

Abstract
Diabetic retinopathy is a neurovascular diabetes complication resulting in vision loss. A wealth of literature reports retinal molecular changes indicative of neural deficits, inflammation, and vascular leakage with chronic diabetes, but the mechanistic causes of disease initiation and progression are unknown. Microvascular mitochondrial DNA (mtDNA) damage leading to mitochondrial dysfunction has been proposed to drive vascular dysfunction in retinopathy. However, growing evidence suggests that neural retina dysfunction precedes and may cause vascular damage. Therefore, we tested the hypothesis that neural mtDNA damage and mitochondrial dysfunction are an early initiating factor of neural diabetic retinopathy development in a rat streptozotocin (STZ)-induced, Type I diabetes model. Mitochondrial function (oxygen consumption rates) was quantified in retinal synaptic terminals from diabetic and non-diabetic rats with paired retinal structural and function assessment (optical coherence tomography and electroretinography, respectively). Mitochondrial genome damage was assessed by identifying mutations and deletions across the mtDNA genome by high depth sequencing and absolute mtDNA copy number counting through digital PCR. Mitochondrial protein expression was assessed by targeted mass spectrometry. Retinal functional deficits and neural anatomical changes were present after 3 months of diabetes and prevented/normalized by insulin treatment. No marked dysfunction of mitochondrial activity, maladaptive changes in mitochondrial protein expression, alterations in mtDNA copy number, or increase in mtDNA damage was observed in conjunction with retinal functional and anatomical changes. These results demonstrate that neural retinal dysfunction with diabetes begins prior to mtDNA damage and dysfunction, and therefore retinal neurodegeneration initiation with diabetes occurs through other, non-mitochondrial DNA damage, mechanisms.
Keywords: Diabetic retinopathy, diabetes, retina, mtDNA, mitochondria, insulin
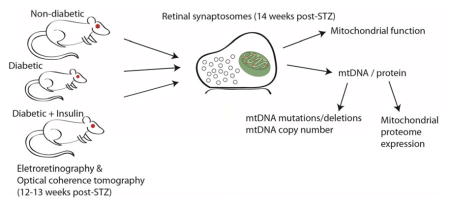
Graphical Abstract
Neuronal dysfunction precedes traditional vascular defects in diabetic retinopathy disease progression. In this study neuronal mitochondrial status was examined across functional, proteomic, and genomic domains in rats characterized for neural retinal function and structure. Mitochondrial deficits were not evident at a disease timepoint where neural retinal deficits are evident demonstrating that diabetic retinopathy disease initiation occurs through non-mitochondrial mechanisms.
Introduction
Diabetic retinopathy (DR) is a neurovascular secondary complication of diabetes that affects the retina, the light-sensitive neural tissue of the posterior eye. While the majority of Type 1 diabetic patients experience some form of vision loss attributed to DR, the etiology of DR and pathophysiological progression remain elusive (Stitt et al. 2016). The combination of inflammation, neurodegeneration, and vascular leakage present clinically likely forms a feed-forward cycle causing tissue dysfunction (Antonetti et al. 2006). Recent evidence supports the idea that neurodegeneration may be the initial insult, setting this cycle in motion (Lynch & Abramoff 2017, Sohn et al. 2016). Early vision deficits are neural in origin and occur in the absence of angiogenesis and other vascular hallmarks of DR (Jackson et al. 2012, Sohn et al. 2016). Diligent glycemic control is a proven approach for DR prevention, but aggressive insulin replacement therapy is often insufficient to prevent disease development (Aiello & Group 2014, Nathan et al. 2015). Treatments aimed at halting neovascularization and angiogenesis are promising for preserving remaining vision in late-stage proliferative DR (Vaziri et al. 2015), but likely have limited utility as preventative treatments for diabetic patients. Thus, there is the opportunity to identify the initial/early DR mechanisms, prior to the development of proliferative retinopathy, which could be targeted in combination with glycemic control to prevent DR disease process initiation. Previously we reported loss of synaptic connections and synaptic proteins within the inner and outer plexiform layers as well as dysregulation of gene expression starting at around 1 month of uncontrolled diabetes (VanGuilder et al. 2008, Bixler et al. 2011). These neural changes occur before vascular dysfunction and increased permeability (Masser et al. 2014, Brucklacher et al. 2008, VanGuilder et al. 2011, Bixler et al. 2011). This sequence of neural changes prior to overt vascular dysfunction is also evident clinically (Sennlaub et al. 2003, Gardner et al. 2002, Lorenzi & Gerhardinger 2001, Sohn et al. 2016). However, the initiating causes of the neural retina DR disease process and neurodegeneration remain unclear.
In the retinal vasculature, extended, uncontrolled diabetes results in mitochondrial deficits including mitochondrial DNA (mtDNA) damage, presumably mutations and deletions, as a result of oxidative damage (Kowluru & Abbas 2003, Kowluru & Mishra 2015). mtDNA are circular genomes present in multiple copies in the inner mitochondrial matrix that encode only necessary subunits of the electron transport chain in addition to rRNA and tRNA for localized mRNA translation (Taylor & Turnbull 2005). Mitochondria are critical for proper neural tissue function by providing cellular energy in the form of ATP through glycolysis, and calcium storage and buffering necessary for proper synaptic transmission (Dupuis 2014, Sheng & Cai 2012). Therefore, mtDNA mutations have the potential to disrupt neural function and result in a loss of mitochondrial homeostasis with diabetes. mtDNA damage and mitochondrial function have been assessed previously, albeit with qualitative methods, after long durations (>6 months) of uncontrolled experimental diabetes with the interpretation that damaged mitochondria in retinal endothelial cells drive DR initiation and progression (Madsen-Bouterse et al. 2010, Santos et al. 2012, Tewari et al. 2012, Zhong & Kowluru 2011). However, the nature and degree of mtDNA damage and functional deficits in the diabetic neural retina is unknown. The scientific premise of this study is: the neural retina damage evident early in diabetes may be mediated by mitochondrial dysfunction. This is supported by the findings that long-term diabetes causes mitochondrial dysfunction in non-neural cells of the retina, and in other retinal diseases neural mitochondrial dysfunction induces neurodegeneration (Barot et al. 2011). Therefore, we sought to examine mitochondrial function and quantify neural mtDNA damage in a rat model (Freeman et al. 2010) at a time point where neuronal changes are occurring but prior to vascular dysfunction (Masser et al. 2014, VanGuilder et al. 2008). Novel quantitative molecular techniques, mtDNA sequencing and absolute copy number, were used to rigorously assess this scientific premise. Chronic insulin treatment was examined to determine if a standard approach to glycemic control could prevent or normalize any changes observed. We hypothesized that neural retinal mitochondria would be dysfunctional and mtDNA damaged with diabetes at a time where neural retina functional and structural deficits are evident. In this study, retinal function and morphology were measured in experimental diabetic rats, and subsequently a quantitative assessment of mitochondrial respiration, mtDNA damage, mtDNA copy number, and mitochondrial protein expression from retinal synaptosomes were performed.
Methods
Animals
Rats were housed from birth in a dimly-lit pathogen-free environment at the Dean McGee Eye Institute Animal Facility, and maintained in accordance with Institutional Animal Care and Use Committee guidelines (IACUC approval #101379-15-101-HC). Light intensity on the top and bottom housing racks were equal. Water and food were provided ad-libitum throughout the study, unless otherwise stated. Sprague-Dawley (RRID: RGD_5508397) breeder pairs were purchased from Charles River. ANOVA power analysis was performed assuming alpha of 0.5, minimum difference of 10% and standard deviation of 5% with three experimental groups and minimal sample size of six gives a power of 0.812. A total of 12 animals per group were therefore chosen to provide more than sufficient power even in the event of loss of animals to humane endpoints. Two cohorts of male rat pups were weaned at 7 days, ear tagged, and placed in cages with littermates (n=36/cohort [N=72]). Data include animals from both cohorts combined, unless otherwise stated. At ~1.5 months of age (~150–250 g), rats were fasted overnight and weighed. Randomization into treatment groups was carried out using block randomization to ensure equal experimental groups (Suresh 2011). A single intraperitoneal injection of streptozotocin (STZ, at 65 mg/kg, Sigma Aldrich) dissolved in 10mM sodium citrate (pH 4.5) was administered to 24 rats in each cohort as previously described (Bixler et al. 2011, Masser et al. 2014, VanGuilder et al. 2008). Aged-matched non-diabetic controls received a single equal volume intraperitoneal injection of sodium citrate buffer. Diabetes induction was confirmed one week post-injection and only STZ-injected animals with non-fasted blood glucose measurements ≥250 mg/dL (groups were blinded from the tester) were retained. This resulted in 93% conversion, or 7% of animals failing diabetes induction. Additionally, animals were excluded if they lost >15% body mass over two consecutive weeks. This resulted in 3 diabetic animals euthanized before the termination of the study. The unconverted animals and animals that were euthanized early for humane endpoints were excluded from all study endpoints. All animals were euthanized humanely by decapitation under pentobarbital anesthesia (100mg/kg) administered by a single intraperitoneal injection.
Blood glucose & glycated hemoglobin
Starting one week post-STZ or sodium citrate vehicle injection blood glucose levels were measured from a drop of blood from a lancet tail-nick (Lifescan One-touch Meter; Johnson & Johnson) (Masser et al. 2014) bi-weekly for diabetic rats and non-diabetic controls and weekly for insulin-treated diabetic rats. At euthanasia, glycated hemoglobin (HbA1c) levels were measured from a drop of blood collected by lancet tail-nick using monoclonal antibody agglutination immunoassay cartridges and a DCA analyzer (PTS Diagnostics) (Masser et al. 2014).
Insulin administration
A subset of diabetic rats (n=12/cohort) received insulin replacement beginning 6 weeks post-STZ injection by subcutaneous implantation (26 mg human insulin pellet, 7 mm x 2 mm; LinShin Canada) via trocar under transient isoflurane vapor anesthesia. These insulin pellets provide a continual dose of 2U of insulin per 24 hours for the duration for the pellet. Endogenous rat c-peptide and exogenous human insulin levels were measured by enzyme-linked immunosorbent assays (Mercodia) (Masser et al. 2014) using plasma collected from trunk blood at the time of euthanization.
Electroretinography and optical coherence tomography
One week prior to euthanasia (12–13 weeks diabetes duration), rats were assessed for retinal function by electroretinography (ERG). Rats were weighed and dark adapted for 12 hours overnight prior to ERG recording. Preparation of the rats is described in detail in the Supplemental Methods. A ColorDome and Espion E3 ERG instrument (Diagnosys LLC) was used to stimulate and record responses. The 11 step protocol consisted of a zero stimulus for the purpose of removing the baseline trace, then a 6 step serial scotopic range of −4.87 log cd*s/m2 to −1.4 log cd*s/m2, and finally a 4 step photopic series ranging from 0.34 log cd*s/m2 to 1.3 log cd*s/m2 paired with a rod suppressing background illumination (10 cd/m2) to isolate cone function. Oscillatory potentials (OP) were recorded for both scotopic and photopic readings using a digital filter of 75 – 300 Hz on virtual channels. Raw data from right and left eyes were isolated for each rat from scotopic and photopic measurement stimuli and responses were averaged for each rat. Time to peak (TTP) for each OP was isolated and averaged per group.
Retinal thickness was measured using optical coherence tomography (OCT) (Bioptigen SD OCT Envisu R4300, Leica Microsystems) immediately following ERG recording. OCT images were analyzed manually via Bioptigen InVivoVue Diver Software 2.4. A 5x5 grid retinal template was used for depth analysis centered on the optic nerve head for each image. Measurements from each layer were averaged across eyes from each animal (Supplemental Figure 1).
Synaptosome isolation
Retinal synaptosomes were isolated as previously described (VanGuilder et al. 2008) with modifications. Right and left retinas were excised, combined per animal, and placed in 15 mL of ice-cold sucrose buffer (0.32 M sucrose, 4 mM HEPES, 1 mM sodium orthovanadate, pH to 7.4). Retinas were homogenized by mechanical dounce homogenization in 3 mL of ice-cold sucrose buffer (approximately 10–15 strokes) and centrifuged at 4 °C (Optima L-80 XP ultracentrifuge, SW32.1 Ti swinging bucket, Beckman Coulter) 800g for 10 minutes to pellet nuclei and large cell fragments. Supernatants were decanted into a clean ultracentrifuge tube and placed on ice and pellets saved. Supernatant fractions were centrifuged at 4 °C and 25,000g for 12 minutes to pellet synaptosomes. Pellets were maintained on ice for subsequent use.
Oxygen consumption rate measurement
Oxygen consumption rates (OCR) for retinal synaptosomes were measured by Seahorse XFe96 extracellular flux analyzer and XF Cell Mito Stress Test (Agilent) as outlined previously (Choi et al. 2009) with modifications and according to manufacturer’s instructions. Isolated retinal synaptosome pellets from each animal were resuspended in 300 μl ionic media (20 mM HEPES, 10 mM D-glucose, 1.2 mM disodium phosphate, 1 mM magnesium chloride, 5 mM sodium bicarbonate, 140 mM sodium chloride, pH to 7.4), and 9 μl of the resuspended synaptosomes were aliquoted (20 μg of protein/well quantified after OCR measurements) in duplicate or triplicate wells containing 50 μl of ice-cold ionic media. Oligomycin, FCCP (Carbonyl cyanide-4-trifluoromethoxy phenylhydrazone), and antimycin A/rotenone drugs were dissolved and diluted in incubation media to final assay well concentrations of 1 μM oligomycin, 2 μM FCCP, and 1 μM rotenone/antimycin a (R/AA). All steps of the Seahorse run protocol were performed at 37 °C.
Immunoblot and mass spectrometry
Protein was isolated from Seahorse microplate wells and pellet fractions. Immuno dot-blots were performed using COXIV primary antibody (1:1000 in 5% BSA, COXIV, abcam, RRID: AB_301443). COXIV dot-blot intensities were determined by densitometry using ImageQuant (GE, RRID: SCR_014246) image analysis software. Immunoblots were performed using Lamin B1 (1:5000 in 5% non-fat dry milk, Lamin B1, abcam, RRID: AB_2616597), COXIV (1:1000 in 5% BSA, COXIV, abcam, RRID: AB_301443), and synaptophysin (1:500 in 5% BSA, synaptophysin, abcam, RRID: AB_2198854) antibodies, and visualized using HRP-conjugated secondary antibodies (1:1000 in 5% BSA, HRP-conjugated goat anti-mouse, Rockland, RRID: AB_2610851; 1:1000 in 5% BSA, HRP-conjugated rabbit anti-rabbit, Rockland, RRID: AB_2610848) and X-ray film exposure.
Mass spectrometry of retinal synaptosomes was carried out as previously described (Nakada et al. 2017). The analyses were carried out on a TSQ Vantage triple quadrupole mass spectrometry system (ThermoFisher). The HPLC was an Eksigent splitless nanoflow system (Eksigent) with a 10cm x 75μm i.d. C18 reversed phase capillary column. The mass spectrometer was operated in the selected reaction monitoring mode. Data were analyzed using SkyLine to determine the integrated peak area of the appropriate chromatographic peaks. The response for each protein was calculated as the geometric mean of the two peptide area. These values were normalized to the response for the BSA standard in pmol/100 μg total protein.
Nucleic acid isolation
DNA and RNA were co-isolated as previously described (Masser et al. 2013, Masser et al. 2016) using silica-spin column purification according to manufacturer’s instructions (Zymo Research). Isolated DNA and RNA was qualified and preliminarily quantified by spectrophotometry (NanoDrop, ThermoFisher), and precisely quantified by fluorometric assay (PicoGreen, ThermoFisher).
Sequencing standard generation
Total genomic rat DNA isolated from two rats with germline base differences at four known locations on the mitochondrial genome. A common forward primer and two reverse primers were used to amplify short and long mtDNA fragments (Supplemental Table 1) containing the base differences. mtDNA amplicons were ligated into vectors (TOPO TA, Life Technologies), transformed into competent E.coli. (One Shot TOP10, Life Technologies). Colonies were picked and grown in SOB medium. Plasmids were isolated using silica-spin column purification (Zymo Research) and sequence identity of inserts confirmed by Sanger sequencing.
mtDNA copy number quantitation
mtDNA was absolutely quantified (copies per ng input DNA) by digital PCR by methods we previously developed (Masser et al. 2016) using a custom mtDNA TaqMan primer-probe copy number assay (RnMtDNA_CCI1MPG, ThermoFisher).
mtDNA sequencing and analysis
mtDNA was amplified using 1 ng total DNA and long-range proofreading PCR (TaKaRa LR DNA Polymerase) with primers specific for the mitochondrial genome (Supplemental Table 1). 500 pg of mtDNA amplicon per sample were used for sequencing library preparation with Nextera XT reagents (Illumina) similar to previously described (Masser et al. 2013). Libraries were diluted to 4 nM and pooled for benchtop next generation sequencing (MiSeq, Illumina) using 600 cycle (2x250bp) reagents (Illumina) at a final library concentration of 12 pM. Detailed bioinformatics analysis methods are provided in the Supplemental Methods.
Statistics
Statistical analyses were carried out in Sigma Plot 12.5. Parametric One-Way ANOVAS with Student-Newman-Keuls post-hoc testing, were performed on data passing equal variance and normality testing (Shapiro-Wilk), while non-parametric analyses, Kruskal-Wallis One-Way ANOVA on Ranks with Dunn’s post-hoc, were carried out on data not meeting equal variance and normality. Significance was set to α = 0.05. P-values were corrected for multiple comparisons as appropriate with a false-discovery rate of q < 0.2 (Benjamini & Hochberg 2000).
Results
Longitudinal blood glucose and body weight monitoring
Timecourse of treatments and endpoints is detailed in Figure 1A. Hyperglycemia (blood glucose ≥250 mg/dL) in diabetic (D) animals was confirmed at one week post STZ injection and in comparison, blood glucose in age-matched non-diabetic (ND) controls were normal (<140 mg/dL) (Figure 1B). Diabetic animals that received insulin replacement (D+I) received subcutaneous insulin pellets starting 6 weeks post-STZ injection. Blood glucose dropped to ND control levels in D+I rats and was maintained by subsequent insulin pellet implants when blood glucose levels reached >250 mg/dL (Figure 1B). At the end of the study, blood glucose levels were equivalent in ND and D+I animals, while D animals had significantly higher blood glucose levels compared to ND and D+I animals (Supplemental Table 2). Body mass was also measured longitudinally. ND animals gained weight, while D animal growth was stunted (Figure 1C). Once diabetic animals received insulin, body mass increased, indicative of insulin action and a resumed growth pattern. At the end of the study, ND and D+I animal weights were significantly greater compared to D animals (Supplemental Table 2). These differences in blood glucose and body weights were consistent across both animal cohorts.
Figure 1.

Longitudinal blood glucose and body mass monitoring. A) Experimental design and timeline of data acquisition. Electroretinography (ERG) and optical coherence tomography (OCT) were performed 12–13 weeks post-STZ, while physiological, molecular, and biochemical end-points were measured 14 weeks post-STZ. B) Weekly B blood glucose monitoring (mg/dL) for the duration of the study (weeks post-streptozotocin (STZ) or vehicle injection) in non-diabetic (ND – white circles), diabetic (D – black circles), and diabetic with insulin replacement (D+I – gray circles) animals. Insulin administration was begun 6 weeks post-STZ injection (arrow). C) Body mass monitoring (g – grams) for the duration of the study before STZ injection (week 0) and for the subsequent study weeks in ND (white circles), D (black circles), and D+I (gray circles) animals. Arrow indicates when D+I animals received insulin implants. n=16–22 animals/group. Error bars – standard error of the mean.
Insulin and HbA1c
Glycated hemoglobin (HbA1c) levels were significantly higher in D animals compared to ND controls (Supplemental Table 2) and in D+I animals compared to ND controls, indicative of the initial 6 week period of hyperglycemia, however they were also significantly lower as compared to D animals (D vs. D+I p < 0.05), indicative of improved glycemic control. In addition to blood glucose levels, diabetes induction was confirmed at the end of the study by measuring serum levels of rat c-peptide, a marker of endogenous insulin production. ND animals had significantly higher c-peptide levels compared to D and D+I (Supplemental Table 2). C-peptide levels were equivalent in D and D+I animals, demonstrating similar levels of pancreatic β-cell depletion with STZ treatment. HbA1c and c-peptide differences were consistent across both animal cohorts. Serum from D+I animals were positive for human insulin, confirming delivery from the insulin pellets (Supplemental Table 2).
Retinal morphology and function
Structural morphology of retinal cell layers was assessed one week prior to the conclusion of the study using non-invasive optical coherence tomography (OCT), which allows for the quantitative measurement of retinal layer morphology. Representative retinal cross section images (middle data point of the 5 x 5 grid) from each experimental group are shown in Supplemental Figure 1. In D animals, the outer nuclear layer (ONL) was significantly thicker compared to ND controls (Supplemental Table 3). D+I animals also had a significantly thicker ONL compared to ND controls, however D+I animals had a significantly thinner ONL compared to D animals. Additionally, D and D+I animals had a significantly thinner retinal pigmented epithelium (RPE) compared to ND controls (Supplemental Table 3). The ganglion cell layer, inner plexiform layer, inner nuclear layer, outer plexiform layer, and photoreceptor layer were not significantly altered with diabetes. In total, diabetes did not significantly alter total retinal thickness (Supplemental Table 3).
Retinal neuronal function was measured by electroretinography (ERG) in dark-(scotopic) and light-adapted (photopic) conditions to assess rod and cone channels, respectively. Scotopic flash intensities were low (see methods)(Pardue et al. 2014), and elicited only a minor a-wave response. Therefore, the final scotopic intensity (−1.4 log cd s/m2) was used to compare a-wave amplitudes across experimental groups, rather than the intensity-response relationship. The scotopic a-wave amplitude at this flash intensity was significantly attenuated in D animals compared to ND controls (Supplemental Table 4) but a-wave implicit time was not. The scotopic b-wave maximal amplitude, half-maximal stimulus intensity and b-wave implicit time were not significantly altered with diabetes (Supplemental Table 4). Scotopic oscillatory potentials (OPs) implicit times were significantly longer in D animals compared to ND controls as demonstrated by representative traces of OP responses from the scotopic −1.4 log(cd s/m2) stimulus (Figure 2A). Comparison of mean implicit times of OP1 through OP4 peaks were significantly longer in D animals for each OP compared to ND controls (Figure 2B). OPs measured from D+I animals demonstrated an intermediate phenotype with OP1 mean implicit time normalized to ND control levels (Figure 2B). OP2 mean implicit time was significantly shorter in D+I animals compared to D animals, but was significantly higher compared to ND animals (Figure 2B). OP3 and OP4 mean implicit times in D+I animals were not significantly different from either D or ND controls.
Figure 2.

Electroretinogram (ERG) scotopic oscillatory potentials (OP) 12–13 weeks post-STZ injection. A) Representative OP traces from non-diabetic (ND - top), diabetic (D – middle), and diabetic with insulin replacement (D+I – bottom) animals showing amplitude ( μV) and time (ms) after dark-adapted light stimulus (−1.4 log(cd s/m2)) at time 0. Numbers (1–4) mark the OPs used for mean and individual implicit time comparison between experimental groups (OP1, OP2, OP3, and OP4). B) Scotopic OP mean implicit time group comparisons for OP1, OP2, OP3, and OP4 between ND (white), D (black), and D+I (gray) animals. * p < 0.05, *** p < 0.001, One-Way ANOVA with Student-Newman-Keuls Method, n=9–11 animals/group. # p < 0.05, Kruskall-Wallis One-Way ANOVA on Ranks with Dunn’s Method, n=9–11 animals/group).
For photopic ERG responses, there were no significant differences with diabetes in a- or b-wave implicit times and mean amplitudes (Supplemental Table 4). Photopic OP implicit times were not different for the first OP as show in representative traces (Supplemental Figure 2A). However, OP2 and OP3 implicit times demonstrated small, but significant, delays in implicit time (Supplemental Figure 2B). In the D+I animals there were no significant differences in OP implicit time compared to either the ND controls or the D animals, demonstrating a partial normalization with insulin replacement (Supplemental Figure 2B).
Mitochondrial respiratory function
In order to quantify mitochondrial function, oxygen consumption rates (OCR) were measured from isolated retinal synaptosomes by extracellular flux analysis (Figure 3) and normalized to the protein expression of Cytochrome c oxidase subunit IV (COXIV). Synaptosomes were isolated from the rest of the retina and enriched for mitochondria (Supplemental Figure 3). Oxygen consumption attributed to basal respiration, ATP production, non-ATP respiration, maximal respiration, spare respiratory capacity, and non-mitochondrial respiration was quantified through the use of sequential pharmacological inhibitors to components of the electron transport chain (Figure 3A and B). Basal OCR was significantly higher in D and D+I animals compared to ND controls (Figure 3C). Additionally, ATP-linked OCR was significantly higher in D and D+I animals compared to ND controls (Figure 3D). However, diabetes did not alter OCRs of non-ATP linked respiration (Figure 3E), maximal respiration (Figure 3F), spare respiration capacity (Figure 3G), or non-mitochondrial respiration (Figure 3H).
Figure 3.

Mitochondrial oxygen consumption measurements from retinal synaptosomes 14 weeks post-STZ injection. Normalized mean oxygen consumption rates (OCR) of isolated retinal synaptosomes from non-diabetic (ND), diabetic (D), and diabetic with insulin replacement (D+I). A) Oxygen consumption rate (OCR) over time with sequential pharmacological inhibitors from isolated retinal synaptosomes. Non-diabetic (ND - white), diabetic (D - black), and diabetic with insulin replacement (D+I - gray) animals (n=10–13 animals/group). Arrows indicate when drug (oligomycin, FCCP, or rotenone/antimycin A (R/AA)) were added. B) Schematic of OCR experiment demonstrating components of the OCR curve attributed to the consumption rate. Oligomycin inhibits complex V, which blocks ATP production, and the loss of oxygen consumption can be attributed to ATP-linked respiration. FCCP uncouples the proton gradient and ATP production of the inner-mitochondrial membrane allowing protons to freely pass into the inner-mitochondrial matrix which disrupts the membrane potential. In response, respiration is increased to re-establish the proton gradient and membrane potential giving a measure of maximal respiratory capacity. Finally, the combination of antimycin A, which inhibits complex III, and rotenone, which inhibits complex I, blocks mitochondrial respiration completely, and the subsequent oxygen consumption is a result of respiration independent of mitochondria. Error bars – standard error of the mean. C) Basal OCR D) OCR linked to ATP production E) OCR from non-ATP F) OCR from maximal respiration G) Spare capacity OCR and H) non-mitochondrial OCR. * p < 0.05, ** p < 0.01, One-Way ANOVA with Student-Newman-Keuls Method, n=10–13 animals/group, # p < 0.05, Kruskall-Wallis One-Way ANOVA on Ranks with Dunn’s Method, Benjamini Hochberg Multiple Testing Correction, n=10–13 animals/group). COXIV – cytochrome c oxidase subunit IV protein expression.
Antioxidant/Beta Oxidation/Krebs Cycle protein quantification
The quantitative expression of 100 proteins directly related to antioxidant status, beta oxidation, and the Krebs/tricarboxylic acid (TCA) cycle was carried out on retinal synaptosome protein by mass spectrometry using a panel of known peptides specific to each protein target (n=6/group) (Figure 4A)(Nakada et al. 2017). Proteins are listed in Supplemental Table 5. The housekeeping protein Hspd1 was quantified and demonstrated no group differences (Supplemental Table 5). Seven of the antioxidant-related proteins were found to be significantly different across groups. Five of these proteins (Pygm, Gsta3, Msra, Pygb, Prdx1) were significantly increased in the D animals compared to ND controls. Pc was significantly decreased in D animals compared to controls (Figure 4B). With the exception of Pc, proteins that were significantly increased with diabetes returned to ND control levels or were significantly lower than D animals in the D+I animals (Figure 4B). Three beta oxidation-related proteins (Acot13, Hadhb, Acsl1) were significantly increased in the D animals compared to ND controls and were subsequently normalized in the D+I animals (Figure 4C). Furthermore, none of the TCA/Krebs cycle proteins were significantly differentially expressed with diabetes. However, of the proteins that passed multiple testing corrections (Slc25a11, Sucgl1, Etfb, Sdhb, Idh3a, Echs1, Idh3g, Idh3b) all were significantly decreased in expression in the D+I animals compared to D or ND controls (Figure 4D).
Figure 4.

Mass spectrometry quantitation of mitochondrial proteins 14 weeks post-STZ injection. A) Heatmap (mean expression relative to BSA standard) of B) antioxidant, C) beta oxidation, and B) TCA/Krebs cycle proteins quantified from isolated retinal synaptosomes in non-diabetic (ND), diabetic (D), and diabetic with insulin replacement (D+I) animals. * p < 0.05, ** p < 0.01, *** p < 0.001, One-way ANOVA with Student-Newman-Keuls Method, Benjamini Hochberg Multiple Testing Correction, n=6 animals/group. # p < 0.05, One-Way ANOVA on Ranks with Dunn’s Method, n=6 animals/group. Pygm – glycogen phosphorylase, muscle associated. Gsta3 – glutathione s-transferase alpha 3. Msra- methionine sulfoxide reductase A. Pygb - glycogen phosphorylase, brain associated. Prdx1 – peroxiredoxin 1. Prdx6 – peroxiredoxin 6. Pc – pyruvate carboxylase. Acot13 – acyl-coa thioesterase 13. Hadhb – hydroxyacyl-coA dehydrogenase beta. Hmgcl – 3-hydroxymethyl-3-methylglutaryl-coA lyase. Hadha – hydroxyacyl-coA dehydrogenase alpha. Acsl1 – acyl-coA synthetase long-chain family member 1. Slc25a20 – Solute carrier family 25 member 20. Slc25a11 – solute carrier family 25 member 11. Suclg1 – succinate-CoA ligase alpha subunit. Etfb – electron transfer flavoprotein beta subunit. Sdhb – succinate dehydrogenase complex iron sulfur subunit b. Idh3a – isocitrate dehydrogenase 3 (NAD+) alpha. Echs1 – enoyl-CoA hydratase, short chain 1. Idh3g – isocitrate dehydrogenase 3 (NAD+) gamma. Idh3b - isocitrate dehydrogenase 3 (NAD+) beta.
Absolute quantitation of mitochondrial DNA copy number
To assess if mitochondrial DNA (mtDNA) copy number in isolated retinal synaptic mitochondria decreased with diabetes, absolute mtDNA number was quantified by digital PCR (dPCR) (Masser et al. 2016). There were no differences in the number of mtDNA genomes present in retinal synaptosomes across the groups (Figure 5A). Furthermore, mtDNA copy number was quantified from whole retinal DNA isolated from an independent animal cohort presented in our previous report (Masser et al. 2014) and no difference in mtDNA copies in D or D+I animals compared to ND controls was observed (Supplemental Figure 4). Differences in absolute levels are due to the inclusion of nuclear DNA in the whole retinal samples that is absent in the synaptosome preparations and represents the bulk of the DNA mass in a cell.
Figure 5.

Mitochondrial genomics 14 weeks post-STZ injection. A) mtDNA absolute copy number was determined from isolated retinal synaptosomes from non-diabetic (ND), diabetic (D), and diabetic with insulin replacement (D+I) animals normalized to ng of DNA. n=13–18 animals/group. B) Relative mtDNA deletion amounts were determined by gapped reads. ND, animals, ratio of gapped/total mapped reads from diabetic (D) and diabetic with insulin replacement (D+I) from mtDNA sequencing from DNA isolated retinal synaptosomes. C) Ideogram of the 16,313 basepair (bp) mtDNA genes and annotations; dark gray – ribosomal RNA genes, red – D-Loop region, light gray – protein coding genes, and black – tRNAs. Blue regions ‘F’ and ‘R’ represent forward and reverse primer binding regions. D) Number of base variants relative to genomic location of mtDNA per non-diabetic control animal (heatmap rows – gray scale). The single column heatmap represents the mean variant frequency total across the mtDNA for each control animal (total mean variant frequency/bp – orange scale) (n=10 animals). E) Number of base variants relative to genomic location of mtDNA per diabetic control animal (heatmap rows – gray scale). The single column heatmap represents the mean variant frequency total across the mtDNA (total mean variant frequency/bp – orange scale) (n=7 animals). F) Number of base variants relative to genomic location of mtDNA per diabetic with insulin animal (heatmap rows – gray scale). The single column heatmap represents the mean variant frequency total across the mtDNA (total mean variant frequency/bp – orange scale) (n=10 animals). G) Box-plot representation of the mean variant frequency/bp per experimental group. ND- non-diabetic, D – diabetic, D+I – diabetic with insulin replacement. ND1 – NADH dehydrogenase subunit 1. ND2 – NADH dehydrogenase subunit 2. COXI – cytochrome c oxidase subunit 1. COXII – cytochrome c oxidase subunit 2. ATP8 – ATP synthase Fo subunit 8. ATP6 – ATP synthase Fo subunit 6. COX III – cytochrome c oxidase subunit 3. ND3 – NADH dehydrogenase subunit 3. ND4L – NADH dehydrogenase subunit 4L. ND4 – NADH dehydrogenase subunit 4. ND5 – NADH dehydrogenase subunit 5. ND6 – NADH dehydrogenase subunit 6. CytB – cytochrome b.
mtDNA sequencing
In order to quantify and map mtDNA damage in the form of large deletions and somatic single nucleotide variants with diabetes, high-depth mtDNA sequencing was performed. Positive controls and quantitative determination of method sensitivity for detection of large deletions and single nucleotide variant frequencies was performed using in vitro generated sequencing standards with and without known deletions and variants (Supplemental File 1 and Supplemental Figure 5A) mixed at known ratios resulting in standard curves for variants and deletions ranging from 0% to 100% deleted/variant frequency. Single nucleotide variants were quantified at the expected variant frequencies (Supplemental Figure 5B and Supplemental Table 6). Large-scale deletions were identified in the form of gapped reads and were found at the expected frequencies relative to the ‘100% deleted’ sequencing standard (Supplemental Figure 5C and Supplemental Table 6). These validation experiments demonstrate that somatic deletions and single nucleotide variants can be quantitatively detected down to frequencies of 0.1% through this deep sequencing approach.
Sequencing was performed on 15,848 bp of the mtDNA genome (primer binding regions were excluded) from base pairs 223 to 16,121 (Supplemental Table 1) of the rat reference mtDNA genome. Average mapped reads per sample was 335,790 in ND controls (n=10), 357,827 in D controls (n=7), and 317,457 in D+I animals (n=10), and the average depth of coverage was 1,757 ± 1,395, 1,837 ± 615, and 1,712 ± 1,394, respectively. No differences in the number of mtDNA deletions between the groups (Figure 5B) were observed in synaptosomal samples. A comprehensive, quantitative analysis of somatic mitochondrial genome variants identified a number of low frequency mutations in every animal (Figure 5C). The total variant frequency per base-pair of the mtDNA (sum of variant frequency/number of variants/15848 bp) was not significantly different when assessing all called variants. Examining only those variants that would cause an amino acid change, or only silent variants (Supplemental Table 7) revealed no differences between groups. Additionally, variants present in the coding regions of each annotated mitochondrially-encoded gene were assessed. No significant differences in variant frequencies (sum of variants frequency across the annotation/number of variants/length of gene in basepairs) (Figure 5G) across any gene regions were detected between experimental groups (Supplemental Table 7). Re-running retinal synaptosome mtDNA sequencing at higher sequencing depths and sequencing mtDNA from whole retinas also demonstrate no differences in variant frequencies or deletions with diabetes (Supplemental Figures 6 and 7, Supplemental Tables 8 and 9). Results for these sequencing runs are presented in detail in the Supplemental Material.
Discussion
This study demonstrates that diabetic changes in neural retinal morphology and function occur within three months, are partially normalized/prevented by systemic insulin treatment, and confirms a number of studies demonstrating early neuronal retinal dysfunction with diabetes (Aung et al. 2013, Pardue et al. 2014, Sohn et al. 2016). Critically, no quantitative changes in mitochondrial function, protein expression, or mtDNA integrity, indicative of mitochondrial dysfunction or damage, occur in the neural retina over this timeframe as determined by novel quantitative approaches to assay the mitochondrial genome directly. These findings provide strong evidence that diabetic mitochondrial dysfunction is not a causative factor in the initial neural retina response.
These findings are contrary to our hypothesis and suggest a difference between the neural retina and retinal vasculature where studies demonstrate endothelial mitochondrial dysfunction as an initiating factor of vascular dysfunction (Kowluru & Abbas 2003, Kowluru & Mishra 2015, Madsen-Bouterse & Kowluru 2008, Madsen-Bouterse et al. 2010, Santos et al. 2012, Tewari et al. 2012, Zhong & Kowluru 2011). No significant deficits in challenged mitochondrial activity were quantified with diabetes at a time point where neural retinal deficits are present. Increased expression of mitochondrial-related beta oxidation and antioxidant proteins with diabetes, indicates a cellular response to systemic hyperglycemia in the retina, but is likely a positively adaptive response. These results are similar to a previous demonstration of protective, positively adaptive mitochondrial protein activity changes in response to short-term diabetes (Osorio-Paz et al. 2015), which results in decreased mitochondrial efficiency but not dysfunction. Base and location specific quantitative assessment of retinal mtDNA damage in the form of somatic deletions and mutations across multiple high-depth mitochondrial sequencing datasets demonstrate that no induction of deletions or mutations occurs with early diabetes. This form of mitochondrial sequencing has not been previously applied to DR research and provides a new level of specificity and quantitation to this analysis. Absolute quantification of retinal mtDNA copy number using dPCR also demonstrates no differences with diabetes. Taken together, our data show that neural retinal functional and morphological changes in early diabetes occur without mitochondrial dysfunction or mtDNA damage. The neural function and anatomy studies provide further validation of the STZ-rat DR model and demonstrates that in addition to developing the similar molecular phenotypes as DR patients (Masser et al. 2014, Brucklacher et al. 2008, Freeman et al. 2010, VanGuilder et al. 2011), this model has early neural functional (Aung et al. 2013, Pardue et al. 2014) and morphological changes (Masser et al. 2014, VanGuilder et al. 2008, Sohn et al. 2016) and shares commonalities with non-proliferative diabetic retinopathy patients (Abcouwer & Gardner 2014).
While our results and interpretation do not support the hypothesis that mitochondrial dysfunction is causative of neural DR initiation and early progression, our findings are buttressed by the threshold effect concept in mitochondrial biology (Stewart & Chinnery 2015, Durham et al. 2007). The threshold effect is the idea that mtDNA damage must pass a critical frequency threshold within a mitochondrion/cell in order for that damage to biochemically and functionally alter the mitochondrion. Put simply, the relationship of mtDNA mutation and deletion rates to impaired mitochondrial function is not linear. In diseases and model systems where mutations or deletions of mtDNA cause functional impairments, mtDNA damage reaches frequencies of greater than 60% before a functional phenotype is present (Stewart & Chinnery 2015). Therefore, if mtDNA damage causes mitochondrial dysfunction in the retina with diabetes and these events initiate the hallmarks of vascular leakage, inflammation, and neurodegeneration observed early with DR, the mtDNA damage induced by diabetes would need to be several orders of magnitude greater than the <0.02% frequency observed here. A recent study using mtDNA sequencing data collected from exome sequencing within the 1000 Genomes Project, found that low frequency variants occurred across a majority of control, non-disease subjects (Ye et al. 2014). These data support the validity of our experimental data demonstrating the same observation in our ND controls. Another recent report detailed the effects of mutagens on the mtDNA and found no increased rates of point mutations and deletions after mutagen treatment (Valente et al. 2016). These data suggest that adduct formation on the mtDNA is not be as readily converted to somatic variants as previously hypothesized.
These data also address a knowledge gap in the field regarding assessment of mtDNA damage with DR. Most prior reports have not demonstrated mtDNA damage directly, but have concluded damage is the initiator (Kowluru & Abbas 2003, Kowluru & Mishra 2015, Madsen-Bouterse & Kowluru 2008, Madsen-Bouterse et al. 2010, Santos et al. 2012, Tewari et al. 2012, Zhong & Kowluru 2011). The methods used have been qualitative assays, such as densitometric analysis of long versus short PCR products potentially indicative of mtDNA deletions or adducts. These approaches are not quantitative, are prone to error (Barchiesi et al. 2017), and do not determine the base location in the mitochondrial genome of the purported mutations/deletions. The quantitatively validated approaches of dPCR and deep sequencing (massively paralleled sequencing – or next-generation sequencing) approaches used here can determine mtDNA damage load and precise location. This is a significant advance in mtDNA analysis and can provide not only more reproducible data but also more specific findings that generate new testable hypotheses, e.g., deletions or mutations in a specific gene.
The duration of experimental diabetes in the present study, (3 months), cannot support or oppose the hypothesis that vascular mitochondrial changes occur later in the disease process. Diabetes-induced retinal vascular mtDNA damage may reach the threshold required to cause functional changes, but currently no data is present in the literature from in vivo retinal endothelium with the quantitative resolution to support that conclusion. Our whole retina data does include endothelial cells, but these endothelial cells are a small minority of cells analyzed and the whole retina data does not show increased damage. Detailed longitudinal studies using quantitative approaches, such as presented here, as opposed to qualitative assessments, and of specific cellular populations could determine if, when, and where mtDNA damage is present in the diabetic retina. However, the importance/relevance of mitochondrial damage and dysfunction late in the neural disease process is unclear, because the deficits in neural retinal function are already manifested. The potential mitochondrial dysfunction in later disease time points may develop secondarily to neuronal dysfunction, where breakdown of the neurovascular retina unit induces endothelial cell dysfunction and capillary cell loss (Lynch & Abramoff 2017). Interestingly, antioxidant treatments and pharmacological interventions that may support mitochondrial function are successful in reducing retinal deficits observed with diabetes (Alam et al. 2015). Our data do not refute the presence of ROS or superoxide elements in the diabetic retina as increases in beta oxidation and antioxidant proteins demonstrate a response to altered metabolism and potential increased oxidative stress, but our data do not support the hypothesis that these are oxidatively damaging neural retinal mtDNA and inducing dysfunction. Longitudinal analysis of mtDNA adducts may be needed to fully assess oxidative damage over-time in the diabetic retina to determine if they manifest as functional mutations. A recent report suggests that short-term diabetes induces mitochondrial activity maintenance through prevention of oxidative stress and not dysfunction (Osorio-Paz et al. 2015). Therefore, it is reasonable to postulate that reducing the amount of potentially damaging molecules through pharmacological inhibition could benefit diabetic retinal function and mitigate DR progression in the absence of mitochondrial dysfunction and mtDNA damage. The benefit of antioxidants and pharmacological boosting of mitochondrial function in the diabetic retina is not evidence of oxidatively damaged mitochondria or mtDNA. If indeed mitochondrial dysfunction does induce capillary cell loss after initial retinal neural deficits, administration of anti-oxidant drugs could inhibit further breakdown of the neurovascular unit, but would not be useful for inhibiting the initial neural deficits of DR that precede the vascular deficits observed with long-term uncontrolled diabetes. Rather, neuroprotective treatments may prevent the initial neural retina dysfunction and possibly prevent subsequent vascular damage (Hernandez et al. 2013). Furthermore, our results demonstrate a significant decrease in the expression of a number of proteins related to the TCA/Krebs cycle in D+I rats compared to ND and D controls. These results suggest that with diabetes the increase in beta oxidation to form acetyl-CoA through fatty acid degradation is reversed with insulin treatment. This response in the D+I animals is potentially related to our previous findings demonstrating increased fat mass in insulin-treated diabetic animals (Masser et al. 2014). However, this seems to be a systemic cellular response as the retinal synaptic terminals do not store fatty acids for energy.
We conclude that neural retinal deficits that occur with 3 months of hyperglycemia are not caused by mtDNA damage-induced mitochondrial dysfunction. Our results leave open the possibility that late-stage vascular DR progression is mediated by mitochondrial damage in retinal microvasculature from long-term diabetes. However, it is not clear that the mtDNA damage reported in these studies meets or exceeds frequencies that would result in functional penetrance as detailed by the threshold effect hypothesis. Rather, the neurodegeneration occurring early in DR progression may be causative of later, classical, vascular dysfunction (Lynch & Abramoff 2017). Therefore, given the interdependence of inflammation and oxidative stress (Du et al. 2013), we postulate that neurodegeneration and inflammation are the mediators of neural DR initiation and progression at the beginning of the DR disease process. Additionally, the presence of inflammation could be contributing to the antioxidant response observed with DR. Future work will be instrumental in deciphering the interplay of neuroinflammation and neurodegeneration in starting the sequence of retinal changes that ultimate result in vascular dysfunction and proliferation.
Supplementary Material
Acknowledgments
The authors declare no potential conflicts of interest. The authors would like to thank: Dr. Allison Gillaspy the Laboratory for Molecular Biology and Cytometry Research Core Director at OUHSC for access to MiSeq sequencing instrument, Dr. Muralidharan Jayaraman the Stephenson Cancer Center Cancer Functional Genomics Core Director for training and access to the Seahorse XFe96 instrument, Dr. Anja Bastian for lending expertise with designing OCR measurement protocol, Dr. Feng Li and Megan Stiles at the Dean McGee Eye Institute Live Animal Imaging Core for assistance and training with ERG and OCT methods, Mark Dittmar Director of the Dean McGee Eye Institute Animal Facility and staff for support with animal housing and procedures, Dr. Robert E. Anderson Director of Research at Dean McGee Eye Institute and Dr. Blake Hopiavuori for gracious allowance of bench space and ultracentrifuge use for synaptosome isolations as well as aid in ERG data analysis, and Steven Glansberg for assistance with figure generation. This work was supported by the National Eye Institute (R21EY024520, R01EY021716, T32EY023202, P30EY021725), National Institute on Aging (T32AG052363), the Donald W. Reynolds Foundation, the Oklahoma Nathan Shock Center of Excellence in the Biology of Aging Targeted DNA Methylation and Mitochondrial Heteroplasmy Core (P30AG050911), and supported in part by an award from Harold Hamm Diabetes Center at the University of Oklahoma.
Abbreviations
- BHMTC
Benjamini-Hochberg Multiple Testing Correction
- D
diabetic
- D+I
diabetic with insulin replacement
- dPCR
digital PCR
- ERG
electroretinography
- ND
non-diabetic
- DR
diabetic retinopathy
- DMMC
Dunn’s Method Multiple Comparisons
- FCCP
carbonyl cyanide-4-trifluoromethoxy phenylhydrazone
- mtDNA
mitochondrial DNA
- OCT
optical coherence tomography
- OCR
oxygen consumption rate
- ONL
outer nuclear layer
- OP
oscillatory potentials
- PBST
phosphate buffered saline with tween
- qPCR
quantitative PCR
- R/AA
rotenone/antimycin a
- ROS
reactive oxygen species
- SNK
Student Newman Keuls
- STZ
streptozotocin
- TCA
tricarboxylic acid cycle
- COXIV
cytochrome c oxidase subunit IV
Footnotes
Data access: All fastq files are available from the NCBI Sequence Read Archive under the accession number PRJNA389563.
Conflicts of interest: none
References
- Abcouwer SF, Gardner TW. Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci. 2014;1311:174–190. doi: 10.1111/nyas.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello LP, Group DER. Diabetic retinopathy and other ocular findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care. 2014;37:17–23. doi: 10.2337/dc13-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam NM, Mills WCt, Wong AA, Douglas RM, Szeto HH, Prusky GT. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech. 2015;8:701–710. doi: 10.1242/dmm.020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonetti DA, Barber AJ, Bronson SK, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- Aung MH, Kim MK, Olson DE, Thule PM, Pardue MT. Early visual deficits in streptozotocin-induced diabetic long evans rats. Invest Ophthalmol Vis Sci. 2013;54:1370–1377. doi: 10.1167/iovs.12-10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchiesi A, Baccarani U, Billack B, Tell G, Vascotto C. [Letter to the Editor] Isolation of mitochondria is necessary for precise quantification of mitochondrial DNA damage in human carcinoma samples. Biotechniques. 2017;62:13–17. doi: 10.2144/000114491. [DOI] [PubMed] [Google Scholar]
- Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36:1069–1077. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. On the adaptive control of the false discovery fate in multiple testing with independent statistics. J Educ Behav Stat. 2000;25:60–83. [Google Scholar]
- Bixler GV, Vanguilder HD, Brucklacher RM, Kimball SR, Bronson SK, Freeman WM. Chronic insulin treatment of diabetes does not fully normalize alterations in the retinal transcriptome. BMC Med Genomics. 2011;4:40. doi: 10.1186/1755-8794-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucklacher RM, Patel KM, VanGuilder HD, et al. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med Genomics. 2008;1:26. doi: 10.1186/1755-8794-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, Nicholls DG. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J Neurochem. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Veenstra A, Palczewski K, Kern TS. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A. 2013;110:16586–16591. doi: 10.1073/pnas.1314575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L. Mitochondrial quality control in neurodegenerative diseases. Biochimie. 2014;100:177–183. doi: 10.1016/j.biochi.2013.07.033. [DOI] [PubMed] [Google Scholar]
- Durham SE, Samuels DC, Cree LM, Chinnery PF. Normal levels of wild-type mitochondrial DNA maintain cytochrome c oxidase activity for two pathogenic mitochondrial DNA mutations but not for m.3243A-->G. Am J Hum Genet. 2007;81:189–195. doi: 10.1086/518901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WM, Bixler GV, Brucklacher RM, et al. A multistep validation process of biomarkers for preclinical drug development. Pharmacogenomics J. 2010;10:385–395. doi: 10.1038/tpj.2009.60. [DOI] [PubMed] [Google Scholar]
- Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253–262. doi: 10.1016/s0039-6257(02)00387-9. [DOI] [PubMed] [Google Scholar]
- Hernandez C, Garcia-Ramirez M, Corraliza L, Fernandez-Carneado J, Farrera-Sinfreu J, Ponsati B, Gonzalez-Rodriguez A, Valverde AM, Simo R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes. 2013;62:2569–2578. doi: 10.2337/db12-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GR, Scott IU, Quillen DA, Walter LE, Gardner TW. Inner retinal visual dysfunction is a sensitive marker of non-proliferative diabetic retinopathy. Br J Ophthalmol. 2012;96:699–703. doi: 10.1136/bjophthalmol-2011-300467. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta. 2015;1852:2474–2483. doi: 10.1016/j.bbadis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44:791–804. doi: 10.1007/s001250100544. [DOI] [PubMed] [Google Scholar]
- Lynch SK, Abramoff MD. Diabetic retinopathy is a neurodegenerative disorder. Vision Res. 2017 doi: 10.1016/j.visres.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen-Bouterse SA, Kowluru RA. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord. 2008;9:315–327. doi: 10.1007/s11154-008-9090-4. [DOI] [PubMed] [Google Scholar]
- Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010;13:797–805. doi: 10.1089/ars.2009.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR, Berg AS, Freeman WM. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenetics Chromatin. 2013;6:33. doi: 10.1186/1756-8935-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR, Clark NW, Van Remmen H, Freeman WM. Loss of the antioxidant enzyme CuZnSOD (Sod1) mimics an age-related increase in absolute mitochondrial DNA copy number in the skeletal muscle. Age (Dordr) 2016;38:323–333. doi: 10.1007/s11357-016-9930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR, VanGuilder Starkey HD, Bixler GV, Dunton W, Bronson SK, Freeman WM. Insulin treatment normalizes retinal neuroinflammation but not markers of synapse loss in diabetic rats. Exp Eye Res. 2014;125:95–106. doi: 10.1016/j.exer.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada Y, Canseco DC, Thet S, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. doi: 10.1038/nature20173. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Diabetes C, Lachin JM, White NH, Hainsworth DP, Sun W, Cleary PA Complications Trial /Epidemiology of Diabetes I, Complications Research G. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes. 2015;64:631–642. doi: 10.2337/db14-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio-Paz I, Uribe-Carvajal S, Salceda R. In the Early Stages of Diabetes, Rat Retinal Mitochondria Undergo Mild Uncoupling due to UCP2 Activity. PLoS One. 2015;10:e0122727. doi: 10.1371/journal.pone.0122727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue MT, Barnes CS, Kim MK, Aung MH, Amarnath R, Olson DE, Thule PM. Rodent Hyperglycemia-Induced Inner Retinal Deficits are Mirrored in Human Diabetes. Transl Vis Sci Technol. 2014;3:6. doi: 10.1167/tvst.3.3.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med. 2012;53:1729–1737. doi: 10.1016/j.freeradbiomed.2012.08.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennlaub F, Valamanesh F, Vazquez-Tello A, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204. doi: 10.1161/01.CIR.0000080735.93327.00. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn EH, van Dijk HW, Jiao C, et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci U S A. 2016;113:E2655–2664. doi: 10.1073/pnas.1522014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- Stitt AW, Curtis TM, Chen M, et al. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res. 2016;51:156–186. doi: 10.1016/j.preteyeres.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Suresh K. An overview of randomization techniques: An unbiased assessment of outcome in clinical research. J Hum Reprod Sci. 2011;4:8–11. doi: 10.4103/0974-1208.82352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012;17:492–504. doi: 10.1089/ars.2011.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente WJ, Ericson NG, Long AS, White PA, Marchetti F, Bielas JH. Mitochondrial DNA exhibits resistance to induced point and deletion mutations. Nucleic Acids Res. 2016;44:8513–8524. doi: 10.1093/nar/gkw716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Bixler GV, Kutzler L, Brucklacher RM, Bronson SK, Kimball SR, Freeman WM. Multi-modal proteomic analysis of retinal protein expression alterations in a rat model of diabetic retinopathy. PLoS One. 2011;6:e16271. doi: 10.1371/journal.pone.0016271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Brucklacher RM, Patel K, Ellis RW, Freeman WM, Barber AJ. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J Neurosci. 2008;28:1–11. doi: 10.1111/j.1460-9568.2008.06322.x. [DOI] [PubMed] [Google Scholar]
- Vaziri K, Schwartz SG, Relhan N, Kishor KS, Flynn HW., Jr New Therapeutic Approaches in Diabetic Retinopathy. Rev Diabet Stud. 2015;12:196–210. doi: 10.1900/RDS.2015.12.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A. 2014;111:10654–10659. doi: 10.1073/pnas.1403521111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Vis Sci. 2011;52:8739–8746. doi: 10.1167/iovs.11-8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.