review describes the recent advances utilizing photosensitizers and visible light to harness the synthetic potential of P450 enzymes. The structures of the photosensitizers investigated to date are first presented along with their photophysical and redox properties. Functional photosensitizers range from organic and inorganic complexes to nanomaterials as well as the biological photosystem I complex. The focus is then on the three distinct approaches that have emerged for the activation of P450 enzymes. The first approach utilizes the in situ generation of reactive oxygen species entering the P450 mechanism via the peroxide shunt pathway. The other two approaches are sustained by electron injections into catalytically competent heme domains either facilitated by redox partners or through direct heme domain reduction. Achievements as well as pitfalls of each approach are briefly summarized.
Keywords: light-driven biocatalysis, cytochrome P450, regio- and stereoselective C-H functionalization, electron transfer, redox partners, photosensitizer
Graphical abstract
1. Introduction
The activation of inert C-H bonds under mild conditions using molecular dioxygen has rendered the cytochrome P450 enzymes of high interest for biotechnological applications.[1–3] This class of heme-thiolate enzymes catalyzes the regio- and stereoselective functionalization of unactivated hydrocarbon bonds, offering major advantages over traditional methods. The catalytic mechanism involves the generation of a ferryl porphyrin radical species called compound I,[4] via the reductive activation of molecular dioxygen at the heme active site. Alternatively, hydrogen peroxide and various organic peroxides can generate the same highly oxidative species via the so-called peroxide shunt, highlighting the peroxygenase activity of some members of the P450 superfamily.[5] Cytochrome P450 enzymes share a conserved tertiary structure despite low sequence homology while displaying great diversity in substrates and redox partners.[5–7]
In terms of biotechnology, cytochrome P450 enzymes show promise toward the synthesis of high value chemicals and natural products[8–10] as well as in the detection and identification of drug metabolites for toxicological studies.[11, 12] With respect to the former, these enzymes often trump their chemical catalyst counterparts on the grounds of reaction rates, specificity and feasibility.[1] In addition, they are amenable to protein engineering, allowing the catalysis of non-canonical chemical reactions as well as the expansion of their substrate repertoire.[13, 14]
One area that has captured the imagination of scientists has been the innovation of alternative methods to promote P450 catalysis. These approaches are diverse in their scope and have already been reviewed.[15] We encourage the reader to refer specifically to those regarding electrochemical reduction,[16] fusion of redox partners and catalytic domains,[17] the use of peroxide[5] as well as cofactor regeneration systems.[18] Arguably, the most limiting factors across these approaches are the timely delivery of electrons and the minimization of uncoupled reactions.[19] Uncoupling resulting from inefficient electron transfer is problematic not only due to the waste of reducing equivalents, but additionally for its generation of reactive oxygen species, which are detrimental to the enzyme.
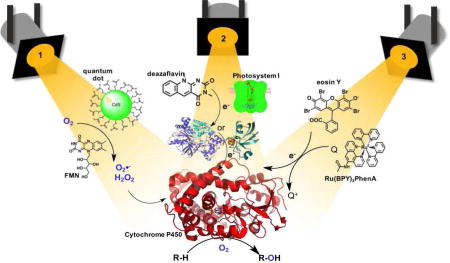
In this review, we would like to turn the spotlight to the emerging approaches that take advantage of light energy to drive P450 reactions. These approaches utilize a light-harvesting unit with the appropriate excited state properties and reduction potential to initiate the P450 catalytic mechanism (See Figure 1). Two separate paths have been pursued: the first relies on the in situ reduction of dioxygen and subsequent peroxide shunt pathway; the second mimics the natural pathway through the stepwise delivery of electrons and activation of dioxygen at the heme active site. While the first approach is limited to applications in hydrogen peroxide tolerant P450 members[5] or evolved strains,[20] widespread use of the second requires the rapid injection of electrons to catalytically competent heme domains. Two unique strategies have been employed for direct heme reduction. One approach is dependent on redox partners to mediate electron transfers from the photosensitizer to the heme domain, whereas the second utilizes a photogenerated reductive species to directly inject electrons to the heme active site, and is thus independent of redox partners (Figure 1B and C). Examples of all three strategies paired with unique photosensitizers are detailed below, each demonstrating the use of light to activate P450 enzymes.
Fig. 1.

Strategies employed to carry out P450 reactions, (A) via the formation of reactive oxygen species (peroxide or superoxide) using the peroxygenase activity of specific P450 enzymes or via heme reduction with redox partners (B) or directly (C).
2. Survey of photosensitizers
The following characteristics are desired of a suitable photosensitizer: 1) water solubility and dioxygen compatibility; 2) light-harvesting capability, preferentially in the visible region of the spectrum and not overlapping with the intense heme Soret band, 3) a photogenerated species with a redox potential suitable to carry on the reduction in the appropriate time scale and 4) compatibility with the P450 enzymes of choice. Regarding the activation of P450 enzymes, various photosensitizers have been investigated ranging from nanomaterials, such as quantum dots,[21] to organic molecules (deazaflavins,[22] isoalloxazine-type flavins, [23] and eosin Y[24]) and inorganic complexes (d6 metal polypyridyl complexes[25, 26]), as well as biological systems through the use of Photosystem I [27, 28]. In addition, their photophysical properties such as absorption, emission and redox properties can be tailored. The properties of quantum dots can be tuned through size variations and different shell capping.[29, 30] Meanwhile, varying the substituents in the organic and inorganic photosensitizers is known to tune their properties in a predictable manner.[31, 32]
One of the main considerations in utilizing highly reducing species to activate P450 enzymes is the tight balance between reductive dioxygen activation at the heme active site in a productive pathway versus dioxygen reduction leading to the generation of reactive oxygen species. This conundrum has been nicely depicted in a recent review.[19] In addition, regarding electron transfer (ET) considerations, the semi-classical Marcus theory lays the theoretical framework and the crucial parameters to consider for the optimization of ET rate.[33] The parameters include the driving force between the donor and the acceptor, the electronic coupling between the two redox centers as well as the distance separating them.
Summarized in Table 1 are the structures of the various photosensitizers successfully functioning in conjunction with P450 enzymes, alongside their photophysical properties and reduction potentials of the photogenerated species. The photosensitizer absorptions span the entire visible range of the spectrum from 400 to 710 nm. Their rich photochemistry enables access to reductive potentials ranging from −0.22 V to about −1.28 V (vs NHE) in comparison to the natural NADPH/NADP+ cofactor redox potential of −0.32 V in standard aqueous solutions.
Table 1.
Summary of the photophysical and redox properties of the photosensitizers used to activate P450 enzymes as well as their respective structures.
| Light-harvesting unit |
Structure | Absorption and Emission max |
Reduction potentiala |
Electron supplierb |
Reference |
|---|---|---|---|---|---|
|
| |||||
| 3-nm CdS-Quantum Dots Capped with mercaptoacetic acid |
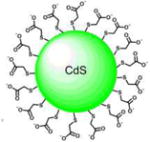
|
λmax(Abs) = 390 nm | 3 eV bandgap | H2O | [34] |
| λmax(Em) = 550 nm | |||||
|
| |||||
| 3,10- dimethyl-5-deazaflavin |
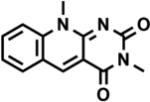
|
λmax(Abs) = 390 nm | −0.770 V | EDTA | [35] |
| λmax(Em) = 480 nm | |||||
|
| |||||
| Photosystem I |
|
λmax(Abs) = 700 nm | −1.2 V | Plastocyanin | [36] |
|
| |||||
| FAD, FMN, RBFd |
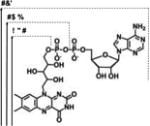
|
λmax(Abs) = 441-43 nm | −0.240 V | EDTA | [35] |
| λmax(Em) = 530-32 nm | |||||
|
| |||||
| Eosin Y |
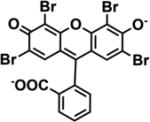
|
λmax(Abs) = 525 nm | −1.16 V | TEOA | [24] |
| λmax(Em) = 543 nm | |||||
|
| |||||
| Ruthenium diimine |
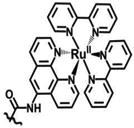
|
λmax(Abs) = 452 nm | −1.28 V | DTC | [37] |
| λmax(Em) = 620 nm | |||||
eported vs NHE
EDTA: ethylenediaminetetraacetic acid, TEOA: triethanolamine; DTC: diethyldithiocarbamate
Mg2+: green sphere
FAD: Flavin adenine dinucleotide; FMN: Flavin mononucleotide; RBF: Riboflavin.
3. Light driven formation of reactive oxygen species
A peculiar class of P450 enzymes grouped primarily into the CYP152 family display peroxygenase activity.[5] The typical candidates include the P450BSβ, CYPC1a and OleTJE proteins. The latter catalyzes the decarboxylation of long chain fatty acid to the corresponding alkene. The peroxide shunt pathway has traditionally been of interest owing to low costs[38, 39] in contrast to the redox partners and cofactors. Despite the capacity for this class of P450 enzymes or engineered P450 BM3 variants to be compatible with hydrogen peroxide induced activity, the high concentrations necessary for productivity are often problematic considering its destabilizing effect on proteins. To maintain an adequate supply of hydrogen peroxide for efficient reactions and still retain enzyme stability, there has been recent interest in the in situ controlled generation of its analogs using light.
3.1 CdS Quantum Dots
The finding that fluorescent semiconductor quantum dots form radical species in aqueous solution upon visible light excitation motivated the investigation of their use to drive P450 peroxygenase activity.[21, 34, 40, 41] The following method pioneered the development of nanohybrids comprised of P450BSβ enzymes adsorbed to the surface of quantum dots.[21] The nanohybrids were prepared via electrostatic attraction of the histidine tagged P450 enzymes and the negatively charged mercaptoacetic acid shell of the Cadmium Sulfide (CdS) quantum dots (See Figure 2). Each quantum dot was saturated with a total of six molar equivalents of enzyme as predicted based on their size. Upon visible light irradiation, due to their highly reductive conduction band potential, CdS quantum dots produce superoxide and hydroxyl radicals from molecular dioxygen in aqueous solutions. These reactive oxygen species (ROS) were utilized by the surface adsorbed P450 enzymes to carry out the hydroxylation of myristic acid.
Fig. 2.

Schematic representation of the nanohybrid approach composed of histidine tagged P450BSβ enzymes adsorbed to the negatively charged mercaptoacetic acid shell of the Cadmium Sulfide (CdS) quantum dots. Excitation of the quantum dots leads to the generation of reactive oxygen species utilized by the P450 enzymes via the peroxide shunt pathway to perform the selective hydroxylation of myristic acid.
The nanohybrids generated hydroxylated products similar to the native enzyme despite a turnover rate that was six times slower. The retention of steady turnover numbers and solubility after continuous irradiation for 20 minutes established the feasibility of the nanohybrid approach.
Potential sources for the slow reactivity included impediment of conformational flexibility or denaturation of the enzymes due to binding with the nanoparticles. The reusability of the enzymes was noted unsuccessful, and this was likely due to aforementioned denaturation. Further investigations included the expansion of the nanohybrid approach to other peroxygenase enzymes[40] as well as additional P450 enzymes including the xenobiotic metabolite generating CYP2D6.[42, 43] Another promising approach to the field is carbon dots, which are soluble in aqueous solutions and can readily be functionalized at their surface, but have yet to be associated with P450 enzymes.[44]
3.2 Isoalloxazine flavins
As an alternative route toward the use of the peroxide shunt pathway, photoexcitable isoalloxazine type flavins such as riboflavin (RBF), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD) have been explored, often in conjunction with the inexpensive electron donor EDTA, to generate hydrogen peroxide in situ. [23, 39] The approach was proven successful by Hollmann and coworkers with chloroperoxidase.[45] The FMN/EDTA approach surpassed the stoichiometric addition of hydrogen peroxide leading to total turnover numbers greater than 20,000 in the catalytic oxidation of thioanisole.
Urlacher et al. then explored the photoreactivity of P450 peroxygenases powered by flavins.[23, 39] Light-driven reactions were carried out using either P450BSβ or P450C1a peroxygenases with each flavin and myristic acid as the substrate (See Figure 3).[23] Using riboflavin, P450BSβ generated a 91% product conversion in 20 minutes. Using FMN, 81% conversion was achieved and only 3% conversion occurred in the FAD-containing system. The typical reaction conditions included 40 µM flavin per µM of P450 enzymes with 1 mM EDTA and 200 µM of myristic acid. The use of P450C1a generated product conversions around half those of P450BSβ with each respective flavin. The reaction was shown to proceed primarily by the peroxide shunt pathway but residual activity in the presence of catalase suggested that partial reduction of the heme domain by the flavins was possible.
Fig. 3.

Schematic representation of isoalloxazine flavin approach (RBF: Riboflavin; FMN: flavin mononucleotide and their reduced forms (RBFH2 and FMNH2, respectively) for the in situ generation of H2O2 to power P450 enzymes (P450BSβ (CYP152A1) or P450C1a (CYP152A2)) with peroxygenase activity.
More recently, the same FMN/EDTA approach was also employed to power OleTJE.[46] Up to 99% yield was obtained in 2 hours for the decarboxylation of stearic acid with a 3.3:1 ratio of 1-alkene to β-hydroxy acid. The reaction yield shows dependence on FMN concentration with a maximum obtained with 10 µM FMN and 50 mM EDTA for 200 µM OleTJE crude cell extracts.
4. Light driven heme reduction mediated by redox partners
4.1 Deazaflavins
In an attempt to induce catalysis in P450 enzymes without the requisite NAD(P)H cofactor, a light-powered flavin cofactor regeneration system was initially employed. Using EDTA as sacrificial electron donor, a flavin mediator was sought to reduce the enzyme-bound FMN cofactor and initiate catalysis, but with negligible product formation. To minimize oxidative damage and regenerate enzyme efficiency under the aerobic conditions necessary for monooxygenases, the use of reduced deazaflavins, which reportedly undergo slow reaction with oxygen, was explored in place of the isoalloxazine flavins.[35] The deazaflavin system was modeled on the prototype holoenzyme P450 BM3[47] with lauric acid as substrate.
When the flavin mediators were compared, FAD generated no appreciable heme reduction, and the deazaflavin approach was clearly superior with the caveat that an input of a catalytic amount of NADP+ was necessary for its function. Whereas proven to not be involved in an alternate pathway, NADP+ induced P450 structural changes required for effective catalysis. Light reactions were performed on this NADP+ supplemented system and generated 74% of hydroxylated lauric acid products after 17 hours, with unaffected regioselectivity with respect to the natural system, demonstrating enzyme catalysis. The final total turnover frequency reported was 117 h−1 suffering in comparison to the 4500 min−1 reported rate in the native enzyme. Nevertheless, the deazaflavin approach had thus far established the proof of concept to reduce the heme domain while balancing the in situ reduction of molecular dioxygen.
4.2 Photosystem I with ferredoxin as electron mediator
The conversion of light energy into chemical energy has been adopted to drive the P450 reactions in the most biological sense, using the photosynthetic machinery.[27, 28, 48] With a redox potential of −1.2 V and a quantum yield near 1, Photosystem I (PSI) is the strongest reductant in nature, establishing its suitability for the replacement of NAD(P)H in the activation of P450 enzymes.[36] To that end, pioneering work by Jensen and coworkers successfully demonstrated that photoreduction of the CYP79A1 heme domain, a P450 enzyme involved in tyrosine oxidation, can be carried out directly by PSI using the electron carrier partner, ferredoxin (Fd), common to both systems.[27] Upon photoactivation, the electron transport chain readily supplies the necessary electrons to power the P450. The electron carrier ferredoxin was crucial to mediating electron transfer and a linear dependence was noted with the highest activity requiring a 1000-fold excess of free ferredoxin. As a proof of concept, L-tyrosine oxidation by CYP79A1 to its corresponding oxime was achieved in a key step along the synthesis pathway that produces the cyanogenic glucoside dhurrin (See Figure 5).
Fig. 5.

Schematic representation of Photosystem I approach for the light-driven CYP79A1 oxidation of L-tyrosine en route to the synthesis of cyanogenic glucoside dhurrin.
As is the theme with the P450 approaches, the necessity for a powerful reductant is paired with the challenge for ensuring that the electrons are successfully delivered to the P450 itself. The requisite for extreme saturation of Fd was a limitation that was combated in the next generation, through the use of a fused Fd/CYP79A1 construct.[28] The most successful fusion construct included a 15 amino acid long linker joining the ferredoxin to the C terminal end of the protein. This fused variant outperformed the CYP79A1 with its natural reductase (CPR) system by 2 fold with a caveat that the CPR was only in a stoichiometric ratio.[28] Following these results, the same research groups evaluated the possibility of powering the entire metabolic pathway using PSI. A tour de force was achieved when the system was successfully introduced in tobacco plants and the generation of dhurrin was quantified.[48, 49]
While showing high potential, the current successes are thus far limited to the natural enzyme substrate and with requirement for the electron mediator ferredoxin. The future of this approach resides in the cost-effective generation of specialized natural metabolites in plants.[50]
5. Direct Light-driven heme reduction
5.1 Eosin Y
The idea of diversifying the light activation of P450s using a common system was explored by Park and coworkers using the fluorescent dye eosin Y (EY) as a photosensitizer.[24] This strategy capitalizes on the whole cell biocatalysis potential of cytochrome P450 for biotechnological applications. In this in vivo approach, P450 holoenzymes or heme domains are expressed in the cytoplasm of E. coli cells. Confocal microscope experiments confirmed the colocalization of the organic dye with the cytochromes. The EY photosensitizer is proposed to use triethanolamine (TEOA) to directly transfer electrons to the heme active site. Spectral binding studies show that the EY molecule binds directly at the enzyme active site, as typical type I and II binding modes were observed with the mutants studied. This could potentially be problematic during catalysis with the photosensitizer competing with substrate binding. Preliminary electron transfer studies showed that 34% of the heme domain can be reduced by the eosin Y/TEOA system in 5 min of continuous irradiation as compared to sodium dithionite reduction.
The approach is marked by its simplicity, where the photosensitizer in solution is readily uptake into cells producing the desired P450 enzymes (See Figure 6). The O-dealkylation of 7-ethoxycoumarin by the BM3m2 mutant (Y51F/F87A) was utilized as a proof of concept for this approach. The EY platform system was then shown to be compatible with multiple P450 BM3 variants known to produce human drug metabolites[12] as well as several human P450 enzymes. Among those enzymes was the human CYP3A4, which functionalizes greater than 50 percent of human drugs. Examples of high impact substrates compatible with the EY photosensitizer include the marketed drugs simvastatin, omeprazole, and lovastatin, and the steroid 17β-estradiol. Turnover numbers for all enzymes in the study are rather low ranging between 8 to 180 (µM) product / (µM) P450 for a 10 to 24h reaction time. Greater activity was obtained with the P450 BM3 mutants than with the human P450 enzymes. In light of the current review, it has, however, not been ruled out that peroxygenase activity contributes to the overall turnover numbers, since EY and TEOA are known to efficiently reduce dioxygen.[51]
Fig. 6.

Schematic representation of the whole cell biocatalysis approach using the eosin Y platform. The organic photosensitizer readily penetrates the cell wall of cytochrome P450 producing E. coli to promote the oxyfunctionalization of several marketed drugs.
The EY platform pioneered the light-driven P450 reaction on the oxyfunctionalization of commercial drugs and steroids in a particularly exciting new development in this field. The straightforwardness and apparent wide applicability would place this whole cell photosensitizer approach at the forefront of the field, but the slow enzyme productivity suggests that improvements are necessary for its wider use and scalability.
5.2 Covalently attached Ru(II)-diimine complexes
In order to overcome the limitations associated with bimolecular processes in the electron transfer machinery, our group has undertaken the covalent attachment of the photosensitizer to P450 heme domains.[25, 26, 52] The photosensitizer of choice used in those studies is the Ru(II)-diimine complex taking advantage of the wealth of knowledge on their photophysical properties as well as their wide application in single electron transfer studies in metalloenzymes and light-driven enzymatic activation of small molecules.[53] The covalent attachment was achieved utilizing the unique nucleophilicity of the cysteine amino acid side chain and the introduction of a sulfhydryl reactive moiety on the Ru(II)-diimine complexes.[25, 26] Under the photocatalytic cycle shown in Figure 7a highly reductive species with the necessary driving force can be photogenerated and was shown to reduce the heme domain with rate three orders of magnitude faster than those observed with the reductase.[54]
Fig. 7.

Schematic representation of the hybrid P450 enzyme containing a Ru(II)-polypyridyl complex covalently attach to P450 BM3 heme domain mutants (PDB entry: 5JTD).[54] The photogenerated reductive species is able to deliver the necessary electrons to the heme domain to catalyze the hydroxylation of various substrates.
Our initial attempts to maximize the electron transfer focused on optimizing the distance between the photosensitizer and the heme domain by varying the site of the single cysteine mutation.[26] An ideal location (L407C) was identified which led to high photocatalytic activity (900 total turnover numbers) and initial reaction rate (120 min−1) in the selective hydroxylation of lauric acid.[55] The covalent attachment has thus enabled high initial reaction rates compared to the bimolecular process with the photosensitizer freely diffusing in solution.[56] Determination of coupling efficiency in the hybrid enzyme has been hampered by the high concentration of reductive quencher, DTC, present.
The recently solved crystal structure of the efficient hybrid enzyme reveals that the photosensitizer is located in a unique position on the proximal side of the heme where the redox partner is thought to bind.[54] The position of the photosensitizer is ideal to take advantage of the natural electron transfer pathway involving highly conserved amino acid residues F393 and Q403. Mutagenesis of the Q403 residue (Q403W) resulted in enhanced electron transfer and a 60% increase in photocatalytic activity using nitrophenoxy chromogenic substrates.[57]
The hybrid enzyme approach requires the post-modification and site-directed mutagenesis of the heme domain for assembly. However, having the photosensitizer covalently attached offers unique opportunities for rapid electron injection into the heme domain and high initial reaction rate as well as the thorough characterization of the hybrid enzyme (e.g. X-ray structure and single electron transfer kinetic studies).[57] Current work in our laboratory focuses on protein engineering of the next generation of hybrid enzymes and on expanding the Ru(II)-diimine approach to other P450 heme domains, taking advantage of their conserved tertiary structure.
6. Conclusion
In summary, each light-driven approach with its respective photosensitizer shows unique promise to power P450 enzymes using visible light energy. These systems utilize either the direct reduction of molecular dioxygen to initiate the P450 catalytic cycle through the peroxide shunt or the injection of the necessary electrons in order to activate molecular dioxygen at the heme active site. While these proofs of concept have been established with various photosensitizers and P450 systems, several challenges still remain in the light-driven powering of P450 enzymes, especially in terms of protein stability, activity and coupling efficiency in the direct heme reduction. The field of light-driven P450 enzymes is still in its infancy and greater efforts are needed in order to improve the photocatalytic activity and rival their natural counterpart.
Fig. 4.

Schematic representation of the oxygen tolerant deazaflavin/EDTA approach using the CYP102A1 (P450 BM3) holoenzyme for the selective hydroxylation of lauric acid.
Highlights.
Light-driven approaches to harness the synthetic potential of P450 enzymes.
Photosensitizers generating reactive oxygen species coupled with P450 peroxygenase activity.
Light-harvesting units to inject electrons and initiate the P450 catalytic mechanism.
Acknowledgments
The authors gratefully acknowledge financial support by the National Institutes of Health through grant number SC3 GM095415 and by the National Science Foundation through grant number 1509924.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Roduner E, Kaim W, Sarkar B, Urlacher VB, Pleiss J, Glaser R, Einicke WD, Sprenger GA, Beifuss U, Klemm E, Liebner C, Hieronymus H, Hsu SF, Plietker B, Laschat S. Selective Catalytic Oxidation of C-H Bonds with Molecular Oxygen. Chemcatchem. 2013;5:82–112. [Google Scholar]
- 2.Roiban GD, Reetz MT. Expanding the toolbox of organic chemists: directed evolution of P450 monooxygenases as catalysts in regio- and stereoselective oxidative hydroxylation. Chem. Commun. 2015;51:2208–2224. doi: 10.1039/c4cc09218j. [DOI] [PubMed] [Google Scholar]
- 3.Lundemo MT, Woodley JM. Guidelines for development and implementation of biocatalytic P450 processes. Appl. Microbiol. Biotechnol. 2015;99:2465–2483. doi: 10.1007/s00253-015-6403-x. [DOI] [PubMed] [Google Scholar]
- 4.Rittle J, Green MT. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 5.Shoji O, Watanabe Y. Peroxygenase reactions catalyzed by cytochromes P450. J. Biol. Inorg. Chem. 2014;19:529–539. doi: 10.1007/s00775-014-1106-9. [DOI] [PubMed] [Google Scholar]
- 6.McLean KJ, Luciakova D, Belcher J, Tee KL, Munro AW. Biological diversity of cytochrome P450 redox partner systems. Adv. Exp. Med. Biol. 2015;851:299–317. doi: 10.1007/978-3-319-16009-2_11. [DOI] [PubMed] [Google Scholar]
- 7.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 8.Lewis JC, Coelho PS, Arnold FH. Enzymatic functionalization of carbon-hydrogen bonds. Chem. Soc. Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fasan R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012;2:647–666. [Google Scholar]
- 10.Girvan HM, Munro AW. Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr. Opin. Chem. Biol. 2016;31:136–145. doi: 10.1016/j.cbpa.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Behrendorff JBYH, Gillam EMJ. Prospects for Applying Synthetic Biology to Toxicology: Future Opportunities and Current Limitations for the Repurposing of Cytochrome P450 Systems. Chem. Res. Toxicol. 2017;30:453–468. doi: 10.1021/acs.chemrestox.6b00396. [DOI] [PubMed] [Google Scholar]
- 12.Di Nardo G, Gilardi G. Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites. Int. J. Mol. Sci. 2012;13:15901–15924. doi: 10.3390/ijms131215901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McIntosh JA, Farwell CC, Arnold FH. Expanding P450 catalytic reaction space through evolution and engineering. Curr. Opin. Chem. Biol. 2014;19:126–134. doi: 10.1016/j.cbpa.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renata H, Wang ZJ, Arnold FH. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Edit. 2015;54:3351–3367. doi: 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hlavica P. Assembly of non-natural electron transfer conduits in the cytochrome P450 system: a critical assessment and update of artificial redox constructs amenable to exploitation in biotechnological areas. Biotechnol. Adv. 2009;27:103–121. doi: 10.1016/j.biotechadv.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Sadeghi SJ, Fantuzzi A, Gilardi G. Breakthrough in P450 bioelectrochemistry and future perspectives. Biochim. Biophys. Acta -Proteins Proteom. 2011;1814:237–248. doi: 10.1016/j.bbapap.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Munro AW, Girvan HM, McLean KJ. Cytochrome P450 - redox partner fusion enzymes. Biochim. Biophys. Acta -Gen. Subjects. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Urlacher VB, Lutz-Wahl S, Schmid RD. Microbial P450 enzymes in biotechnology. Appl. Microbiol. Biotechnol. 2004;64:317–325. doi: 10.1007/s00253-003-1514-1. [DOI] [PubMed] [Google Scholar]
- 19.Holtmann D, Hollmann F. The Oxygen Dilemma: A Severe Challenge for the Application of Monooxygenases? Chembiochem. 2016;17:1391–1398. doi: 10.1002/cbic.201600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cirino PC, Arnold FH. A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew. Chem. Int. Edit. 2003;42:3299–3301. doi: 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- 21.Ipe BI, Niemeyer CM. Nanohybrids composed of quantum dots and cytochrome P450 as photocatalysts. Angew. Chem. Int Edit. 2006;45:504–507. doi: 10.1002/anie.200503084. [DOI] [PubMed] [Google Scholar]
- 22.Zilly FE, Taglieber A, Schulz F, Hollmann F, Reetz MT. Deazaflavins as mediators in light-driven cytochrome P450 catalyzed hydroxylations. Chem. Commun. 2009:7152–7154. doi: 10.1039/b913863c. [DOI] [PubMed] [Google Scholar]
- 23.Girhard M, Kunigk E, Tihovsky S, Shumyantseva VV, Urlacher VB. Light-driven biocatalysis with cytochrome P450 peroxygenases. Biotechnol. Appl. Bioc. 2013;60:111–118. doi: 10.1002/bab.1063. [DOI] [PubMed] [Google Scholar]
- 24.Park JH, Lee SH, Cha GS, Choi DS, Nam DH, Lee JH, Lee JK, Yun CH, Jeong KJ, Park CB. Cofactor-Free Light-Driven Whole-Cell Cytochrome P450 Catalysis. Angew. Chem. Int. Edit. 2015;54:969–973. doi: 10.1002/anie.201410059. [DOI] [PubMed] [Google Scholar]
- 25.Dwaraknath S, Tran NH, Dao T, Colbert A, Mullen S, Nguyen A, Cortez A, Cheruzel L. A facile and versatile methodology for cysteine specific labeling of proteins with octahedral polypyridyl d(6) metal complexes. J. Inorg. Biochem. 2014;136:154–160. doi: 10.1016/j.jinorgbio.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran NH, Huynh N, Chavez G, Nguyen A, Dwaraknath S, Nguyen TA, Nguyen M, Cheruzel L. A series of hybrid P450 BM3 enzymes with different catalytic activity in the light-initiated hydroxylation of lauric acid. J. Inorg. Biochem. 2012;115:50–56. doi: 10.1016/j.jinorgbio.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen K, Jensen PE, Moller BL. Light-Driven Cytochrome P450 Hydroxylations. ACS Chem. Biol. 2011;6:533–539. doi: 10.1021/cb100393j. [DOI] [PubMed] [Google Scholar]
- 28.Mellor SB, Nielsen AZ, Burow M, Motawia MS, Jakubauskas D, Moller BL, Jensen PE. Fusion of Ferredoxin and Cytochrome P450 Enables Direct Light-Driven Biosynthesis. ACS Chem. Biol. 2016;11:1862–1869. doi: 10.1021/acschembio.6b00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith AM, Nie SM. Semiconductor Nanocrystals: Structure, Properties, and Band Gap Engineering. Acc. Chem. Res. 2010;43:190–200. doi: 10.1021/ar9001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burda C, Chen XB, Narayanan R, El-Sayed MA. Chemistry and properties of nanocrystals of different shapes. Chem. Rev. 2005;105:1025–1102. doi: 10.1021/cr030063a. [DOI] [PubMed] [Google Scholar]
- 31.Walsh JD, Miller AF. Flavin reduction potential tuning by substitution and bending. J. Mol. Struc. Theochem. 2003;623:185–195. [Google Scholar]
- 32.Campagna S, Puntoriero F, Nastasi F, Bergamini G, Balzani V. Photochemistry and photophysics of coordination compounds: Ruthenium. Top. Curr. Chem. 2007;280:117–214. [Google Scholar]
- 33.Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 34.Ipe BI, Lehnig M, Niemeyer CM. On the generation of free radical species from quantum dots. Small. 2005;1:706–709. doi: 10.1002/smll.200500105. [DOI] [PubMed] [Google Scholar]
- 35.Hemmerich P, Massey V, Fenner H. Flavin and 5-Deazaflavin - Chemical Evaluation of Modified Flavoproteins with Respect to Mechanisms of Redox Biocatalysis. Febs Lett. 1977;84:5–21. doi: 10.1016/0014-5793(77)81047-8. [DOI] [PubMed] [Google Scholar]
- 36.Webber AN, Lubitz W. P700: the primary electron donor of photosystem I. Biochim. Biophys. Acta-Bioenergetics. 2001;1507:61–79. doi: 10.1016/s0005-2728(01)00198-0. [DOI] [PubMed] [Google Scholar]
- 37.Kalyanasundaram K. Photophysics, Photochemistry and Solar-Energy Conversion with Tris(Bipyridyl)Ruthenium(Ii) and Its Analogs. Coord. Chem. Rev. 1982;46:159–244. [Google Scholar]
- 38.Bormann S, Baraibar AG, Ni Y, Holtmann D, Hollmann F. Specific oxyfunctionalisations catalysed by peroxygenases: opportunities, challenges and solutions. Catal. Sci. Technol. 2015;5:2038–2052. [Google Scholar]
- 39.Paul CE, Churakova E, Maurits E, Girhard M, Urlacher VB, Hollmann F. In situ formation of H2O2 for P450 peroxygenases. Bioorg. Med. Chem. 2014;22:5692–5696. doi: 10.1016/j.bmc.2014.05.074. [DOI] [PubMed] [Google Scholar]
- 40.Fruk L, Rajendran V, Spengler M, Niemeyer CM. Light-induced triggering of peroxidase activity using quantum dots. Chembiochem. 2007;8:2195–2198. doi: 10.1002/cbic.200700594. [DOI] [PubMed] [Google Scholar]
- 41.Gandubert VJ, Torres E, Niemeyer CM. Investigation of cytochrome P450-modified cadmium sulfide quantum dots as photocatalysts. Mater. Chem. J. 2008;18:3824–3830. [Google Scholar]
- 42.Qian J, Zhu W, Mi L, Xu X, Yu JC, Cui DM, Xue YC, Liu SQ. Nanohybrids of quantum dots and cytochrome P450 for light-driven drug metabolism. J. Electroanal. Chem. 2014;733:27–32. [Google Scholar]
- 43.Jiang L, Li Y, Wei W, Liu AR, Zhang YJ, Liu SQ. Confining nanohybrid of CdTe quantum dots and cytochrome P450 2D6 in macroporous ordered siliceous foam for drug metabolism. J. Electroanal. Chem. 2016;781:345–350. [Google Scholar]
- 44.Hutton GAM, Reuillard B, Martindale BCM, Caputo CA, Lockwood CWJ, Butt JN, Reisner E. Carbon Dots as Versatile Photosensitizers for Solar-Driven Catalysis with Redox Enzymes. J. Am. Chem. Soc. 2016;138:16722–16730. doi: 10.1021/jacs.6b10146. [DOI] [PubMed] [Google Scholar]
- 45.Perez DI, Grau MM, Arends IW, Hollmann F. Visible light-driven and chloroperoxidase-catalyzed oxygenation reactions. Chem. Commun. 2009:6848–6850. doi: 10.1039/b915078a. [DOI] [PubMed] [Google Scholar]
- 46.Zachos L, Gassmeyer SK, Bauer D, Sieber V, Hollmann F, Kourist R. Photobiocatalytic decarboxylation for olefin synthesis. Chem. Commun. 2015;51:1918–1921. doi: 10.1039/c4cc07276f. [DOI] [PubMed] [Google Scholar]
- 47.Whitehouse CJC, Bell SG, Wong LL. P450(BM3) (CYP102A1): connecting the dots. Chem. Soc. Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 48.Nielsen AZ, Ziersen B, Jensen K, Lassen LM, Olsen CE, Moller BL, Jensen PE. Redirecting Photosynthetic Reducing Power toward Bioactive Natural Product Synthesis. ACS Synth. Biol. 2013;2:308–315. doi: 10.1021/sb300128r. [DOI] [PubMed] [Google Scholar]
- 49.Gnanasekaran T, Karcher D, Nielsen AZ, Martens HJ, Ruf S, Kroop X, Olsen CE, Motawie MS, Pribil M, Moller BL, Bock R, Jensen PE. Transfer of the cytochrome P450- dependent dhurrin pathway from Sorghum bicolor into Nicotiana tabacum chloroplasts for light-driven synthesis. J. Exp. Bot. 2016;67:2495–2506. doi: 10.1093/jxb/erw067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Renault H, Bassard JE, Hamberger B, Werck-Reichhart D. Cytochrome P450-mediated metabolic engineering: current progress and future challenges. Curr. Opin. Plant Biol. 2014;19:27–34. doi: 10.1016/j.pbi.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 51.Gorner H. Oxygen uptake induced by electron transfer from donors to the triplet state of methylene blue and xanthene dyes in air-saturated aqueous solution. Photoch. Photobio. Sci. 2008;7:371–376. doi: 10.1039/b712496a. [DOI] [PubMed] [Google Scholar]
- 52.Ener ME, Lee YT, Winkler JR, Gray HB, Cheruzel L. Photooxidation of cytochrome P450-BM3. Proc. Natl. Acad. Sci. USA. 2010;107:18783–18786. doi: 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lam Q, Kato M, Cheruzel L. Ru(II)-diimine functionalized metalloproteins: From electron transfer studies to light-driven biocatalysis. Biochim. Biophys. Acta. 2016;1857:589–597. doi: 10.1016/j.bbabio.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spradlin J, Lee D, Mahadevan S, Mahomed M, Tang L, Lam Q, Colbert A, Shafaat OS, Goodin D, Kloos M, Kato M, Cheruzel LE. Insights into an efficient light-driven hybrid P450 BM3 enzyme from crystallographic, spectroscopic and biochemical studies. Biochim. Biophys. Acta. 2016;1864:1732–1738. doi: 10.1016/j.bbapap.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tran NH, Nguyen D, Dwaraknath S, Mahadevan S, Chavez G, Nguyen A, Dao T, Mullen S, Nguyen TA, Cheruzel LE. An Efficient Light-Driven P450 BM3 Biocatalyst. J. Am. Chem. Soc. 2013;135:14484–14487. doi: 10.1021/ja409337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tran NH, Huynh N, Bui T, Nguyen Y, Huynh P, Cooper ME, Cheruzel LE. Light-initiated hydroxylation of lauric acid using hybrid P450 BM3 enzymes. Chem. Commun. 2011;47:11936–11938. doi: 10.1039/c1cc15124j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lam Q, Cortez A, Nguyen TT, Kato M, Cheruzel L. Chromogenic nitrophenolate-based substrates for light-driven hybrid P450 BM3 enzyme assay. J. Inorg. Biochem. 2016;158:86–91. doi: 10.1016/j.jinorgbio.2015.12.005. [DOI] [PubMed] [Google Scholar]