

Abstract
Chondroitin sulfate proteoglycans (CSPGs) are components of the extracellular matrix that inhibit the extension and regeneration of axons. However, the underlying mechanism of action remains poorly understood. Mitochondria and endoplasmic reticulum (ER) are functionally inter-linked organelles important to axon development and maintenance. We report that CSPGs impair the targeting of mitochondria and ER to the growth cones of chicken embryonic sensory axons. The effect of CSPGs on the targeting of mitochondria is blocked by inhibition of the LAR receptor for CSPGs. The regulation of the targeting of mitochondria and ER to the growth cone by CSPGs is due to attenuation of PI3K signaling, which is known to be downstream of LAR receptor activation. Dynactin is a required component of the dynein motor complex that drives the normally occurring retrograde evacuation of mitochondria from growth cones. CSPGs elevate the levels of p150Glu dynactin found in distal axons, and inhibition of the interaction of dynactin with dynein increased axon lengths on CSPGs. CSPGs also decreased the membrane potential of mitochondria, and pharmacological inhibition of mitochondria respiration at the growth cone independent of manipulation of mitochondria positioning impaired axon extension. Combined inhibition of dynactin and potentiation of mitochondria respiration further increased axon lengths on CSPGs relative to inhibition of dynactin alone. These data reveal that the regulation of the localization of mitochondria and ER to growth cones is a previously unappreciated aspect of the effects of CSPGs on embryonic axons.
Keywords: mitochondria, axon, growth cone, dynein, dynactin
INTRODUCTION
CSPGs are components of the extracellular matrix of the nervous system with roles in the development and regeneration of axonal projections (Silver and Silver, 2014). CSPGs impair axon extension and induce the growth cone to become “dystrophic”. Dystrophic growth cones are characterized by decreased and unstable protrusive activity and altered membrane turnover (Tom et al., 2004). The LAR and PTP receptors mediate most inhibitory effects of CSPGs (Sharma et al., 2012). Binding of CSPGs to either receptor decreases PI3K signaling and activates RhoA-ROCK signaling (Fisher et al., 2011; Silver et al., 2014; Ohtake et al., 2016), and both restoration of PI3K signaling and inhibition of ROCK signaling partially counter the effects of CSPGs on axon extension (Monnier et al., 2003; Jain et al., 2004; Silver et al., 2014). CSPGs also attenuate Erk signaling in neurons (Ohtake et al., 2016). While CSPGs are considered to affect the cytoskeleton, if and how CSPGs affect axonal organelles has not been investigated.
Mitochondria are a major source of ATP production, regulate cytoplasmic calcium levels and are sources of reactive oxygen species generation. In neurons mitochondria undergo bidirectional transport along axons and accumulate at the growth cone (Saxton and Hollenbeck, 2012; Sheng et al., 2014). Recent studies have identified the transport of mitochondria to distal axons as an important component of axon regeneration after injury (Cartoni et al., 2016; Han et al., 2016; Zhou et al., 2016). Conversely, the stalling of axons correlates with retrograde displacement of mitochondria from the axon tip (Morris and Hollenbeck, 1993). A previous study found that CSPGs decrease mitochondria respiration along the axon shaft, thereby contributing to the suppression of branching along embryonic sensory axons (Sainath et al., 2016). Mitochondria and the endoplasmic reticulum (ER) are functionally and structurally linked organelles that cooperate in calcium signaling and traffic metabolites (Filadi et al., 2017). Both organelles are considered to contribute to axon morphogenesis (Winkle et al., 2016), and the respiration of axonal mitochondria can be controlled by extracellular signals (Verburg and Hollenbeck, 2008; Pacelli et al., 2015; Sainath et al., 2016). However, the regulation of the subcellular distribution of axonal organelles by extracellular signals is poorly understood. We report that the targeting of mitochondria and ER to the distal axon is negatively regulated by CSPGs and provide evidence that this effect of CSPGs is mediated by the LAR receptor and suppression of PI3K signaling. Dynactin associated with polymerizing microtubule tips serves to activate the normally occurring retrograde transport of organelles in distal axons resulting in their evacuation from growth cones (Moughamian and Holzbaur, 2012; Moughamian et al., 2013; Nirschl et al., 2016). The decreased targeting of mitochondria to distal axons on CSPGs are similarly mediated by a dynactin and microtubule dynamics based mechanism, and CSPGs elevate the levels of dynactin in distal axons.
METHODS
Culturing, substrata and transfection
Chicken embryonic (E) day 7 dorsal root ganglion neurons were cultured in defined medium containing 20 ng/mL nerve growth factor as previously detailed (Lelkes et al., 2006). Transfection of dissociated neurons was performed prior to plating using Amaxa Nucleofector (Lonza) on setting G013 and 10 μg of each plasmid in Amaxa transfection solution following the manufacturer’s protocol. Substrates were prepared as described in Sainath et al (2016). Control substrates were coated with polylysine (100 μg/mL; Sigma) and laminin (15 μg/mL; Invitrogen), and experimental substrates further coated with CSPG mixtures from E14 chicken brains (EMD Millipore; 5 μg/mL was used unless otherwise noted). The CC1-mCherry plasmid was as in Moughamian and Holzbaur (2012), the NPYss-mCherry as in Sainath and Gallo (2015), the eYFP-β-actin plasmid as in Ketschek and Gallo (2010), the mtDsRed and mt-GFP were as in Spillane et al (2013). Chondoitinase ABC (Sigma) treatment was performed as previously described (Lang et al., 2015) using 0.033 units/mL for 1 hour, followed by washing away the enzyme prior to plating neurons.
Microfluidic chambers were designed by the Thomas Lab (Holland et. al, 2016) using AutoCAD software, based on a prior description (Park et. Al, 2006). Master molds (fabricated at Stanford University Microfluidics Foundry and Missouri State University Jordan Valley Innovation Center) consisted of three chambers (width of middle chamber: 1,000 μm; width of adjacent chambers: 1,500 μm) connected by 450-μm-long microgrooves. Microfluidic chambers devices were cast using Sylgard 184 (Dow Corning). DRG neurons were dissociated and cultured on laminin and polylysine substrata in media containing 20 ng/mL NGF in the microfluidic chambers for 3 days in vitro with a media change on day 2 in vitro. The fluidically isolated axonal compartment contained either control media containing DMSO or Antimycin A (Sigma).
Pharmacological reagents and peptides
Extracellular LAR peptide (ELP) and random peptide (RP, control) were prepared and used as described in Fisher et al (2011). PI3Kpep and PI3KpepAla peptides (custom synthesized by American Peptide Corporation) were used as previously described (Silver et al., 2014). LY294002, Y27632 and PD325901 were purchased from Sigma and dissolved in DMSO at 1000× the final concentration.
Immunostaining
Cultures were fixed with 0.25% glutaraldehyde for 15 min and subsequently permeabilized using (0.1% TX-100) for 15 min. Glutaraldehyde autofluorescence was then quenched with 2 mg/mL sodium borohydride in calcium magnesium-free phosphate buffered saline (PBS) for 15 min. Samples were then blocked using 10% goat serum in PBS with 0.1% Triton X-100 (GST) and stained with primary antibodies for 45 min, washed in GST 3 times, and when needed (i.e., p150Glu staining) secondary antibodies were applied for 45 min, washed in deionized water, and mounted in no-fade mounting medium. The p150Glu dynactin antibody was used as detailed in Sainath and Gallo (2015). Tubulin was stained using anti-α-tubulin conjugated to fluorescein (DM1A-FITC; Sigma). Rhodamine phalloidin was obtained from Invitrogen and used per the manufacturer’s protocol.
Organelle dye labeling
All organelles dyes were purchased from Invitrogen. Organelles were stained by a 30 min treatment with 25 nM Mitotracker Green, 1 nM Mitotracker Red, 500 nM ER tracker red or 75 nM Lysotracker red, followed by three washes in medium and a 30 min recovery period prior to imaging.
Imaging and image analysis
Imaging of fluorescently labeled cells was performed on a Zeiss 200M Axiovert microscope using a 100× (1.3 N.A) objective, a 40× objective for imaging of the rate of axon extension, and a 20× objective for acquisition of data addressing axon lengths in neurons expressing fluorescent constructs. Zeiss Axiovision software was used for image acquisition using an Orca ER (Hamamatsu) camera, and analysis using the built in functions for area, length and intensity measurements. All images used for quantification of fluorescence intensities were acquired using parameters that did not result in pixel saturation and maintained constant in any given experiment with all data acquired during the same microscopy session.
Analysis of mitochondria and ER positioning
Mito-Tip measurements were obtained by tracing the distance between the distal-most extent of the distal-most mitochondrion in the axon to the distal-most portion of the growth cone excluding filopodia. Similarly, the distance between the highest peak of ER staining in the distal 50 μm of the axon and the distal most part of the axon was determined, as with Mito-Tip measurements. Peaks were determined using the NIH ImageJ plot profile function with a line scan through the axon.
Analysis of growth cone dynamics
The frequencies of the initiation of protrusive events were determined from time lapses of growth cones from using either phase contrast optics or epifluorescent imaging of eYFP-β-actin expressing neurons. A protrusive event was defined as the initiation of a filopodium or a lamellipodium from a stretch of the perimeter of the growth cone which did not exhibit protrusive activity in the prior frames. The continued extension of a lamellipodium of filopodium in subsequent frames was not considered a protrusive event, but rather the continuation of the previously initiated event. If a lamellipodium or filopodium underwent retraction into the perimeter of the growth cone but was then reinitiated after more than 20 seconds this was then considered a new independent protrusive event.
Analysis of axon lengths and growth cone morphology
Overall axon morphology and length were analyzed from 20× images of neurons, with montage of multiple images if the axons did not fit in a single image. Sensory neurons generate 1–2 axons in our culturing conditions. Since axon extension is growth cone mediated and each axon has a growth cone, two separate axon measurements were made for neurons with two axons. When axons bifurcate through growth cone splitting the ensuing branches of the main axon shaft grow at an approximate 60–70° angle and exhibit similar axon calibers. If an axon bifurcated, as determined by this definition, the lengths of each emergent branch was considered in the total axon length for that axon. In contrast, axon collateral branches arise at an approximate 90° angle from the axon shaft and have a much smaller caliber than the main axon. Measurements of collateral lengths were not considered in the determination of total axon length.
For growth cones with clearly defined “neck” regions, reflective of where the growth cone enlarged relative to uniform caliber of the axon shaft, the proximal most extent of the growth cone was considered to be the neck. For growth cones that did not have necks (i.e., did not spread out and exhibited a fusiform morphology) we defined the growth cone as the distal 10 μm of the axon. This distance was used because it approximates the proximo-distal length of growth cones with necks in our culturing system.
Statistical analysis
Analysis was performed using Graphpad Instat software, which automatically checks the normalcy of the data distribution. Sets containing non-normally distributed data were compared using the Mann-Whitney test. Non-normally distributed data sets are shown in graphs using the mean, SEM and median. Normally distributed data sets were compared using Welch t-tests. Categorical data sets were compared using the Fisher’s exact test performed on the raw categorical data, but categorical data are presented as percentages in figures. For all experiments data were collected from 4–6 replicate cultures per group.
RESULTS
CSPGs promote the retrograde displacement of mitochondria away from the tip of the axon
The extension of axons on uniform CSPGs substrata, or up a gradient of CSPGs, is inhibited and growth cones generate few filopodial and lamellipodial protrusions and exhibit altered membrane dynamics (Tom et al., 2004; Silver et al., 2014; Lang et al., 2015). Growth cones of adult rat dorsal root ganglia (DRG) extending up a gradient of CSPGs exhibit the formation of varicosities along the distal axon that subsequently undergo retrograde movement (Lang et al., 2015). Time-lapse analysis of the extension of chicken embryonic DRG axons on uniform CSPG substrates revealed that the tip of the axon similarly undergoes repeated formation of varicosities which subsequently undergo retrograde displacement along the axon shaft (Figure 1A). Unless otherwise noted the remainder of this study was conducted using chicken embryonic DRGs. Varicosities found in the vicinity of the axon tip exhibited accumulation of mitochondria (Figure 1B). The distance between the distal-most mitochondrion and the distal most extent of the growth cone excluding filopodia but including lamellipodia (hereafter referred to as Mito-Tip) was increased in both axons growing from sensory explants and dissociated neurons cultured overnight on CSPG coated substrata, and in axons growing from explants cultured on the control substratum following acute treatment with soluble CSPGs (Figure 1B,C). The increase in Mito-Tip was observed both through labeling mitochondria with dyes or expression of genetically encoded mitochondrially targeted GFP (Figure 1C). On CSPGs, the distal 10 μm of the axon representing the growth cone contained fewer mitochondria than on the permissive control substratum (Figure 1D). Analysis of the number of mitochondria along the more proximal axon (20–60 μm behind the tip) in the same population did not reveal differences between CSPG coated and control substrata (1.6+0.1 and 1.7+0.1 mitochondria/10 μm respectively, p>0.05).
Figure 1.

CSPGs impair the targeting of mitochondria and the ER to the distal axon. (A) Example of the dynamics of axons on CSPGs. Varicosities formed along the distal axon that subsequently underwent retrograde displacement (yellow arrows). Although periods of growth cone elaboration were observed (e.g., 15–24 min) the axon failed to advance significantly. (B) Example of localization of mitochondria (green; distal most denoted by green arrow) relative to the distal most axon (denoted by red arrow) in dissociated neurons. The inset shows the accumulation of mitochondria in a varicosity proximal to the axon tip. (C) Quantification of the number of mitochondria in distal axons of dissociated neurons. Bars on right show analysis considering only axons with mitochondria targeted to distal 10 μm in both control and CSPG groups. (D) Quantification of Mito-Tip under different culturing conditions, on CSPG substrata or treatment with soluble CSPGs (20 mg/mL, 1 hr), and labeled with mitotracker green (MT-green) or genetically encoded GFP targeted to mitochondria (mt-GFP). (E) Example of timelapse imaging of mitochondria in distal axons of dissociated neurons. White arrows in first panels denote distal and the blue dots the outline of the growth cones (time in sec). In CSPG panels, green arrow denotes a mitochondrion which moved distally, and yellow arrows denote retrograde movement of mitochondria away from the axon tip. While the number of mitochondria increased in the control axon, 2/3 of mitochondria evacuated on CSPGs. (F) Quantification of the percentage of mitochondria that underwent net retrograde or anterograde/distal movement during the imaging periods (360 sec, 10 sec intervals). (G) Measurements of Mito-Tip from dual channel phase contrast and green movies. Note that the Y-axis has approximately twice the range for the CSPG group relative to the control due to the increase Mito-Tip measurements in the former. Each line represents one axon. Arrows denote brief periods of increased Mito-Tip in controls. (H) Example of intermittent increase in Mito-Tip in control axons. Between 0–6 min the axon advances but the mitochondria do not. At 7–9 min some of the mitochondria advance and return in proximity to the tip of the axon. (I) Examples of ER Tracker labeled axons and line scans. White and yellow arrowheads denote distal axon tip and accumulation of ER. Line scans of the intensity of ER-tracker labeling along the axons are shown below the images. The tip of the axon and the distal-most highest peak of labeling intensity are denoted by white and yellow arrows, respectively. (J) Quantification of the mean distance between the axon tip and the distal-most highest peak of ER-tracker staining intensity.
Timelapse imaging of mitochondria dynamics, focusing on axons that contained mitochondria in the distal 20 μm at the beginning of imaging on both control and CSPG coated substrata, revealed that on CSPGs mitochondria initially located to the axon tip exhibited a greater tendency to undergo retrograde evacuation from the tip and decreased anterograde movements toward the tip on CSPG substrata (Figure 1E, F). Analysis of the proportions of mitochondria that remained stalled or underwent net anterograde or retrograde movement in the more proximal axons (20–60 μm from tip) in the same set of axons did not reveal any difference (p=0.54, Chi2 test for independence). Consideration of the dynamics of axon extension relative to mitochondria motility in distal axons, by measuring the Mito-Tip in timelapses, revealed that on the control substratum mitochondria overall remained targeted to the distal axon (Figure 1G). On the control substratum, Mito-Tip occasionally briefly increased and this was due to the forward advancement of the axon without coordinated advance of the mitochondria (Figure 1G,H), which however “caught up” with the distal end of the axon within 2–4 minutes. In contrast, on CSPG coated substrata the mitochondria continuously exhibited elevated Mito-Tip measurements and, consistent with prior observations (Figure 1E,F), when Mito-Tip increased it was due to the evacuation of mitochondria from the distal axon.
CSPGs impact the positioning of the ER in distal axons but not lysosomes or Golgi-derived vesicles
To study the distribution of the ER in axons and growth cones we labeled the ER with ER-tracker, a dye that selectively incorporates into the ER. On the control substratum, the ER accumulates at the growth cone (Figure 1I). Imaging of the distribution of the ER along axons revealed that, similar to mitochondria, on CSPGs the distal accumulation of the ER was displaced away from the tip of the axon and also colocalized to varicosities along axons (Figure 1I,J), although some ER was retained at the tips of axons. The mean maximal intensity of the distal-most ER accumulations did not differ between control and CSPG substrata (p=0.15). In contrast to the ER and mitochondria, analysis of the distribution of lysosomes, labeled using LysoTracker, along axons did not reveal any difference between the control and CSPG-coated substrata (Supplemental Figure 1A,B). Similarly, analysis of the number of NPYss-mCherry puncta, reflective of Golgi-derived vesicles (El Meskini et al., 2001), along distal axons did not show any difference between the control and CSPG substrata (Supplemental Figure 1C,D). Collectively, the data indicate that CSPGs impair the targeting of mitochondria and ER to the distal axon, but lysosomes and Golgi-derived vesicles do not appear to be affected.
Involvement of the LAR receptor and suppression of PI3K signaling in the effects of CSPGs on the targeting of mitochondria to growth cones
The majority of the effects of CSPGs on neuronal function are due to the chondroitin sulfate glycosaminoglycan (GAG) chains attached to the core protein (Sharma et al., 2012). Chondroitinase ABC (ChABC) is an enzyme that cleaves the GAG chains and blocks the axon growth inhibitory effects of CSPGs in vivo and in vitro (Bardbury and carter, 2011). The determine if GAG chains mediate the effects of CSPGs on mitochondria positioning and growth cone morphology CSPG substrata were treated with ChABC prior to plating neurons following established protocols (Lang et al., 2015). Treatment with ChABC restored mitochondria positioning to distal axons, increased the number of growth cone filopodia and the percentage of growth cones exhibiting lamellipodia (Supplemental Figure 2). These data indicate that the GAG chains of CSPGs are required for the effects of CSPGs on mitochondria positioning and growth cone morphology.
The LAR and PTPσ receptors account for the majority of the inhibitory effects of CSPGs on axon extension and regeneration (Ohtake and Li, 2015). A 2 hr treatment of cultures with the ELP peptide, which blocks the interaction of CSPGs with the LAR receptor and promotes axon extension in vitro and regeneration in vivo (Fisher et al., 2011), decreased Mito-Tip on CSPGs (Figure 2A,B), and also expectedly promoted elaboration of growth cones as reflected by increases in their area and the number of filopodia relative to treatment with the inactive control RP peptide (Figure 2A,C,D). The LAR receptor mediates its effects through a suppression of PI3K-Akt signaling (Fisher et al., 2011; Ohtake et al., 2016). Pharmacological inhibition of PI3K (1 hr 25 μM LY294002) on permissive substrata increased Mito-Tip (Figure 2E,F). Direct activation of PI3K signaling using the cell permeable peptide PI3Kpep partially restores the extension of axons and growth cone morphology on CSPG coated substrata (Silver et al., 2014). A 1 hr treatment with PI3Kpep, but not the inactive control PI3KpepAla peptide (Ketschek and Gallo, 2010; Silver et al., 2014), of neurons cultured on CSPG coated substrata overnight decreased Mito-Tip and restored mitochondria into distal axons (Figure 2G). We next considered the effects of activating and inhibiting PI3K on the localization of the ER to distal axons. PI3Kpep treatment promoted the distal localization of the ER on CSPG substrata (Figure 3A,B) and inhibition of PI3K resulted in the retrograde displacement of the ER away from the axon tip on the control substratum (Figure 3C,D).
Figure 2.

Evidence for the involvement of the LAR receptor and suppression of PI3K signaling in mediating the effects of CSPGs on mitochondria positioning and growth cone morphology. (A) Examples of axons on CSPGs +/− treatment with the LAR inhibitory ELP peptide or the control inactive RP peptide(2 hrs). Phase contrast and Mitotracker green labeled mitochondria are shown. Labeling as in Figure 1B. (B) Graph showing Mito-Tip measurements under conditions of RP (control) and ELP treatment on CSPG substrata. Sample sizes showing the bars also apply to panels C and D. (C) Graph showing the effects of RP and ELP treatment on growth cone size on CSPG substrata. (D) Graph showing the effects of RP and ELP treatment on the number of growth cone filopodia on CSPG substrata. (E) Examples of growth cones on the control substratum treated for 30 min with DMSO or the PI3K inhibitor LY294002. Presentation as in panel (A). (F) Graph of Mito-Tip measurements for axons on the control substratum as a function of treatment with DMSO or LY294002. (G) Graph showing the effects of treatment with the PI3K activating PI3Kpep, and its inactive control PI3KpapAla, on Mito-Tip measurements. PI3KpepAla does not affect Mito-Tip measurements on CSPGs. However, treatment with PI3Kpep treatment decreases Mito-Tip measurements relative to RP treatment, and with PI3Kpep treatment Mito-Tip measurements are not different from the control substratum. Samples sizes are denoted in the bars.
Figure 3.

Regulation of ER positioning at growth cones on CSPGs by PI3K signaling. (A) Examples of the distribution of the ER, as revealed by ER tracker labeling, in distal axons on CSPGs treated with the PI3K activating peptide PI3Kpep or the inactive control peptide PI3KpepAla. Arrows denote the distal most ER accumulation. (B) Quantification of the distance of the distal most ER peak relative to the tip of the axon, as in Figure 1J, on CSPGs with PI3Kpep or PI3KpepAla treatment. (C) Quantification of the distance of the distal most ER peak relative to the tip of the axon on the control substratum following treatment with DMSO or the PI3K inhibitor LY294002. (D) Quantification of the distance of the distal most ER peak relative to the tip of the axon and Mito-Tip on CSPGs following treatment with the ROCK inhibitor Y27632. Samples sizes are denoted in the bars.
CSPGs also activate RhoA-ROCK signaling and inhibition of ROCK promotes axon extension of CSPG substrata. In contrast to the manipulation of PI3K signaling, treatment with an inhibitor of ROCK signaling (25 μM Y27632, 1 hr) failed to affect either Mito-Tip or the localization of the distal most accumulation of the ER on CSPG substrata (Figure 3E). CSPGs also decrease Erk signaling (Ohtake et al., 2016). However, inhibition of Mek-Erk signaling using PD325901 (1 μM, 1 hr) also did not affect either Mito-Tip (p=0.75, n=166 and 144) or the number of mitochondria in growth cones (p=0.97) on the control substratum. However, as a positive control the treatment with PD325901 was effective in decreasing the levels of phosphorylated Erk (not shown).
CSPGs depolarize the membrane potential of mitochondria in distal axons
Actin is an ATPase and the turnover of actin filaments is a major sink for ATP utilization in developing neurons (Bernstein and Bamburg, 2003). We previously reported that CSPGs depolarized the membrane potential of mitochondria along the axon shaft of sensory neurons contributing to CSPG-mediated suppression of axon branching by decreasing axonal actin dynamics (Sainath et al., 2016). In sensory neurons, and other cell types, the mitochondrial dye Mitotracker Red incorporates into mitochondria in a manner dependent on the membrane potential (Buckman et al., 2001; Pendergrass et al., 2004; Sainath et al., 2016). Analysis of the labeling of Mitotracker Red in the distal-most mitochondria in axons similarly showed that mitochondria membrane potential is depolarized on CSPGs relative to the control substratum (Figure 4A). The intensity of mitotracker red staining in mitochondria on CSPGs was decreased by 47% relative to the control substratum (Figure 4A; p<0.001, n=102 and 116).
Figure 4.

Inhibition of mitochondria membrane potential to a similar extent as observed on CSPGs impairs axon extension and growth cone protrusive dynamics. (A) Examples of axons stained with mitotracker red. Top panels show phase contrast images of axonal morphology on control and CSPG coated substrata. Bottom panels show the mitotracker red grayscale staining intensity. The false colored insets show the distal most mitochondria (shown in white boxes in the gray scale images). Line plots of mitotracker red intensity are shown below each of the false colored insets. (B) Examples of the extent of axon extension on the control substratum following a 30 min treatment with either DMSO of 2.5 μM antimycin-A. The white arrows in the 30 min panels show the position of the growth cone at time 0. (C) Examples of growth cone dynamics in DMSO and Antimycin-A treatment conditions. The control DMSO treated growth cone exhibits many protrusions. Antimycin-A treatment suppresses protrusive activity, but as denoted by the arrows some degree of protrusion persists. (D) Quantification of the total number of protrusive events at growth cones (lamellipodia or filopodia) in DMSO and Antimycin-A treated samples during a 6 min imaging period starting at 30 min post-treatment. (E) Examples of the extent of axon extension on the control substratum at 24 hrs and 72 hrs representing pre-treatment (24 hrs) and 48 hrs post-treatment with either DMSO or 2.5 μM antimycin-A. The schematic shows the time course of the experiment and treatments. Note that the DMSO treated control (72 hrs) exhibits axons much longer than the 24 hr pre-treatment example. In contrast, the antimycin-A treated example (72 hrs) shows minimal advance of axons beyond the extent evident at 24 hrs. The boxed region in the antimycin-A panel is magnified below showing that the axons do not exhibit signs of degeneration and maintain a uniform linear morphology, as revealed by tubulin staining. (F) The schematic shows the setup of the microfluidic chambers. The examples below show axons that extended through the channels and into the DMSO containing distal axon chamber in the control group and stalled at the entry of the distal axon chamber containing antimycin-A. Samples sizes are denoted in the bars.
We sought an approach to mimic the effect of CSPGs on mitochondria membrane potential so as to investigate how this might impact axon extension and growth cone protrusion dynamics. Antimycin-A is an inhibitor of cytochrome C reductase that results in the disruption of the proton gradient, depolarization of the mitochondrial membrane potential and decreased ATP production. Therefore, we carried out a dose response curve analysis of the effects of varied concentration of antimycin-A on the membrane potential of mitochondria in distal axons (not shown). Treatment with 2.5 μM antimycin-A decreased Mitotracker red staining intensity by 53% on the control substratum (Sainath et al., 2016), an effect similar to the 47% decrease observed on CSPGs. The effect of antimycin-A treatment on mitotracker red intensity is established by 30 min following treatment (Sainath et al., 2016). Analysis of the response of growth cones on the control substratum to acute treatment with 2.5 μM antimycin-A yielded a 39% suppression of the rate of axon extension during a 30 min post-treatment period relative to control treatment with DMSO (Figure 4B; p<0.05, n=18, 23 axons). Treatment with antimycin-A also suppressed the initiation of filopodia and lamellipodia at growth cones sampled at 30 min post-treatment for 6 min (Figure 4C,D). After antimycin-A treatment the frequency of formation of protrusive structures was decreased by approximately 70%. Treatment of sensory explants with antimycin-A starting at 24 hrs following culturing and persisting for an additional 48 hrs showed that antimycin-A treated axons underwent minimal further extension without exhibiting overt retraction or degeneration (Figure 4E), the latter evidenced by the absence of the formation of axonal swellings or loss of tubulin staining (see boxed in region of Figure 4E). Finally, culturing neurons in microfluidic chambers containing antimycin-A in the distal compartment, into which axons extend after having gone through the microfluidic channels connecting the cell body and distal compartment, blocked extension into the distal compartment (Figure 4F). While 63% of axons within 20 μm of the entry into the distal chamber containing DMSO penetrated the chamber by >20 μm, only 7% did so when the chamber contained antimycin-A (p<0.0001; n=7 and 4 chambers respectively; the position of axon tips denoted by arrows in Figure 4F). Collectively, these observations indicate that suppression of mitochondrial membrane potential at levels analogous to those observed on CSPGs can contribute to the impairment of axon extension and growth cone dynamics.
Treatment with acetyl-L-carnitine (ALC) promotes mitochondrial respiration (reviewed in Onofrj et al., 2013). ALC partially restores mitochondria-dependent axon branching on CSPG substrata and rescues the dynamics of the axonal actin filament cytoskeleton locally associated with mitochondria from the inhibitory effects of CSPGs (Sainath et al., 2016). To determine if ALC could promote protrusive activity at growth cones, neurons were transfected with eYFP-β-actin and mt-DsRed and then treated for four hrs with medium + ALC (500 μM final concentration; the same concentration as in Sainath et al. (2016) that promotes branching and restores actin dynamics along the axon shaft). ALC treatment did not alter the Mito-Tip distance on CSPGs (Figure 5A,B), indicating it had no effect on the targeting of mitochondria into distal axons. We next analyzed the rate of formation of protrusive structures, represented by the initiation of new lamellipodia or filopodia, at the growth cones of neurons transfected with eYFP-β-actin. ALC treatment promoted the initiation of growth cone protrusive activity during the 6 min imaging period (Figure 5C,D). However, the effect of ALC on the promotion of protrusive active was partial. Growth cones treated with ALC exhibited 48% of the relative rates of protrusive activity of growth cones on the control substratum without additional treatments (n=21 growth cones; p=<0.001). Considering filopodia existing at the growth cone at the beginning of imaging we observed that treatment with ALC increased the percentage of these filopodia that underwent one or more bouts of tip elongation >1 μm during the imaging period (Figure 5E). Furthermore, the growth cones treated with ALC also exhibited more filopodial and lamellipodial retractions (Figure 5C; data not shown). Collectively these observations indicate that ALC treatment promotes actin filament polymerization and turnover at the growth cone. The observations on growth cone dynamics are consistent with a morphological analysis of growth cones. CSPGs decreased the number of growth cone filopodia and the percentage of growth cones exhibiting lamellae (Figure 5F–H). Treatment with ALC partially recovered both the number of filopodia and percentage of growth cones exhibiting lamellae (Figure 5F–H). In both cases, the CSPG+ALC groups trended upward toward the control values but were not statistically different from either the control or CSPG, indicating that ALC treatment resulted in partial recovery.
Figure 5.

Treatment with acetyl-L-carnitine (ALC) partially restores growth cone dynamics and morphology on CSPGs. (A) Examples of axons expressing eYFP-β-actin and mtDsRed on CSPG and CSPG+ALC. (B) Graph showing Mito-Tip measurements on CSPG and CSPG+ALC. (C) Examples of the dynamics of growth cones on CSPG + ALC. The two growth cones shown are the same as depicted in panel A. The growth cone on CSPGs exhibits very little dynamics as reflected by minimal remodeling. In contrast, the growth cone treated with ALC is much more dynamic. Examples of lamellar protrusion (30–60 sec, yellow arrows at 60 sec) and filopodial protrusion (30–60 sec, green arrow at 60 sec) are shown. Also note that not only was protrusive activity increased at growth cones but also retraction of filopodia and lamellae (white arrows at 0 sec). (D) Graph showing de novo protrusive events at growth cones in the form of filopodia or lamellipodia on CSPG and CSPG+ALC. (E) Graph showing the percentage of filopodia existing at the beginning of the video sequence that undergo > 1 μm protrusion on CSPG and CSPG+ALC. (F) Examples of growth cones stained with rhodamine phalloidin to reveal actin filaments on the control substratum, CSPG and CSPG+ALC. (G) Graph showing the number of filopodia at growth cones on the control substratum, CSPG and CSPG+ALC. (H) Graph of the percentage of growth cones exhibiting one or more lamellae on the control substratum, CSPG and CSPG+ALC. Bars are color coded as in panel G. Samples sizes are denoted in the bars.
Decreasing the number of mitochondria targeting to growth cones impacts growth cone morphology
CSPGs result in decreased number of mitochondria in growth cones (Figure 1D). In order to individually mimic this aspect of the effects of CSPGs on mitochondria we used mDivi-1, an inhibitor of the Drp1 GTPase that mediates mitochondria fission, to decrease the targeting of mitochondria into axons. Chronic treatment with mDivi-1 suppresses the transport of mitochondria into axon from the cell body (Steketee et al., 2012; Spillane et al., 2013), presumably because many mitochondria attain lengths not permissible for fast transport and because mitochondria length is inversely proportional to their frequency of transport (Narayanareddy et al., 2014). Consideration of a dose response to chronic treatment with mDivi-1 starting at the time of culturing and analyzed at 24 hrs of culturing showed that 1 μM mDivi-1 decreased the length of axons extending from sensory explants by approximately half (not show) and decreased the number of mitochondria at growth cones by 50% relative to DMSO treatment (Figure 6A,B), similar to the the range of the effects of CSPGs (Figure 1D). mDivi-1 did not alter Mito-Tip measurements relative to DMSO controls (p=0.15), indicating that although fewer mitochondria are present at growth cones they are similarly targeted relative to the distal edge. However, consistent with the effects of mDivi-1 on mitochondria fission and in turn length, relative to the DMSO group the length of the most distal two mitochondria in axons cultured in 1 μM mDivi-1 was slightly increased (median lengths of 1.3 and 1.5 μm for DMSO and mDivi-1 respectively, p=0.03, n=160 and 170). Overnight culturing in 1 μM mDivi-1, decreased growth cone area (Figure 6C) and the number of filopodia at growth cones (Figure 6D). These observations provide proof of concept evidence that decreasing the number of mitochondria that target to distal axons impacts both the extension and morphology of growth cones. However, as with any other approach that decreases the transport of mitochondria in axons, we cannot rule out possible effects of decreased mitochondria density along the axon shaft or effects due to increased mitochondria density in cell bodies.
Figure 6.

Decreasing the number of mitochondria at the growth cone affects growth cone morphology. (A) Examples of growth cones treated with DMSO or 1 μM mDivi-1 for the entirety of the culturing period. Phase contrast with over-layered mitochondria (green) is shown. Note the simplification of growth cone morphology in the mDivi-1 treatment condition. (B) Graph of the number of mitochondria in growth cones as a function of DMSO or mDivi-1 treatment. The color scheme and sample sizes also apply to panels (B–C). (C) Graph of growth cone area as a function of DMSO or mDivi-1 treatment. (D) Graph of the number of filopodia present at growth cones as a function of DMSO or mDivi-1 treatment. Samples sizes are denoted in the bars of panel B and apply to B–D.
CSPGs elevate the levels of dynactin p150-Glued in the distal axon
Dynactin is a required component and activator of the dynein retrograde motor complex (Liu, 2017). Dynactin targets to the distal axon and promotes the evacuation of mitochondria and organelles from the distal axon during normally occurring axon extension (Moughamian and Holzbaur, 2012; Moughamian et al., 2013). In contrast, manipulation of dynactin does not greatly affect retrograde transport along the proximal axon (Moughamian and Holzbaur, 2012). The established role of dynactin in mediating the retrograde transport of organelles from the distal axon is shown schematically in Figure 7A. Importantly, dynactin-mediated initiation of retrograde transport in the distal axons involves dynactin being recruited to the actively polymerizing tips of microtubules (Figure 7A; Moughamian et al., 2013). The previously described role of dynactin in mediating the retrograde transport of organelles selectively from the distal axon is an observation similar to the effects of CSPGs on mitochondria, which are also specific to the distal axon. The levels of the required p150-Glued subunit of dynactin were elevated in the distal 20 μm of the axons of neurons cultured on CSPGs in a concentration dependent manner (Figure 7B,C), as determined by quantitative immunochemistry. Treatment with ELP during the entire 24 hr culturing period attenuated the increase in p150-Glued levels relative to treatment with the control RP peptide (Figure 7D). Inhibition of PI3K signaling with LY294002 for one hr on the control substratum increased the levels of p150-Glued by approximately 35% (Figure 7E), indicating that PI3K signaling negatively regulates the levels of p150-Glued in distal axons.
Figure 7.

Elevated levels of p150-Glued in distal axons on CSPG substrata. (A) Schematic of the mechanism that drives the initiation of retrograde transport away from distal axons. Briefly, microtubule plus tips undergoing polymerization recruit EB3 that in turn serves to scaffold a complex consisting of CLIP-170, dynactin and dynein. This complex then promotes the activation of retrograde organelle evacuation from growth cones. Thus, impairing microtubule plus tip dynamics, or the function of dynein-dynactin, prevents to initiation of retrograde transport from the distal axon. (B) Examples of p150glu dynactin staining in growth cones on the control and CSPG (10 μg/mL) substrata. (C) Quantification of the staining intensity of p150Glu staining in growth cones as a function of CSPG concentrations used to coat substrata. Comparison to the control substratum. (D) Culturing in the presence of ELP decreases the staining intensity of p150Glu in growth cones on 5 μg/mL CSPGs relative to the control treatment with RP. (E) Inhibition of PI3K activity using LY294002 (25 μM) increases the staining intensity of p150Glu in growth cones on the control substratum. (F) Suppression of microtubule plus tip dynamics using 3 nM vinblastine (VB; 60 min) increases the number of mitochondria in the distal 10 μm of axons on control substrata. (G) Treatment with vinblastine (60 min) on CSPG substrata decreased Mito-Tip. (H) Treatment with vinblastine on CSPG substrata increased the number of mitochondria in distal axons. Samples sizes are denoted in the bars.
Role of microtubule plus tip dynamics in regulating the number of mitochondria in growth cones
Microtubule dynamics in the distal axon are a required component of the dynactin mediated retrograde displacement of organelles from the distal axon (Figure 7A; Moughamian et al., 2013). As expected from the prior literature, suppression of microtubule dynamics for 1 hr using 3 nM vinblastine (as characterized in our prior work; Ketschek et al., 2016) on the control substratum resulted in increased numbers of mitochondria in distal axons (Figure 7F), but did not affect Mito-TIP measurements (not shown). Analysis of the effects of treatment with vinblastine on CSPG coated substrata revealed that a 1 hr treatment with 3 nM VB decreased Mito-TIP and increased the number of mitochondria populating the distal axons (Figure 7G,H). The effects of vinblastine are thus consistent with the microtubule dynamics and dynactin dependent mechanism for the evacuation of mitochondria from distal axons on both control and CSPG substrata.
Inhibition of the dynactin-dynein interaction increases axon lengths on CSPGs
The observation that suppressing microtubule tip dynamics increased the number of mitochondria in growth cones on CSPGs is generally consistent with the previously detailed mechanism for initiating the evacuation of mitochondria from growth cones summarized in Figure 7A. To further test this mechanism we expressed the CC1 fragment of p150Glu dynactin that results in dissociation of dynactin from dynein (Quintyne et al., 1999) and has been shown to block the initiation of organelle retrograde movements away from growth cones (Moughamian and Holzbaur, 2012). Expression of CC1 in neurons cultured on CSPGs increased median axon lengths by 42% (Figure 8A,B). However, in the same population of axons CC1 expression did not alter the lengths of axon collateral branches (p=0.27, n=52 and 19, Man-Whitney test), the proportion of axons exhibiting collateral branches (p=0.41, Fisher’s exact test) or the proportion of axons exhibiting bifurcations (p=0.85, Fisher’s exact test). Moreover, 77% of the distal axons of CC1 expressing neurons exhibited a swollen appearance (Figure 8A), likely reflective of the accumulation of organelles in distal axons as also previously observed for inhibition of dynactin function on permissive substrata (Ahmad et al., 2006), relative to 2% in the control group. In contrast, 72% of the control axons on CSPGs exhibited varicosities along the distal 50 μm (Figure 8A), as described in Figure 1.
Figure 8.
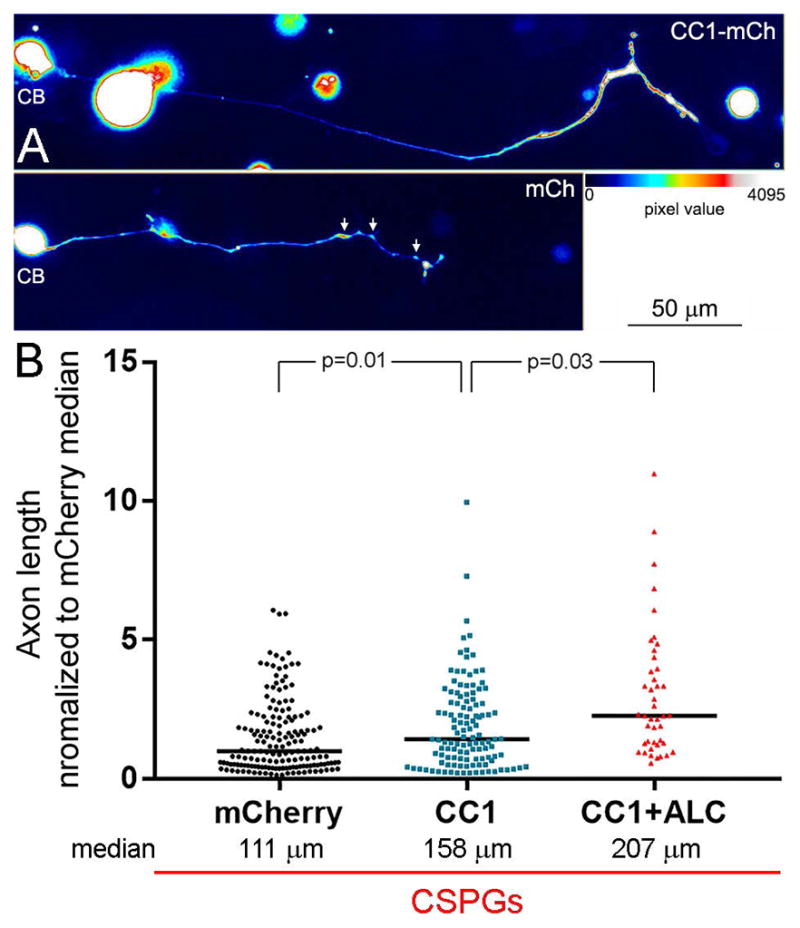
Disruption of the interaction between dynactin and dynein increases axon lengths on CSPG substrata. (A) False colored examples of neurons (CB = cell body) and their axons on CSPGs expressing CC1-mCherry (mCh) or mCh alone. The distal axon of the CC1-mCh expressing neurons exhibits distal thickening as represented by warmer colors in the false colored image (see color bar). The arrows in the mCh panel show the presence of varicosities along the distal axon, as previously detailed in Figure 1, also evidenced by warmer colors. (B) Graph of axon length measurements for mCh, CC1-mCh and CC1-mCh plus 500 μM acetyl-L-carnitine (ALC). Black lines in bars denote the medians as on CSPGs axon length measurements were not normally distributed. Samples sizes are denoted in the bars.
We previously reported that culturing neurons in the presence of 500 μM ALC did not increase axon lengths on CSPG coated substrata, although it increased axon lengths on the control substratum and 500 μM represents a saturating dose (Sainath et al., 2016). In retrospect, this lack of an effect may be due to the fact that ALC does not restore Mito-Tip (Figure 5A,B) and thus promoting mitochondrial respiration when mitochondria are not properly targeted to distal axons on CSPGs may not have an effect. We therefore sought to determine if treatment with ALC would further enhance axon lengths on CSPGs in neurons expressing CC1-mCh. Neurons expressing CC1-mCh and treated with ALC exhibited axon lengths 31% greater than those only expressing CC1-mCh (Figure 8B). However, consideration of the restoration of axon lengths relative to neurons on the control substratum (n=88) indicates that the combination of CC1+ALC treatments only restores 37% of axon length relative to control levels (Figure 9B).
Figure 9.

Working model for the contribution of CSPGs effects on growth cone mitochondria to the suppression of axon elongation. Under control conditions growth cones contain a normal content of normally respiring mitochondria resulting in sufficient ATP production to maintain steady axon elongation. CSPGs result in a decrease in the number of mitochondria targeting to growth cones and a suppression of their respiration. Inhibition of the interaction between dynein and dynactin (using CC1) increases the number of mitochondria at growth cones, but is not expected to affect their respiration, and results in partial increases in axons lengths on CSPGs (see bottom graph). When mitochondria respiration is promoted (ALC treatment) in conjunction with CC1 expression the effects of CSPGs on axon elongation further suppressed. The arrows in the graph below CC1 and CC1+ALC denote the detected increases in axon lengths relative to CSPGs. However, as CSPGs are expected to continue inhibiting pathways that promote elongation, and drive the activity of pathways that inhibit elongation, the restoration of mitochondrial parameters only has partial effects on axon elongation, as shown by the bottom graph depicting the overall effects of treatments on axon lengths.
DISCUSSION
Although CSPGs are established as important regulators of neurodevelopment and axon regeneration, the underlying cellular mechanism of action is not fully understood. This report provides evidence that CSPGs affect the targeting of mitochondria and the ER into the distal axon, the site of axon elongation. Multiple recent reports have emphasized the importance of the axonal transport and targeting of mitochondria to distal axons in axon regeneration (Cartoni et al., 2016; Han et al., 2016; Zhou et al., 2016). The current study addressing CSPGs is consistent with this emerging literature. CSPGs decreased the number of mitochondria in distal axons by approximately 50% and increases the Mito-Tip by approximately 100% relative to control. Actin is an ATPase and the turnover of actin filaments can consume up to 50% of neuronal ATP (Bernstein and Bamburg, 2003). The growth cone can thus be considered a major site of ATP utilization due to the high levels of local actin turnover. Actin filaments are a major contributor to the rate of axon extension and absolutely necessary for the maintenance of the growth cone. Thus, the mis-targeting of mitochondria to growth cones on CSPGs is likely to contribute to the observed dystrophy of the growth cone by removing mitochondria from the subcellular domain where their generation of ATP is in greatest demand. Consistent with this notion, inhibition of the mitochondrial membrane potential impairs axon extension and suppresses growth cone dynamics. Additionally, calcium is a major regulator of the growth cone cytoskeleton (Zheng and Poo, 2007), and mitochondria and the ER contribute to the regulation of cytosolic calcium (Filadi et al., 2017). The modulation of ER function and its role in calcium signaling impacts axon extension and the growth cone (Valenzuela et al., 2011; Shim et al., 2013; Raiborg et al., 2015; Rao et al., 2016; Wada et al., 2016; Watanabe et a;., 2016). CSPGs elevate calcium levels in distal axons (Snow et al., 1994). The mistargeting of these organelles by CSPGs may thus affect the growth cone through changes in calcium signaling. However, the role of calcium and calcium sources in the effects of CSPGs remains to be further elucidated.
The LAR receptor inhibits PI3K and Erk signaling and activates RhoA-ROCK (Fisher et al., 2011; Ohtake et al., 2016) and is localized to the growth cones of the chicken sensory neurons used in this study (Ketschek et al., 2012). The data indicate a sufficient and necessary role for suppression of PI3K signaling, but not ROCK or Erk, downstream of LAR in the effects of CSPGs on mitochondria positioning at growth cones. Similarly, PI3K activity regulates the accumulation of the ER at the growth cone. The function of PI3K in the regulation of organelle transport/localization in axons is not well appreciated. However, a role of PI3K in the anterograde targeting of vesicles during calcium-mediated growth cone guidance has been reported (Akiyama and Kamiguchi, 2010), and is generally consistent with the current observations. Alternatively, depolymerization of actin filaments has also been reported to result in evacuation of mitochondria from distal axons (Morris and Hollenbeck, 1993) and along the axon actin filament based mechanisms locally capture mitochondria (Chada and Hollenbeck, 2004). Thus, altered levels or organization of actin filaments in growth cones in response to the suppression of PI3K activity by CSPGs may also be contributing to the observed aberrant targeting of mitochondria.
The effects of CSPGs on axon growth are mediated through multiple receptor systems including LAR, PTPσ and Nogo receptors (reviewed in Sharma et al., 2012). We did not analyze the possible contribution of CSPG receptors beyond LAR in the current study and cannot exclude that alternative receptors may also contribute to the observed effects of CSPGs on mitochondria. However, the observation that inhibition of LAR alone suffices to restore mitochondria positioning at growth cones indicates that other receptors may have a minor, if any, role in this aspect of the effects of CSPGs on mitochondria. The current study used a mixture of CSPGs purified from embryonic chicken brains that contains a variety of CSPG species. Thus, it is possible that one or more specific species of CSPG acting through LAR may be impacting mitochondria, while other CSPGs may impact different aspects of the mechanism of axon extension through different receptors (Ohtake et al., 2016). Genetic knock out of LAR and PTPσ have partial effects on reversing the inhibition of axon growth by CSPGs (Shen et al., 2009; Fisher et al., 2011), indicating that signals through multiple receptors are responsible for the full effect of CSPG mixtures on axon growth. Alternatively, even different forms of the same CSPG (e.g., aggrecan) can have disparate effects on growth cone filopodia (Beller et al., 2013). Thus, the effects of the CSPG mixture on axonal mitochondria may be due to a specific species of CSPG in the brain derived mixture used in the current study. The role of additional CSPGs receptors and CSPG species in mediating the effects on ER positioning will need to be considered in future investigations.
In sensory neurons, dynactin in growth cones serves to promote the dynein-dependent retrograde transport of mitochondria and additional cargoes away from the growth cone (Moughamian and Holzbaur, 2012; Moughamian et al., 2013). The observation that on CSPGs there is a further enrichment of dynactin in the distal axon provides insight into the mechanism responsible for the mistargeting of mitochondria. Dynein controls ER localization in non-neuronal cells (Wang et al., 2003; Woźniak et al., 2009), but whether the ER is also subject to this dynactin based mechanism in axons is not known. The dynactin mechanism also promotes the evacuation of lysosomes and vesicles from distal axons, which were not found to be affected on CSPGs, indicating an additional level in the mechanism determining which cargoes are affected. The cargo specific regulation may occur through adaptor proteins for cargoes and motors (Maday et al., 2014), and will require further investigation. Recent work has identified Actr10 as a mitochondria specific regulator of retrograde transport (Drerup et al., 2017), not affecting lysosomes, which may be involved the specificity of the effects of CSPGs. How CSPGs elevate dynactin levels in distal axons will require further analysis but may involve regulation of the CAP-gly domain or the microtubule proteins involved in this interaction (Moughamian and Holzbaur, 2012; Moughamian et al., 2013). Similarly, how PI3K signaling may regulate the levels of p150Glu in growth cones will require further consideration. It is possible that the increase in p150Glu levels could, at least in part, arise through the regulation of the axonal translation of p150Glu, which was recently shown to occur in sensory axons (Villarin et al., 2016).
The investigations aimed at dissecting and isolating the effects of CSPGs on mitochondria (e.g., decreasing mitochondria number at growth cones and impairing their respiration) indicate that both the suppression of respiration and impairment of targeting of mitochondria can impact growth cones dynamics and axon extension. The combination of these individual effects of CSPGs on mitochondria is likely to contribute to the suppression of growth cones dynamics and extension. Promoting mitochondria respiration using ALC partially rescued protrusive dynamics at growth cones on CSPGs, but as previously reported has no effect on axon extension (Sainath et al., 2016). Under these conditions ALC did not impact the targeting of mitochondria to growth cones. Thus, the partial effect of ALC is likely attributable to the persistent absence of proper targeting of mitochondria to growth cones, even under conditions of promoted respiration.
The mechanism underlying the evacuation of mitochondria from growth cones on CSPGs appears to be similar to that mediating the normally occurring evacuation of organelles from growth cones on permissive substrata (Figure 7A; Moughamian and Holzbaur, 2012; Moughamian et al., 2013). The elevated levels of p150Glu in growth cones on CSPGs likely contribute to the increased frequency of mitochondria evacuation from growth cones on CSPGs. Consistent with this notion, expression of CC1, which impairs the interaction between dynactin and dynein, increased axon lengths on CSPGs. Moreover, when CC1 expression was combined with ALC treatment, to promote mitochondrial respiration, there was a further increase in axon lengths on CSPGs. Collectively, these data indicate that both the targeting and respiration of mitochondria must be restored on CSPGs in order to obtain increases in axon lengths (Figure 9A,B). However, on CSPGs axons length was approximately 20% of the length on the control substratum (Figure 9B). The combined expression of CC1 and ALC treatment restored axon lengths to approximately 37% of the control lengths. Thus, while relative to the baseline length on CSPGs both CC1 and CC1+ALC had pronounced effects, even with CC1+ALC axon length was only restored to 37% of control axon lengths. This partial effect is not surprising as CSPGs are expected to continue to impair other aspects of the mechanism of axon extension through suppression of growth promoting signaling pathways (e.g., PI3K and Erk) and activation of inhibitory pathways (e.g., RhoA-ROCK). It will be of interest to determine in future studies if combinatorial experimental manipulations that reactivate the suppressed pathways and inhibition of the inhibitory pathways, along with manipulation of mitochondria, will result in further enhanced axon extension.
Supplementary Material
Acknowledgments
RS and LAW contributed equally to this project and share first authorship. Supported by awards to GG (NIH NS095471 and NS078030; the Craig H Neilsen Foundation; Morton Cure Paralysis Foundation) and SL (NIH 1R01NS079432 and 1R01EY024575). We thank Dr. E. Holzbaur (University of Pennsylvania) and Dr. G. Thomas (Temple University) for the gift of CC1 plasmid, and Dr. G. Banker (Oregon Health and Science University) for the gift of the NPYss-mCherry plasmid. We also thank Dr. G. Thomas for assistance setting up the microfluidic chamber system. None of the authors declare a conflict of interest.
REFERENCED LITERATURE
- Ahmad FJ, He Y, Myers KA, Hasaka TP, Francis F, Black MM, Baas PW. Effects of dynactin disruption and dynein depletion on axonal microtubules. Traffic. 2006;7:524–37. doi: 10.1111/j.1600-0854.2006.00403.x. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kamiguchi H. Phosphatidylinositol 3-kinase facilitates microtubule-dependent membrane transport for neuronal growth cone guidance. J Biol Chem. 2010;285:41740–8. doi: 10.1074/jbc.M110.156489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beller JA, Kulengowski B, Kobraei EM, Curinga G, Calulot CM, Bahrami A, Hering TM, Snow DM. Comparison of sensory neuron growth cone and filopodial responses to structurally diverse aggrecan variants, in vitro. Exp Neurol. 247:143–57. doi: 10.1016/j.expneurol.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci. 2003;23:1–6. doi: 10.1523/JNEUROSCI.23-01-00002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Carter LM. Manipulating the glial scar: chondroitinase ABC as a therapy for spinal cord injury. Brain Res Bull. 2011;84:306–16. doi: 10.1016/j.brainresbull.2010.06.015. [DOI] [PubMed] [Google Scholar]
- Buckman JF, Hernandez H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, He Z. The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron. 2016;92:1294–1307. doi: 10.1016/j.neuron.2016.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–6. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Drerup CM, Herbert AL, Monk KR, Nechiporuk AV. Regulation of mitochondria-dynactin interaction and mitochondrial retrograde transport in axons. Elife. 2017:e22234. doi: 10.7554/eLife.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Meskini R, Jin L, Marx R, Bruzzaniti A, Lee J, Emeson R, Mains R. A signal sequence is sufficient for green fluorescent protein to be routed to regulated secretory granules. Endocrinology. 2001;142:864–73. doi: 10.1210/endo.142.2.7929. [DOI] [PubMed] [Google Scholar]
- Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium. 2017;62:1–15. doi: 10.1016/j.ceca.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Fisher D, Xing B, Dill J, Li H, Hoang HH, Zhao Z, Yang XL, Bachoo R, Cannon S, Longo FM, Sheng M, Silver J, Li S. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J Neurosci. 2011;31:14051–66. doi: 10.1523/JNEUROSCI.1737-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M. Mitochondria Localize to Injured Axons to Support Regeneration. Neuron. 2016;92:1308–1323. doi: 10.1016/j.neuron.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, Collura KM, Ketschek A, Noma K, Ferguson TA, Jin Y, Gallo G, Thomas GM. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc Natl Acad Sci. 2016;113(3):763–8. doi: 10.1073/pnas.1514123113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Brady-Kalnay SM, Bellamkonda RV. Modulation of Rho GTPase activity alleviates chondroitin sulfate proteoglycan-dependent inhibition of neurite extension. J Neurosci Res. 2004;77:299–307. doi: 10.1002/jnr.20161. [DOI] [PubMed] [Google Scholar]
- Ketschek A, Gallo G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J Neurosci. 2010;30:12185–97. doi: 10.1523/JNEUROSCI.1740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek AR, Haas C, Gallo G, Fischer I. The roles of neuronal and glial precursors in overcoming chondroitin sulfate proteoglycan inhibition. Exp Neurol. 2012;235:627–37. doi: 10.1016/j.expneurol.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek A, Spillane M, Dun XP, Hardy H, Chilton J, Gallo G. Drebrin coordinates the actin and microtubule cytoskeleton during the initiation of axon collateral branches. Dev Neurobiol. 2016;76:1092–110. doi: 10.1002/dneu.22377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. 2015;518:404–8. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelkes PI, Unsworth BR, Saporta S, Cameron DF, Gallo G. Culture of neuroendocrine and neuronal cells for tissue engineering. In: Vunjak-Novakovic G, Freshney RI, editors. Culture of Cells for Tissue engineering. Chapter 14. Wiley; New York, NY: 2006. [Google Scholar]
- Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. 2015;518:404–8. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ. Regulation of dynein-dynactin-driven vesicular transport. Traffic. 2017;18:336–347. doi: 10.1111/tra.12475. [DOI] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci. 2033;22:319–30. doi: 10.1016/s1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–27. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Moughamian AJ, Holzbaur EL. Dynactin is required for transport initiation from the distal axon. Neuron. 2012;74:331–43. doi: 10.1016/j.neuron.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moughamian AJ, Osborn GE, Lazarus JE, Maday S, Holzbaur EL. Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J Neurosci. 2013;33:13190–203. doi: 10.1523/JNEUROSCI.0935-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanareddy BR, Vartiainen S, Hariri N, O’Dowd DK, Gross SP. A biophysical analysis of mitochondrial movement: differences between transport in neuronal cell bodies versus processes. Traffic. 2014;15:762–71. doi: 10.1111/tra.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirschl JJ, Magiera MM, Lazarus JE, Janke C, Holzbaur EL. α-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 2016;14:2637–52. doi: 10.1016/j.celrep.2016.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Li S. Molecular mechanisms of scar-sourced axon growth inhibitors. Brain Res. 2015;1619:22–35. doi: 10.1016/j.brainres.2014.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Wong D, Abdul-Muneer PM, Selzer ME, Li S. Two PTP receptors mediate CSPG inhibition by convergent and divergent signaling pathways in neurons. Sci Rep. 2016;6:37152. doi: 10.1038/srep37152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofrj M, Ciccocioppo F, Varanese S, di Muzio A, Calvani M, Chiechio S, Osio M, Thomas A. Acetyl-L-carnitine: from a biological curiosity to a drug for the peripheral nervous system and beyond. Expert Review of Neurotherapeutics. 2013;13:925–936. doi: 10.1586/14737175.2013.814930. [DOI] [PubMed] [Google Scholar]
- Pacelli C, Giguère N, Bourque MJ, Lévesque M, Slack RS, Trudeau LÉ. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr Biol. 2015;25:2349–60. doi: 10.1016/j.cub.2015.07.050. [DOI] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1(4):2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Pendergrass W, Wolf N, Poot M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A. 2004;61:162–169. doi: 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- Quintyne NJ, Gill SR, Eckley DM, Crego CL, Compton DA, Schroer TA. Dynactin is required for microtubule anchoring at centrosomes. J Cell Biol. 1999;147:321–34. doi: 10.1083/jcb.147.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, Johansen T, Stenmark H. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature. 2015;520:234–8. doi: 10.1038/nature14359. [DOI] [PubMed] [Google Scholar]
- Rao K, Stone MC, Weiner AT, Gheres KW, Zhou C, Deitcher DL, Levitan ES, Rolls MM. Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol Biol Cell. 2016;27:3245–3256. doi: 10.1091/mbc.E16-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath R, Gallo G. The dynein inhibitor Ciliobrevin D inhibits the bidirectional transport of organelles along sensory axons and impairs NGF-mediated regulation of growth cones and axon branches. Dev Neurobiol. 2015;75:757–77. doi: 10.1002/dneu.22246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath R, Ketschek A, Grandi L, Gallo G. CSPGs inhibit axon branching by impairing mitochondria-dependent regulation of actin dynamics and axonal translation. Dev Neurobiol. 2017;77:454–473. doi: 10.1002/dneu.22420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Selzer ME, Li S. Scar-mediated inhibition and CSPG receptors in the CNS. Exp Neurol. 2012;237:370–8. doi: 10.1016/j.expneurol.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–6. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–98. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Zheng JQ, Ming GL. A critical role for STIM1 in filopodial calcium entry and axon guidance. Mol Brain. 2013;6:51. doi: 10.1186/1756-6606-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L, Michael JV, Goldfinger LE, Gallo G. Activation of PI3K and R-Ras signaling promotes the extension of sensory axons on inhibitory chondroitin sulfate proteoglycans. Dev Neurobiol. 2014;74:918–33. doi: 10.1002/dneu.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DJ, Silver J. Contributions of chondroitin sulfate proteoglycans to neurodevelopment, injury, and cancer. Curr Opin Neurobiol. 2014;27:171–178. doi: 10.1016/j.conb.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow DM, Atkinson PB, Hassinger TD, Letourneau PC, Kater SB. Chondroitin sulfate proteoglycan elevates cytoplasmic calcium in DRG neurons. Dev Biol. 1994;166:87–100. doi: 10.1006/dbio.1994.1298. [DOI] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013;5:1564–75. doi: 10.1016/j.celrep.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee MB, Moysidis SN, Weinstein JE, Kreymerman A, Silva JP, Iqbal S, Goldberg JL. Mitochondrial dynamics regulate growth cone motility, guidance, and neurite growth rate in perinatal retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci. 2012;53:7402–11. doi: 10.1167/iovs.12-10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24:6531–9. doi: 10.1523/JNEUROSCI.0994-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela JI, Jaureguiberry-Bravo M, Couve A. Neuronal protein trafficking: emerging consequences of endoplasmic reticulum dynamics. Mol Cell Neurosci. 2011;48:269–77. doi: 10.1016/j.mcn.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008;28:8306–15. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarin JM, McCurdy EP, Martínez JC, Hengst U. Local synthesis of dynein cofactors matches retrograde transport to acutely changing demands. Nat Commun. 2016;7:13865. doi: 10.1038/ncomms13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada F, Nakata A, Tatsu Y, Ooashi N, Fukuda T, Nabetani T, Kamiguchi H. Myosin Va and Endoplasmic Reticulum Calcium Channel Complex Regulates Membrane Export during Axon Guidance. Cell Rep. 2016;15:1329–44. doi: 10.1016/j.celrep.2016.04.021. [DOI] [PubMed] [Google Scholar]
- Wang S, Romano FB, Field CM, Mitchison TJ, Rapoport TA. Multiple mechanisms determine ER network morphology during the cell cycle in Xenopus egg extracts. J Cell Biol. 2013;3:801–14. doi: 10.1083/jcb.201308001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Bizen N, Sato N, Takebayashi H. Endoplasmic Reticulum-Localized Transmembrane Protein Dpy19L1 Is Required for Neurite Outgrowth. PLoS One. 2016;11:e0167985. doi: 10.1371/journal.pone.0167985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkle CC, Taylor KL, Dent EW, Gallo G, Greif KF, Gupton SL. Beyond the cytoskeleton: The emerging role of organelles and membrane remodeling in the regulation of axon collateral branches. Dev Neurobiol. 2016;76:1293–1307. doi: 10.1002/dneu.22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woźniak MJ, Bola B, Brownhill K, Yang YC, Levakova V, Allan VJ. Role of kinesin-1 and cytoplasmic dynein in endoplasmic reticulum movement in VERO cells. J Cell Sci. 2009;122:1979–89. doi: 10.1242/jcs.041962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Poo MM. Calcium signaling in neuronal motility. Annu Rev Cell Dev Biol. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol. 2016;214:103–19. doi: 10.1083/jcb.201605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.