

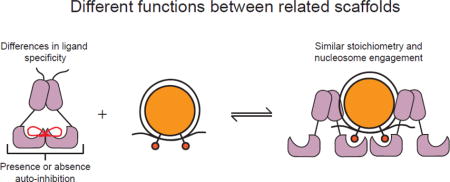
Abstract
Heterochromatin protein 1 (HP1) family proteins are conserved chromatin binding proteins involved in gene silencing, chromosome packaging, and chromosome segregation. These proteins recognize histone H3 lysine 9 methylated tails via their chromodomain (CD) and recruit additional ligand proteins with diverse activities through their dimerization domain, the chromoshadow domain (CSD). Species that have HP1 proteins possess multiple paralogs that perform non-overlapping roles in vivo. How different HP1 proteins, which are highly conserved, perform different functions is not well understood. Here, we use the two Schizosaccharomyces pombe HP1 paralogs, Swi6 and Chp2, as model systems to compare and contrast their biophysical properties. We find that Swi6 and Chp2 have similar dimerization and oligomerization equilibria, and that Swi6 binds slightly (~3-fold) more strongly to nucleosomes than Chp2. Further, while Swi6 binding to the H3K9me3 mark is regulated by a previously described auto-inhibition mechanism, the binding of Chp2 to the H3K9me3 mark is not analogously regulated. In the context of CSD interactions, we show using a newly identified peptide sequence from the Clr3 histone deacetylase and a previously identified sequence from the protein Shugoshin that the Swi6 CSD binds both ligands more strongly than the Chp2. Overall, our findings uncover quantitative differences in how Swi6 and Chp2 interact with nucleosomal and non-nucleosomal ligands and qualitative differences in how their assembly on nucleosomes is regulated. These findings provide a biochemical framework to explain the varied functions of Chp2 and Swi6 in vivo.
Keywords: Schizosaccharomyces pombe, heterochromatin, nuclear magnetic resonance (NMR), analytical ultracentrifugation
Graphical abstract
Introduction
Large regions of eukaryotic genomes are organized into heterochromatin, which is generally inaccessible to RNA polymerase and, therefore, is transcriptionally silenced. In addition to gene silencing, heterochromatin is also important for proper centromere function, repression of recombination, sister chromatid cohesion, and telomere stability1–4. One specific type of heterochromatin is dependent on di- and tri-methylation of histone H3 at lysine 9 (H3K9me2/3), and this type of heterochromatin is conserved from fission yeast to humans5.
In the fission yeast Schizosaccharomyces pombe, heterochromatin is assembled on pericentromeric repeats, subtelomeric regions, the silent mating type locus, and rDNA1. Establishment and maintenance of this silencing is dependent on several proteins including the H3K9 methyltransferase Clr4, histone deacetylases such as Clr3, and the heterochromatin protein 1 family proteins Swi6 and Chp26–8. HP1 proteins contain two globular domains: an N-terminal chromodomain (CD) that specifically recognizes H3K9me2/3 via an aromatic cage and a C-terminal chromoshadow domain (CSD) that dimerizes and recognizes various binding partners. These structured domains are connected by an unstructured, charged hinge region (H) that nonspecifically binds DNA and RNA8–11.
Organisms that possess HP1 proteins generally have multiple paralogs that perform non-overlapping functions in vivo. For example, humans have three HP1 paralogs: HP1α, HP1β, and HP1γ. HP1α and HP1β localize to heterochromatic loci and are involved in gene silencing. In contrast, HP1γ localizes to euchromatic regions and has roles in transcriptional elongation and RNA processing12–15. This supports the idea that these proteins have evolved to be general chromatin regulators involved in multiple nuclear processes. In S. pombe, both Swi6 and Chp2 are involved in the regulation of silent, H3K9me2/3-dependent heterochromatin, but they appear to have different roles; neither protein is able to rescue the loss of silencing phenotype in a deletion strain of the other8. In addition, establishing de novo heterochromatin by artificially recruiting the methyltransferase Clr4 requires Chp2 but not Swi616. Some of the differences in the biological roles of Swi6 and Chp2 could arise from differences in their intrinsic biophysical properties. Previous biochemical work identified an auto-inhibition based mechanism that regulates the binding of Swi6 to the H3K9me mark. This auto-inhibition arises because a loop with the sequence ARK in one chromodomain binds to the aromatic cage of the chromodomain in the other monomer. It is not known if binding of Chp2 to the H3K9me mark is similarly regulated. Swi6 and Chp2 are also known to have different binding partners, and it is possible that some of the biological differences can be explained by different binding preferences. Published IP-MS data showed Swi6 associating with numerous nuclear proteins, whereas Chp2 primarily pulled down components of the Snf2/HDAC-containing repressor complex (SHREC)7,8,17. By in vitro pulldown assays with full-length proteins, it was shown that both Swi6 and Chp2 physically associate with the putative demethylase Epe1 and the histone deacetylase Clr3, albeit with different affinities8. Additionally, Swi6, but not Chp2, has been shown to bind the meiosis-specific protein Sgo1, and the interacting region was narrowed down by yeast two-hybrid18.
To directly test if Swi6 and Chp2 display differences in ligand specificity we compared the ability of their CSD domains to bind specific sequences from the Sgo1 and Clr3 proteins by NMR spectroscopy. Additionally, while much is known about the biochemical and biophysical properties of Swi6, little is known about the biophysical properties of the Chp2 paralog. We therefore also compared the nucleosome binding and oligomerization properties of Chp2 to those of Swi6. Together, these results indicate substantial differences in ligand specificity between Swi6 and Chp2 and suggest that Chp2 binding to H3K9me nucleosomes is not subject to Swi6-like auto-regulation. These intrinsic biophysical differences provide starting points to explain the different biological roles of Chp2 and Swi6.
Results
The chromoshadow domains of Swi6 and Chp2 show specificity differences for non-nucleosomal ligands
HP1 proteins have been proposed to act as platform molecules that recruit diverse activities to heterochromatic regions1,17,19–22. The best-characterized HP1 domain that is known to recruit non-nucleosomal ligands is the CSD. Previously, the CSD domain of D. melanogaster HP1a was found to bind a “PXVXL” consensus motif, and this motif was found in a number of known HP1 interacting partners23. To date, the binding motifs between different HP1 paralogs have not been compared. The center position valine was originally proposed to be strictly required, but a recent study has proposed relaxing this motif to ΦX(V/P)XΦ based on binding of the human HP1 paralogs to a peptide from histone H3, where Φ denotes α-amino acids without side chain nitrogen or oxygen atoms24. In S. pombe, one of the known binding partners of Swi6 is the Shugoshin 1 protein (Sgo1), and yeast two-hybrid experiments narrowed this interaction to the CSD of Swi6 and a peptide in Sgo1 resembling the ΦX(V/P)XΦ motif, VCVCI. Mutations in either the C-terminal extension (CTE) of the CSD of Swi6 (F324, Figure 1A, Supplemental Figure 1A) or the VCVCI motif abrogated this interaction18. To investigate whether additional sequences beyond those described by the ΦX(V/P)XΦ motif could be accommodated within the CSD-CSD interface, we performed phage display with the CSDs of both Swi6 and Chp2.
Figure 1.
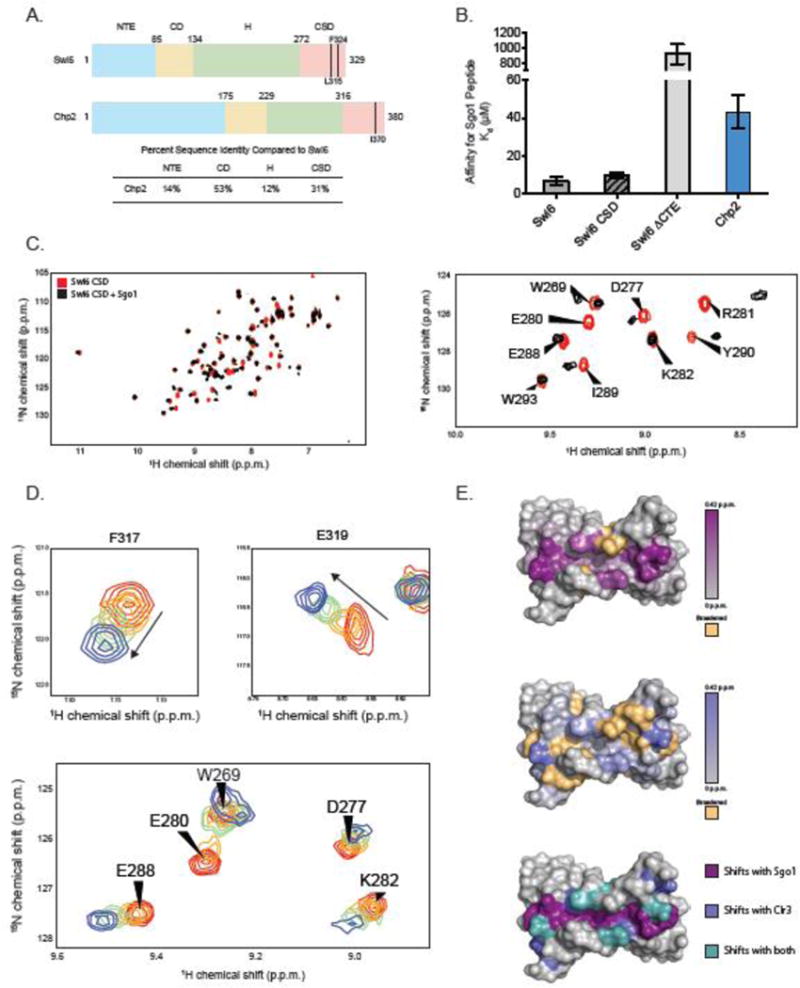
(A) Domain schematic of Swi6 and Chp2 showing residue numbers and percent identity between the two proteins. Mutants used in these studies are indicated. Sequence identities were calculated using EMBOSS Needle. (B) Dissociation constants (Kd) for the Sgo1 peptide measured by fluorescence anisotropy. Experiments were carried out in triplicate at room temperature. (C) (Left) A superimposition of 1H-15N HSQC spectra of Swi6 CSD with (black) and without (red) Sgo1 peptide. (Right) A zoom of a subset of chemical shift perturbations. (D) 1H-15N HSQC spectra overlay of residues F317 and E319 while being titrated by the Clr3 peptide. Concentrations of peptide shown are 0 μM (red), 20 μM (orange), 80 μM (green), 500 μM (blue). (E) The Swi6 CSD crystal structure colored by chemical shift perturbation upon the addition of 2X molar ratio Sgo1 peptide (Top) and 5X molar ratio Clr3 peptide (Middle). Orange indicates resonances that were broadened beyond detection upon peptide addition (PDB 1E0B). (Bottom) The Swi6 CSD crystal structure colored by shifts observed with Sgo1 peptide (purple), Clr3 peptide (blue), and with both peptides (teal).
We used a randomized nonapeptide library presented on the pVIII capsid protein of the filamentous phage M13 to compare recognized motifs between Swi6 and Chp2. We used Multiple EM for Motif Elicitation (MEME) analysis to discover sequence motifs in the identified peptides25. Analysis of these motifs did not strongly indicate specific residues at any position. Rather, the results suggested a degenerate motif skewed toward hydrophobic and aliphatic residues (Supplemental Figure 2A and 2B). We then searched the S. pombe proteome for proteins containing this degenerate motif, specifically focusing on known physical and genetic interactors with Swi6 and Chp2. We tested binding of 24 peptides from selected proteins by fluorescence anisotropy. While the majority of the peptides did not show binding at the highest Swi6 and Chp2 concentrations that we tested (900 μM), we identified one binding partner for Swi6: a peptide from the type II histone deacetylase, Clr3. This peptide is contained in the C-terminal region of Clr3, which appears unstructured in a recent crystal structure of Clr3 (PDB ID 5IKK)26. The sequence of this peptide contains “LLHLL”, which is distinct from the PXVXL motif and conforms to the ΦX(V/P)XΦ more loosely than the Sgo1 peptide. We then compared affinities of Swi6 and Chp2 for the Sgo1 and Clr3 derived peptides.
To date, there is no evidence that Chp2 directly interacts with Sgo1. To assess the binding of Swi6 and Chp2 to this region of Sgo1, we measured binding to a peptide containing the “VCVCI” sequence in Sgo1 using fluorescence anisotropy (Figure 1B). Swi6 bound to this Sgo1 peptide with a Kd of 6.6 μM ± 2.1. The Swi6 CSD alone bound the peptide with the same Kd within error (9.8 ± 1.4), indicating that the major interaction of this peptide is with the CSD. Consistent with previous studies, saturable binding was not observed with a Swi6 CTE deletion even up to a concentration of 200 μM18. Additionally, the V to E mutation previously tested by yeast two-hybrid greatly reduced Swi6 binding. In comparison, Chp2 bound 8.8-fold weaker to the Sgo1 peptide with a Kd of 58.5 μM.
To examine the structural basis for this interaction, we applied NMR spectroscopy. NMR is a powerful tool for binding studies because of its sensitivity in monitoring changes in the chemical environment of amide bonds corresponding to the protein backbone and sidechains upon ligand binding. This allows us to examine even weak protein-protein interactions at high resolution and evaluate structural, thermodynamic, and kinetic aspects of the binding reaction. To this end, we first determined backbone assignments for the Swi6 CSD using standard triple resonance methods and the NMRFAM PINE Server27.
Following the backbone assignment of the CSD, we performed 1H-15N HSQC (Heteronuclear Single Quantum Correlation) NMR experiments with Swi6 and Chp2 CSDs in the presence of unlabeled Sgo1 peptide. Binding of the Sgo1 peptide showed numerous chemical shift perturbations (CSP) in the Swi6 CSD (Figure 1C, Figure 1E, Supplemental Figure 1A, Supplemental Figure 2C). Since the crystal structure of Swi6 CSD is available (PDB ID 1E0B)10, we were able to map the CSPs on the CSD structure and observe that they localize to the dimer interface, regions flanking this interface, and to residues in the CTE. The observed localization in chemical shift changes along a cleft formed by the CSD dimer is consistent with where interacting peptides have been shown to bind in other HP1 proteins28–30. Importantly, as previously observed by yeast two-hybrid, mutating the peptide motif to “VCECI” did not show any resonance changes by NMR (Supplemental Figure 2E). We next examined binding of the Sgo1 peptide to the Chp2 CSD. In contrast to the shifts observed with the Swi6 CSD, resonances were predominantly broadened with the addition of peptide to the Chp2 CSD (Supplemental Figure 3A). We observed some resonances that broaden below detection at low peptide concentration and reappear at a different chemical shift when a large excess of peptide was added. This precluded quantification of the Kd. The increased concentrations required for saturation compared to Swi6 suggest a weaker binding of the Sgo1 peptide to Chp2, which is consistent with our fluorescent anisotropy binding results (Figure 1B).
We next analyzed the Clr3 peptide derived from our phage display experiments. Both Swi6 and Chp2 were previously shown to physically interact with Clr3 by IP studies8,31, but the region of Clr3 that interacts with Swi6 or Chp2 was not identified. To characterize the interaction of the Clr3 peptide with Swi6 and Chp2, we again turned to 1H-15N HSQC NMR. Similar to binding of the Sgo1 peptide, binding of this Clr3 peptide to Swi6 caused chemical shift perturbations in residues mapping to the dimer interface, the regions flanking this interface, and the CTE (Figure 1D and 1E, Supplemental Figure 2C). Similar to the Sgo1 peptide, these residues overlay with the region known to interact with binding partners. These results directly demonstrate the specific region of Clr3 that physically interacts with Swi6. A Kd for this interaction was obtained by performing a series of titrations with increasing concentrations of Clr3 peptide. The observed chemical shift changes were plotted as a function of Clr3 concentration and fitted to a hyperbolic function to determine a Kd of 68 ± 15 μM, roughly 10.2-fold weaker than the affinity for the Sgo1 peptide (Figure 1D, Supplemental Figure 2D). This comparison suggests that the binding motifs recognized by HP1 proteins can be degenerate and that these sequence differences can lead to a broad range of binding affinities. Indeed, several CSD residues in this binding region are uniquely perturbed by either the Sgo1 or Clr3 peptide, suggesting different regions of the CSD are used for binding and this is dependent on ligand sequence (Figure 1E).
We next looked at the binding of the Clr3 peptide to the CSD of Chp2 and performed titration experiments with increasing concentration of peptide. Similar to the addition of the Sgo1 peptide, resonances broadened rather than shifted, indicating intermediate exchange. Although the Kd of this interaction cannot be reliably determined because of the major broadening, we were able to estimate the Kd by plotting the chemical shifts changes of two resonances as a function of Clr3 concentration (Supplemental Figure 3D). The preliminary fit to a hyperbolic function indicates a Kd in the high μM–low mM range. We also observed that binding saturation was achieved at higher concentrations of Clr3 peptide with the Chp2 CSD as compared to the Swi6 CSD. These observations indicate that the Chp2 binding to the Clr3 peptide is weaker than that of Swi6. Chp2 was previously suggested to have a higher affinity for full length Clr3 compared to Swi6 by IP8. It is possible that the weaker affinity of the peptide does not recapitulate binding of the full-length proteins, and that full-length Chp2 makes additional interactions with Clr3.
Together the results above suggest that differences in specificities of the Chp2 and Swi6 CSDs may contribute to their different biological roles.
Chp2 binding to the H3K9me3 mark is not regulated by auto-inhibition
In addition to differences in binding non-nucleosomal ligands, differences in interactions with chromatin could further inform on the varied roles of Chp2 and Swi6 in vivo. Previously, it was shown that the CD alone of both Swi6 and Chp2 binds the H3K9me3 tail peptide with affinities in the low μM range, although Chp2 bound the peptide tighter8. However, a detailed comparison of the interactions of full-length Swi6 and Chp2 with both tail peptide and nucleosomes has not been performed. Comparisons with full-length proteins are critical, especially considering mechanisms such as the observed Swi6 auto-inhibited state that is not present in the CD alone. The full-length Swi6 dimer has been shown to exist in both an open, binding competent state and a closed, auto-inhibited state32. This closed conformation is stabilized ~10-fold over the open state by an ARK loop in one chromodomain binding to the aromatic cage of the chromodomain in the other monomer. As the H3K9me3 peptide also binds in a similar location on the CD, binding of the ARK loop is mutually exclusive with binding to the H3 tail.
We wanted to determine if the Chp2 dimer also adopts an auto-inhibited, closed state. The chromodomains of Swi6 and Chp2 show a high degree of homology, with a sequence identity of 53% and a sequence similarity of 66.7%. Importantly, one of the sites of divergence in sequence lies within the ARK loop of Swi6; Chp2 instead has a KKD sequence (Supplemental Figure 1A). It is possible that the presence of this acidic residue prevents the domain swapping behavior of the Swi6 chromodomain. For Swi6, the presence of auto-inhibition in the Swi6 dimer was inferred, in part, by the finding that disrupting dimerization via the CSD-CSD interface resulted in increased binding for an H3K9me peptide32. This is because disrupting the CSD-CSD interface results predominantly in a monomer thereby disrupting the ARK loop-CD interaction. To test if Chp2 displays analogous auto-inhibition, we compared the affinity of WT and Chp2 I370E, a mutant that disrupts CSD dimerization, for H3K9me peptides. The CSD mutant resulted in Chp2 being predominantly a monomer up to 70 μM (Data not shown), yet this mutant bound the H3K9me peptide with comparable affinity as the WT dimeric protein (Supplemental Figure 6A), suggesting the absence of a Swi6 like auto-inhibitory mechanism in the WT Chp2 dimer.
In the context of Swi6, another way that auto-inhibition was previously disabled was via mutating the ARK loop32. This mutation increased the affinity of Swi6 for the H3K9me3 tail peptides but decreased the affinity for H3Kc9me3 nucleosomes32. This is because the ARK loop plays two roles: an auto-inhibitory role by directly competing with the H3K9me3 mark for binding the CD, and a stabilizing role via interactions with the nucleosome once it is displaced from the CD by the H3K9me3 mark32. Chp2 binds the H3K9me3 tail peptide with a Kd of 1.85 μM, ~5.5-fold tighter than Swi6 (Figure 2A). The Swi6 ARK loop mutant binds this peptide with a similar affinity to that of wild-type Chp2, consistent with the idea that Chp2 does not adopt an auto-inhibitory conformation32. Despite the differences in affinity for the H3K9me3 tail peptide, both Swi6 and Chp2 show substantial specificity for the H3K9me3 mark as they both bind the unmethylated H3 tail peptide at least 10-fold more weakly than the H3K9me3 tail peptide (Figure 2A).
Figure 2.
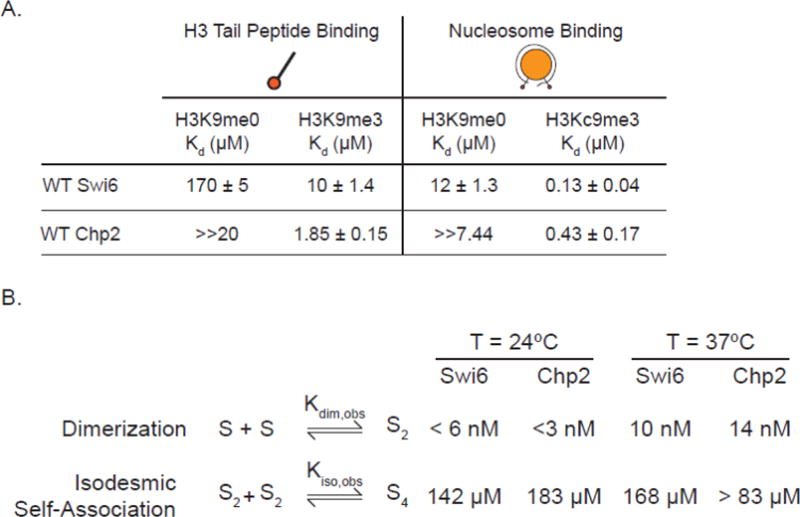
(A) Characterization of H3 tail peptide binding and nucleosome binding with full-length Swi6 and Chp2 by fluorescence anisotropy. The 18 amino acid H3 tail peptide was N-terminally fluorescein labeled and had bona fide methylation at the K9 position. The nucleosomes were labeled on the 5′ end of the 601 DNA. H3Kc9me3 nucleosomes were made by methyl lysine analog chemistry. All experiments were carried out in triplicate at room temperature. (B) Characterization of the self-association properties of WT Chp2. All experiments were performed at 24°C. Swi6 oligomerization values were previously reported in Canzio et al. 2013.
We next asked how the specificity for the H3K9 methyl mark and affinity are affected in the context of a nucleosome. To measure binding to nucleosomes we used a previously developed fluorescence anisotropy based assay (Supplemental Figure 4)32,33. Similar to previous observations, we found that Swi6 binds H3Kc9me3 nucleosomes more strongly (>90-fold) than unmethylated nucleosomes (Figure 2A). We further found that Chp2 also binds H3Kc9me3 nucleosomes more strongly than unmethylated nucleosomes (Figure 2A, >17-fold). These results indicate that like Swi6, Chp2 also displays substantial specificity for the H3K9 methyl mark in the context of nucleosomes. In terms of absolute affinity, Chp2 binds H3Kc9me3 nucleosomes ~3-fold more weakly than Swi6 (Kd= 430 nM vs. 130 nM respectively). This weaker binding is comparable to the affinity of the Swi6 ARK loop mutant for H3Kc9me3 nucleosomes, further consistent with the conclusion that Chp2 lacks a Swi6-like auto-inhibition mechanism32.
In previous work, negative stain electron microscopy (EM) of CFP-tagged Swi6 showed 2D class averages consistent with both a closed, auto-inhibited conformation and an open, spreading-competent conformation32. To see if different conformation states of the Chp2 dimer could be observed, we visualized Chp2 by negative stain EM. 1,806 particles were manually picked and 2D class averages were calculated using RELION. The 2D class averages obtained are consistent a single predominant state that appears elongated (Supplemental figure 6). However, given the low resolution of negative stain EM, we cannot rule out the possibility that alternate conformations exist that we cannot detect. Our attempts to obtain corresponding EM data with untagged Swi6 were inconclusive due to the smaller size of Swi6 dimers (85.9 vs. 74.4 KDa for Chp2 vs. Swi6 dimers respectively).
Together, the tail peptide and nucleosome binding results presented here are consistent with the lack of a Swi6-like auto-inhibition mechanism in the context of Chp2.
The dimerization interfaces of Swi6 and Chp2 are important for nucleosome interactions
The affinity of both Swi6 and Chp2 for an H3Kc9me3 nucleosome is 0.13 μM and 0.43 μM, respectively (Figure 2A). Therefore, Swi6 binds H3Kc9me3 nucleosomes ~77-fold tighter than H3K9me3 tail peptide alone, and Chp2 binds H3Kc9me3 nucleosomes at least 4-fold tighter than for the H3K9me3 tail peptide alone. This suggests that regions of the protein other than the CD are also important for nucleosome binding. The hinge region of HP1 proteins has been shown to nonspecifically bind both DNA and RNA32,34, suggesting that this region of Swi6 and Chp2 could bind the nucleosomal DNA. It is possible that other domains recognize the nucleosome as well. As mentioned in the first section, the CSD dimer can interact with other proteins through a cleft formed between the two monomers. To test whether an intact CSD dimer is important for nucleosome binding by Swi6 and Chp2, we measured nucleosome binding using two mutants, Swi6 L315D and Chp2 I370E, which substantially disrupt dimerization8,32,33. Swi6 L315D and Chp2 I370E displayed a 8.5-fold and 3-fold weaker Kd for mononucleosomes, respectively (Figure 3B). These data suggest that an intact CSD-CSD interface is important for recognizing the nucleosome.
Figure 3.
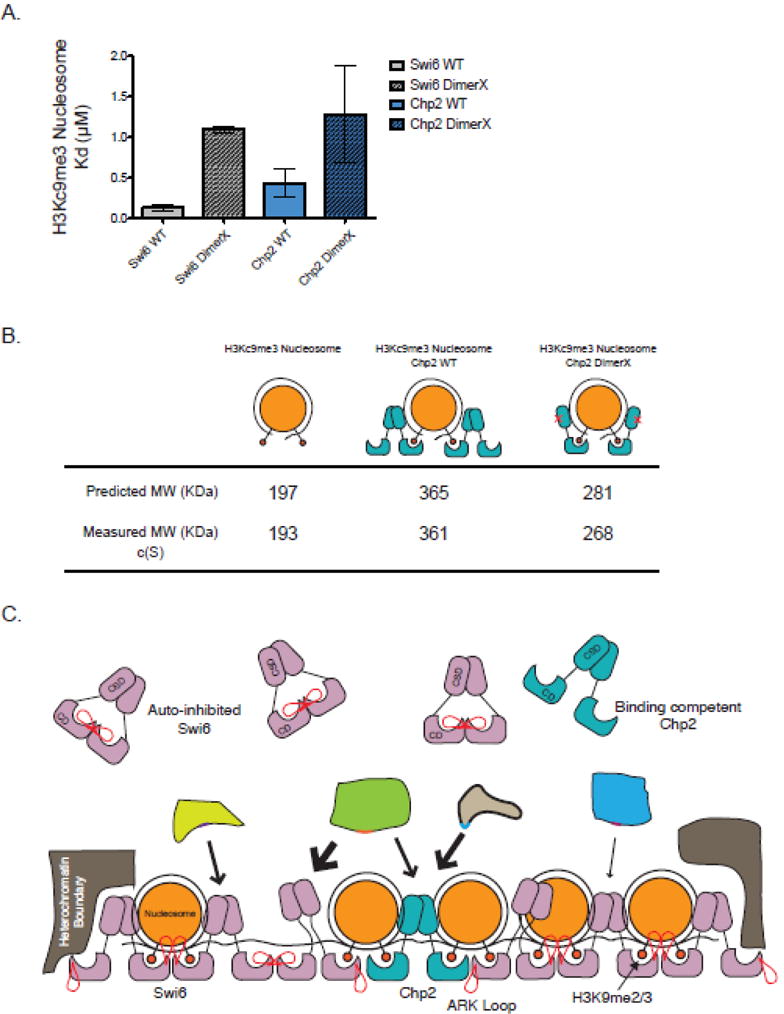
(A) Characterization of H3Kc9me3 core nucleosome binding with WT Swi6 and Chp2 as well as Swi6 and Chp2 dimerization mutants measured by fluorescence anisotropy. All experiments were carried out in triplicate at room temperature. (B) Average calculated masses and theoretical masses for H3Kc9me3 nucleosomes alone, with WT Chp2, and with Chp2 I370E as determined by SV-AUC using a continuous function c(s) and an f/f0 value of 1.6. (C) Model depicts differences in roles between the HP1 paralogs Swi6 (rose) and Chp2 (aqua). Paralogs recruit different effectors to chromatin due to differences in chromoshadow peptide specificity and affinity. An excess of unbound Swi6 is regulated as an auto-inhibited conformation, whereas this does not exist in the lower abundant Chp2.
The dimerization and oligomerization equilibria for Chp2 and Swi6 are similar
A core property of heterochromatin is its ability to spread. It has been suggested that the spreading of heterochromatin arises in part due to the oligomerization of HP1 proteins1,17,19,22,31,33. Previous work using analytical ultracentrifugation (AUC) has shown that Swi6 forms tight dimers (Kdim < 6 nM at 24°C) and weak higher order oligomers (Kiso= 151 μM at 24°C)32,33. Similar measurements have not been carried out with Chp2 although previous qualitative comparisons using elution over a size-exclusion column have suggested that Chp2 is a weaker dimer than Swi68.
To quantify the self-association of Chp2, we used sedimentation velocity analytical ultracentrifugation (SV-AUC). It is well known that the dimerization of HP1 proteins is mediated through the CSD, whereas it was previously shown that the tetramerization interface of Swi6 is through the N-terminal chromodomain33. Similar to Swi6, Chp2 exhibits a tight association of two monomers with a dissociation constant Kdim of <3 nM at 24°C. Further isodesmic association of Chp2 dimers has a dissociation constant Kiso of 183 μM at 24°C (Figure 2A). A comparison at 37 °C showed similar values for Kiso for Swi6 and Chp2 as well as a comparable Kdim value for Chp2 (Figure 2B, Supplemental figure 5). Thus overall, the dimerization and oligomerization equilibrium constants for Chp2 are comparable to those for Swi6.
Previously, it was shown by size exclusion chromatography that Chp2 elutes as two peaks with the slower peak corresponding to a monomer8. Given that we find that the equilibrium constants for dimerization are similar for Chp2 and Swi6, one possibility is that the on and off rates of Chp2 monomers differ from those of Swi6, resulting in Chp2 eluting as multiple peaks by SEC.
Chp2 binds nucleosomes with a stoichiometry of 4:1 similar to Swi6
In addition to the intrinsic ability of Swi6 to oligomerize, its stoichiometry on nucleosomes has suggested models for how Swi6 molecules can spread across chromatin. Specifically, previous work using sedimentation velocity analytical ultracentrifugation (SV-AUC) has showed that two Swi6 dimers bind a single nucleosome, leading to a “sticky end” model in which Swi6 is able to bridge adjacent nucleosomes33. As Chp2 is lower in abundance in S. pombe8 and seems to have a less significant role in heterochromatin spreading8,16, we wondered if only a single dimer of Chp2 binds a mononucleosome.
To determine the stoichiometry of the Chp2:Nucleosome complex, we used the same SV-AUC approach used previously for assessing the stoichiometry of the Swi6:Nucleosome complex33. We performed SV-AUC with H3Kc9me3 nucleosomes alone, with wild-type Chp2, and with Chp2 I370E, a mutant that is unable to dimerize. Each experiment was analyzed using a c(s) distribution with a fixed frictional coefficient (f/f0) value as well as a bimodal f/f0 distribution, where the frictional coefficient is a general measure of molecule shape.
These experiments showed that, analogous to Swi6, four Chp2 molecules bind a single nucleosome implying two dimers of Chp2. Consistent with this model, the Chp2 dimerization mutant bound with a stoichiometry of 2:1. As was hypothesized with Swi6, these data suggest that Chp2 may also form “sticky ends” on mononucleosomes and bridge adjacent nucleosomes.
Discussion
Although much is known about the biochemical properties of individual HP1 proteins, less is known about the biochemical differences between paralogs and how these differences contribute to non-overlapping roles in vivo. In order to understand how paralogs vary in their intrinsic molecular behaviors, we characterized the biophysical properties of S. pombe HP1 protein Chp2 in comparison with Swi6. Both proteins are involved in H3K9me2/3-dependent heterochromatin, but genetically have been shown to have non-overlapping roles.
While the chromoshadow domains of Swi6 and Chp2 are homologous (40% sequence identity, 69% sequence similarity), they have been shown to bind different effector proteins. Indeed, one striking difference is that chimeras swapping the CSDs of Swi6 and Chp2 result in a loss of silencing phenotype8. Here we show that the Swi6 and Chp2 CSDs bind to peptides from Sgo1 and Clr3, although the affinities vary. Chp2, which is not known to bind Sgo1 in vivo, binds 6-fold more weakly to the Sgo1 peptide than Swi6. In contrast to Sgo1, Chp2 has been suggested to directly interact with Clr3, a histone deacetylase involved in heterochromatin formation. Yet compared to Swi6, Chp2 binds the Clr3 peptide at least 12-fold more weakly. It is possible that additional interactions with the full-length Clr3 protein stabilize binding between Chp2 and Clr3 in vivo. Importantly, this work identifies a specific region of Clr3 that directly interacts with Swi6 and Chp2. Together, these data are also consistent with recent studies that imply that the CSD binding motif is more degenerate than the PXVXL motif identified in early studies23,24. These recent studies have suggested a relaxed motif of ΦX(V/P)XΦ24. We speculate, given the peptides derived from our study, that the S. pombe HP1 proteins may tolerate additional deviations from the relaxed ΦX(V/P)XΦ motif. It is also possible that the preferred binding motif differs across species. Further, amino acid differences between the CSDs of HP1 paralogs within a species may also contribute to different binding affinities for the same ligands. Such differences could explain in part why Swi6 and Chp2 interact with different proteins in vivo and in turn why they have different biological functions.
We also find that Chp2 binds ~3-fold more weakly to H3Kc9me3 nucleosomes than Swi6. It is unknown which portions of the Swi6 and Chp2 proteins specifically lead to this small binding difference. There are significant differences in sequence between both the NTE and the hinge between these proteins. It is known that the NTE of Swi6 contributes to histone H3 tail binding. The role of the Chp2 NTE, which is roughly half the size of the whole protein, is not known. Additionally, we show here and in previous work that dimerization of the CSDs of both Swi6 and Chp2 is important for nucleosome binding. This observation raises the possibility that the CSD-CSD dimer interface that interacts with non-nucleosomal ligands like Clr3 also plays a role in nucleosome binding. Indeed, it has recently been shown that CSDs of human HP1α, HP1β, and HP1γ bind a peptide with the sequence PGTVAL from the globular portion of histone H3 with a Kd in the low μM range24. However, we find that the Swi6 CSD binds this peptide with a substantially weaker affinity (Supplemental Figure 3E, Kd > 1 mM). It is thus possible that the CSDs of Swi6 and Chp2 recognize a different portion of the histone octamer. It is also unclear if the same region of the CSD that binds effector proteins also binds the nucleosome. This leads to interesting possible mechanisms with how Swi6 and Chp2 engage with the nucleosome and other proteins. For example, if the same region binds both the nucleosome and effectors, the CSD may not be able to simultaneously engage with both substrates. In this context, the ~3-fold weaker binding of Chp2 may make it slightly more prone than Swi6 to dissociate from nucleosomes if its CSD is bound by a non-nucleosomal ligand, raising the possibility that Swi6 is better suited than Chp2 to act as a platform for recruiting diverse activities while remaining bound to chromatin.
Our data also suggest that Chp2, unlike Swi6, does not adopt an auto-inhibitory conformation. Chp2’s H3K9me3 peptide and nucleosome affinity parallel the properties of the uninhibited Swi6 ARK loop mutant. Yet, like Swi6, Chp2 can assemble as a tetramer on a mononucleosome and lead to a sticky ends architecture. This raises interesting questions about why Chp2 may not be subject to the same auto-regulation as Swi6. Swi6 is much more abundant in vivo than Chp2 (Roughly 20,000 molecules compared to 200 per cell)8. As it is involved in numerous nuclear processes, it is easy to imagine that multiple levels of regulation are required to prevent the ectopic assembly and spread of Swi6. The auto-inhibitory state of Swi6, which is mutually exclusive with higher-order oligomerization and nucleosome binding, provides a means for such regulation. Chp2 is much less abundant and seems to have a more specific role in heterochromatin establishment16. Therefore, we hypothesize it may not be necessary to regulate Chp2 binding and oligomerization to the same extent as Swi6, suggesting one explanation for the lack of a Swi6-like auto-inhibitory state. The lower nuclear concentration of Chp2 also suggests that, unlike with Swi6, the sticky ends architecture may not be used for spreading, but instead may be used for bridging across a proximal nucleosome, for interacting with Swi6 molecules, or for interacting with other binding partners.
HP1 proteins form a dynamic chromatin platform that can recruit effector proteins, and these proteins have likely evolved to suit their different roles within a given species. However, Swi6 and Chp2, as well as other HP1 proteins, interact with many different proteins, and most of these interfaces are unknown. Further studies will lead to a better understanding of which of these interactions are mediated via the CSD, which are mediated by other HP1 domains, how they regulate nucleosome binding and how they differ between paralogs and species.
Materials and Methods
Protein Cloning and Purification
Swi6 was purified from Escherichia coli as previously described32,33. Chp2 was cloned into pET30a at the BamHI and NotI sites with a TEV cleavage site separating the 6xHis tag and the Chp2 coding sequence. Swi6 and Chp2 full-length proteins were purified from E. coli Rosetta (DE3) cells. Cells were grown to OD 0.6 at 37°C in LB medium with 50 μg/μL Kanamycin. Isopropyl-β-D-thiogalactopyranoside was added to a final concentration of 0.5 mM to induce protein expression, and cells were incubated at 18°C for 16 hours. Harvested cells were resuspended in lysis buffer (1X PBS buffer pH 7.8, 300 mM KCl, 10% glycerol, 7.5 mM imidazole, and protease inhibitors: phenylmethanesulfonyl fluoride, pepstatin A, aprotinin, and leupeptin). Following lysis in a C3 Emulsiflex (Avestin), cell debris was removed by centrifugation at 25,000g for 45 minutes. Clarified lysate was incubated with Cobalt-NTA affinity resin (Clontech) for 1 hour at 4°C. Resin was then washed and proteins were eluted with 20 mM Hepes pH 7.5, 100 mM KCl, 10% Glycerol, and 500 mM Imidazole. Proteins were cleaved overnight with 3 mg/mL TEV protease while dialyzing into 20 mM Hepes pH 7.5, 300 mM KCl, 1 mM DTT, and 10% glycerol. Protein was then purified by size-exclusion chromatography on a Superdex 200 26/60 column (GE Healthcare) with storage buffer (20 mM Hepes pH 7.5, 300 mM KCl, 1 mM DTT, 10% glycerol). For the CSDs of Swi6 and Chp2, a Superdex 75 16/60 column (GE Healthcare) was used for the size exclusion step. All N-terminal tags were cleaved using TEV protease except for NMR binding experiments involving the Chp2 CSD since the tag improved stability at NMR concentrations. Protein concentrations were measured by ultraviolet absorption at 280 nm and calculated using the extinction coefficient. Concentrations were measured by UV absorbance at 280 nM using the following calculated extinction coefficients: 40,910 M−1 cm−1 for full length Swi6, 8,480 M−1 cm−1 for Swi6 CSD, 55,350 M−1 cm−1 for full length Chp2, and 16,960 M−1 cm−1 for Chp2 CSD. To ensure that there was little to no DNA contamination, the 260/280 ratio was measured for each purified protein, which was 0.55 on average.
Nucleosome Reconstitution
Gradient salt dialysis was used to assemble mononucleosomes on DNA templates containing the 147 bp 601 sequence. Labeled nucleosomes were modified with a fluorescein tag on the 5′ upstream end of the DNA. All nucleosomes were prepared using recombinant Xenopus leavis histones and assembled as previously described35,36. Histone H3 containing methyl lysine analogue at position 9 (H3Kc9me3) were prepared as previously described37.
Fluorescence Polarization
Fluorescence polarization binding assays to peptides and nucleosomes were performed in binding buffer (20 mM Hepes, 150 mM KCl, 1 mM DTT) supplemented with 0.02% NP-40 at 24°C. Substrates were used at a final concentration of 5 nM, and Swi6 and Chp2 concentrations were varied. The binding reaction was incubated for 30 minutes at 24°C. Fluorescence polarization was measured using an AnalystHT (Molecular Devices) with excitation and emission wavelengths of 480 and 530 nm, respectively.
The binding data were fit using the following equation:
In which FPobs is the fluorescence polarization signal observed, FPmin is the fluorescence polarization signal for the probe alone, and FPmax is the fluorescence polarization signal at saturating protein concentration. The obtained Kd values were averaged over three or more independent sets of data.
Sedimentation Velocity Analytical Ultracentrifugation
Swi6 and Chp2 proteins were independently dialyzed into reaction buffer overnight at 4°C. All sedimentation velocity experiments were performed using an analytical ultracentrifuge (Beckman Coulter) equipped with an absorption optical scanner (Optima XLA). Data were acquired with ProteomeLab data acquisition software 5. Global analysis of sedimentation velocity isotherm data was performed using the SEDPHAT software. Partial-specific volume (v), solution density (ρ), and solution viscosity (η) were calculated in SEDNTERP.
Sample volumes of 100 μL or 400 μL at an overall final optical density (OD) between 0.1 and 1 were pipetted into double-sector centerpieces and places in an eight-hole rotor, which was then placed in a temperature equilibrated AUC chamber. An additional incubation of 1 to 2 hours was added with the rotor at rest and under vacuum for temperature equilibration. Runs were performed at a speed of 50,000 rpm. Scans were collected by following ultraviolet absorption at 230 nm, 250 nm, and 280 nm with a radial step size of 0.003 cm in continuous mode. Data were analyzed using a c(s) continuous distribution of Lamm equation solutions with the SEDFIT software, following by integration and assembly into an isotherm of weighted-average s values. The isotherm was modeled in SEDPHAT with mass-action-based models for the weighted-average s value.
Nuclear Magnetic Resonance
For backbone assignment of the Swi6 CSD, protein was expressed in M9 minimal media containing 15N-ammonium chloride and 13C-glucose as the sole nitrogen and carbon source, respectively. Proteins were purified as previously described (above) with exchange into a final buffer containing 20 mM Hepes pH 7.8, 150 mM KCl, and 2 mM DTT. Backbone assignments were obtained from nitrogen HSQC and triple-resonance (HNCA, CBCA(CO)NH) experiments recorded at 303K on either a Bruker Avance DRX500 or Bruker Avance 800 MHz spectrometer equipped with cryogenic probes. For binding experiments, unlabeled peptides were added to either 100 μM 15N-labeled Swi6 CSD or Chp2 His-tagged CSD and nitrogen HSQC spectra were recorded at 303K on a Bruker Avance DRX500 spectrometer. Chemical shift perturbations were calculated from the equation:
where the factor of 0.2 is used as a scaling factor for the nitrogen spectral width. Titration data were only fitted to obtain Kd values if the chemical shift perturbation between apo and final peptide concentration for a residue was greater than the mean plus one standard deviation.
Negative Stain Electron Microscopy and Image Processing
Chp2 protein was dialyzed overnight into sample buffer. 2.5 μL of Chp2 protein at 0.1 μM was adsorbed onto a glow discharged copper grid for 30 seconds followed by conventional negative stain with uranyl formate. Images were collected using a Tecnai T12 microscope (FEI company) with an LaB6 filament and operated at 120-kV accelerating voltage. All images were collected at a magnification of 52kx with an UltraScan 4096 × 4096 pixel CCD camera (Gatan). All images were 2×2 pixel binned to the final pixel size of 4.42 Å. A total of 1,806 particles were selected and processed using Relion38.
Phage Display
A randomized nonapeptide library (X9) fused to the pVIII protein of the phagemid vector pC89 was used for the phage display procedure39. Screening of the library was performed as follows: 1 μM of biotinylated Swi6 or Chp2 CSD was bound to streptavidin magnetic beads in PBST (phosphate-buffed saline 0.2% BSA. After washing 3 times, the protein and phage were eluted with TEV protease. Logarithmic phase E. coli XL-1 Blue cells were infected by the eluate and the phage amplified using the helper phage M13KO7. The third and fourth panning rounds were used to infect E. coli, and 96 individual colonies from each round were picked and sequenced.
Peptides
Sgo1: EKAKTSNVCVCIPCKSAEQ
Mutant Sgo1: EKAKTSNVCECIPCKSAEQ
Clr3: RAVTQYLLHLLQKARPTSQ
H3 Tail Peptide: ARTKQTARKSTGGKA
Supplementary Material
Supplemental Figure 1: (A) Sequence alignment of Swi6 and Chp2 generated by X showing the general domain architecture of the two HP1 proteins. The alignment of the ARK loop present in the CD of Swi6 is outlined by a red box. Residues shifted with Sgo1 peptide binding (purple), Clr3 peptide binding (orange), and shifted with both peptides (teal) are indicated for the Swi6 CSD.
Supplemental Figure 2: The consensus sequences of the Swi6 CSD (A) and Chp2 CSD (B) displayed in logo format. Amino acid frequency is illustrated by relative letter size at each position. (C) Differences in chemical shifts for assigned resonances between Swi6 CSD alone and with the addition of the Sgo1 (blue) and Clr3 (orange) peptides. (D) Quantification of the normalized chemical shift perturbations (CSP) for F317, E280, and H321 across peptide titration is shown. The curves are hyperbolic fits with dissociation constants (Kd) measured for each residue of 68.02 ± 14.98, 62.48 ± 15.39, and 51.86 ± 7.84, respectively. (E) A superimposition of 1H-15N HSQC spectra of Swi6 CSD with (black) and without (red) mutant Sgo1 peptide.
Supplemental Figure 3: (A) A superimposition of 1H-15N HSQC spectra of the Chp2 CSD with (black) and without (red) Sgo1 peptide. (B) A superimposition of 1H-15N HSQC spectra of the Chp2 CSD with (black) and without (red) Clr3 peptide. (C) 1H-15N HSQC spectra overlay of two residues while being titrated by the Clr3 peptide. Concentrations of peptide shown are 0 μM (Red), 240 μM (Yellow), 480 μM (Pink), 960 μM (Green), 1,440 μM (Purple). (D) Quantification of the normalized chemical shift perturbations (CSP) for the two residues in B. The calculated Kd is at least 745 μM. (E) Log plot of the binding of the Swi6 CSD to the PGTVAL peptide from histone H3 by fluorescence polarization. Experiment was performed in triplicate at room temperature.
Supplemental Figure 4: (A) Schematic for the fluorescence polarization-based nucleosome binding assay. The 601 nucleosomal DNA is labeled on one end with fluorescein. Binding of Swi6/Chp2 near the entry/exit site restricts movement of the fluorophore, resulting in an increase in polarization. Sample raw binding curves of full-length Swi6 (B) and Chp2 (C) binding to fluorescein labeled H3Kc9me3 nucleosomes. Experiments were performed in triplicate at room temperature. (D) Log plot of the overlay of normalized polarization of Swi6 and Chp2 binding to fluorescein labeled H3Kc9me3 nucleosomes from (B) and (C).
Supplemental Figure 5: Sedimentation velocity AUC analysis of WT Chp2 at 24°C (A) and 37°C (B). Sedimentation velocity AUC analysis of WT Swi6 at 24°C (C) and 37°C (D). Best fits for global analysis using an isodesmic self-association model are shown.
Supplemental Figure 6: (A) Characterization of WT Chp2 and Chp2 I370 binding to H3K9me3 tail peptide by fluorescence anisotropy. Affinity constants (Kd) were calculated to be 3.3 ± 1.4 and 4.6 ± 2.8, respectively. (C) Representative two-dimensional class averages of full-length Chp2. (C) Example raw negative stain electron micrograph of Chp2.
Highlights.
Swi6 and Chp2 play different roles in heterochromatin formation and maintenance
Swi6 binds peptides from Sgo1 and Clr3 more strongly than Chp2
Chp2 is not regulated by a Swi6-like auto-inhibition based mechanism
Chp2 and Swi6 biophysical differences found here help explain in vivo differences
Acknowledgments
We thank J. Tretyakova for the preparation of histone proteins and C. Stoddard for help with EM grid preparation, Swi6 EM studies, and RELION processing. We thank C. Stoddard, S. Johnson, R. Almeida for helpful discussion and feedback. This work was supported by National Institutes of Health (NIH) grants R01GM108455 to G.J.N., 5P41CA196276 to M.H., and R01GM104659 to C.S.C.
Abbreviations
- SV-AUC
sedimentation velocity analytical ultracentrifugation
- CD
chromodomain
- CSD
chromoshadow domain
- H
Hinge Domain
- CTE
C-terminal extension
- HP1
heterochromatin protein 1
- H3K9me3
histone H3 lysine 9 trimethylation
- IP-MS
Immunoprecipitation mass spectrometry
- SHREC
Snf2/HDAC-containing repressor complex
- EM
electron microscopy
- NMR
nuclear magnetic resonance
- HSQC
Heteronuclear Single Quantum Correlation spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: R.S.I. and G.J.N. conceived of the core questions of this study. S.S. and R.T. performed the NMR experiments. R.T. and J.D.G. determined backbone assignments of the Swi6 CSD. R.S.I. performed the bulk of the biochemical experiments. R.S.I. and M.H. performed the phage display experiments. R.S.I. and M.R. carried out peptide synthesis and purification. R.S.I. and G.J.N. wrote the manuscript with significant contributions from S.S., R.T., and J.D.G.
Accession Numbers
Backbone assignments of the Swi6 CSD have been deposited in the Biological Magnetic Resonance Data Bank, accession number 27145. NMR chemical perturbations were mapped onto the Swi6 chromoshadow crystal structure with the PDB ID code 1E0B.
References
- 1.Grewal SIS, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 2.Dillon N. Heterochromatin structure and function. Biol Cell. 2004;96:631–637. doi: 10.1016/j.biolcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Peng JC, Karpen GH. Epigenetic regulation of heterochromatic DNA stability. Current Opinion in Genetics & Development. 2008;18:204–211. doi: 10.1016/j.gde.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gartenberg M. Heterochromatin and the cohesion of sister chromatids. Chromosome Res. 2009;17:229–238. doi: 10.1007/s10577-008-9012-z. [DOI] [PubMed] [Google Scholar]
- 5.Hall IM. Establishment and Maintenance of a Heterochromatin Domain. Science. 2002;297:2232–2237. doi: 10.1126/science.1076466. [DOI] [PubMed] [Google Scholar]
- 6.Thon G, Verhein-Hansen J. Four chromo-domain proteins of Schizosaccharomyces pombe differentially repress transcription at various chromosomal locations. Genetics. 2000;155:551–568. doi: 10.1093/genetics/155.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motamedi MR, et al. HP1 Proteins Form Distinct Complexes and Mediate Heterochromatic Gene Silencing by Nonoverlapping Mechanisms. Molecular Cell. 2008;32:778–790. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadaie M, et al. Balance between Distinct HP1 Family Proteins Controls Heterochromatin Assembly in Fission Yeast. Molecular and Cellular Biology. 2008;28:6973–6988. doi: 10.1128/MCB.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 10.Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol. 2000;10:517–525. doi: 10.1016/s0960-9822(00)00467-x. [DOI] [PubMed] [Google Scholar]
- 11.Zhao T, Heyduk T, Allis CD, Eissenberg JC. Heterochromatin protein 1 binds to nucleosomes and DNA in vitro. J Biol Chem. 2000;275:28332–28338. doi: 10.1074/jbc.M003493200. [DOI] [PubMed] [Google Scholar]
- 12.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 Lysine 9 Methylation and HP1γ Are Associated with Transcription Elongation through Mammalian Chromatin. Molecular Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 13.Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y. Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci. 2003;116:3327–3338. doi: 10.1242/jcs.00635. [DOI] [PubMed] [Google Scholar]
- 14.Smallwood A, et al. CBX3 regulates efficient RNA processing genome-wide. Genome Res. 2012;22:1426–1436. doi: 10.1101/gr.124818.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billur M, Bartunik HD, Singh PB. The essential function of HP1 beta: a case of the tail wagging the dog? Trends in Biochemical Sciences. 2010;35:115–123. doi: 10.1016/j.tibs.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Kagansky A, et al. Synthetic Heterochromatin Bypasses RNAi and Centromeric Repeats to Establish Functional Centromeres. Science. 2009;324:1716–1719. doi: 10.1126/science.1172026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer T, et al. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. PNAS. 2009;106:8998–9003. doi: 10.1073/pnas.0813063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamagishi Y, Sakuno T, Shimura M, Watanabe Y. Heterochromatin links to centromeric protection by recruiting shugoshin. Nature. 2008;455:251–255. doi: 10.1038/nature07217. [DOI] [PubMed] [Google Scholar]
- 19.Haldar S, Saini A, Nanda JS, Saini S, Singh J. Role of Swi6/HP1 Self-association-mediated Recruitment of Clr4/Suv39 in Establishment and Maintenance of Heterochromatin in Fission Yeast. Journal of Biological Chemistry. 2011;286:9308–9320. doi: 10.1074/jbc.M110.143198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aygun O, Grewal SIS. Assembly and Functions of Heterochromatin in the Fission Yeast Genome. Cold Spring Harbor Symposia on Quantitative Biology. 2011;75:259–267. doi: 10.1101/sqb.2010.75.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang K, Mosch K, Fischle W, Grewal SIS. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- 22.Sugiyama T, et al. SHREC, an Effector Complex for Heterochromatic Transcriptional Silencing. Cell. 2007;128:491–504. doi: 10.1016/j.cell.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Smothers JF, Henikoff S. The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr Biol. 2000;10:27–30. doi: 10.1016/s0960-9822(99)00260-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, et al. Peptide recognition by heterochromatin protein 1 (HP1) chromoshadow domains revisited: Plasticity in the pseudosymmetric histone binding site of human HP1. Journal of Biological Chemistry. 2017;292:5655–5664. doi: 10.1074/jbc.M116.768374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. 1994 [PubMed] [Google Scholar]
- 26.Job G, et al. SHREC Silences Heterochromatin via Distinct Remodeling and Deacetylation Modules. Molecular Cell. 2016;62:207–221. doi: 10.1016/j.molcel.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic Interaction Network of Evidence Algorithm and its Application to Complete Labeling of Peak Lists from Protein NMR Spectroscopy. PLoS Comp Biol. 2009;5:e1000307–15. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brasher SV, et al. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 2000;19:1587–1597. doi: 10.1093/emboj/19.7.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nietlispach D, Laue E, Thiru A, et al. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004;23:489–499. doi: 10.1038/sj.emboj.7600088. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang J, et al. Mitotic centromeric targeting of HP1 and its binding to Sgo1 are dispensable for sister-chromatid cohesion in human cells. 2011;22:1181–1190. doi: 10.1091/mbc.E11-01-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada T, Fischle W, Sugiyama T, Allis CD, Grewal SIS. The Nucleation and Maintenance of Heterochromatin by a Histone Deacetylase in Fission Yeast. Molecular Cell. 2005;20:173–185. doi: 10.1016/j.molcel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Canzio D, et al. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature. 2013;496:377–381. doi: 10.1038/nature12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canzio D, et al. Chromodomain-Mediated Oligomerization of HP1 Suggests a Nucleosome-Bridging Mechanism for Heterochromatin Assembly. Molecular Cell. 2011;41:67–81. doi: 10.1016/j.molcel.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller C, et al. HP1Swi6 Mediates the Recognition and Destruction of Heterochromatic RNA Transcripts. Molecular Cell. 2012:1–13. doi: 10.1016/j.molcel.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 36.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Meth Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 37.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheres SHW. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felici F, Castagnoli L, Musacchio A, Jappelli R, Cesareni G. Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J Mol Biol. 1991;222:301–310. doi: 10.1016/0022-2836(91)90213-p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) Sequence alignment of Swi6 and Chp2 generated by X showing the general domain architecture of the two HP1 proteins. The alignment of the ARK loop present in the CD of Swi6 is outlined by a red box. Residues shifted with Sgo1 peptide binding (purple), Clr3 peptide binding (orange), and shifted with both peptides (teal) are indicated for the Swi6 CSD.
Supplemental Figure 2: The consensus sequences of the Swi6 CSD (A) and Chp2 CSD (B) displayed in logo format. Amino acid frequency is illustrated by relative letter size at each position. (C) Differences in chemical shifts for assigned resonances between Swi6 CSD alone and with the addition of the Sgo1 (blue) and Clr3 (orange) peptides. (D) Quantification of the normalized chemical shift perturbations (CSP) for F317, E280, and H321 across peptide titration is shown. The curves are hyperbolic fits with dissociation constants (Kd) measured for each residue of 68.02 ± 14.98, 62.48 ± 15.39, and 51.86 ± 7.84, respectively. (E) A superimposition of 1H-15N HSQC spectra of Swi6 CSD with (black) and without (red) mutant Sgo1 peptide.
Supplemental Figure 3: (A) A superimposition of 1H-15N HSQC spectra of the Chp2 CSD with (black) and without (red) Sgo1 peptide. (B) A superimposition of 1H-15N HSQC spectra of the Chp2 CSD with (black) and without (red) Clr3 peptide. (C) 1H-15N HSQC spectra overlay of two residues while being titrated by the Clr3 peptide. Concentrations of peptide shown are 0 μM (Red), 240 μM (Yellow), 480 μM (Pink), 960 μM (Green), 1,440 μM (Purple). (D) Quantification of the normalized chemical shift perturbations (CSP) for the two residues in B. The calculated Kd is at least 745 μM. (E) Log plot of the binding of the Swi6 CSD to the PGTVAL peptide from histone H3 by fluorescence polarization. Experiment was performed in triplicate at room temperature.
Supplemental Figure 4: (A) Schematic for the fluorescence polarization-based nucleosome binding assay. The 601 nucleosomal DNA is labeled on one end with fluorescein. Binding of Swi6/Chp2 near the entry/exit site restricts movement of the fluorophore, resulting in an increase in polarization. Sample raw binding curves of full-length Swi6 (B) and Chp2 (C) binding to fluorescein labeled H3Kc9me3 nucleosomes. Experiments were performed in triplicate at room temperature. (D) Log plot of the overlay of normalized polarization of Swi6 and Chp2 binding to fluorescein labeled H3Kc9me3 nucleosomes from (B) and (C).
Supplemental Figure 5: Sedimentation velocity AUC analysis of WT Chp2 at 24°C (A) and 37°C (B). Sedimentation velocity AUC analysis of WT Swi6 at 24°C (C) and 37°C (D). Best fits for global analysis using an isodesmic self-association model are shown.
Supplemental Figure 6: (A) Characterization of WT Chp2 and Chp2 I370 binding to H3K9me3 tail peptide by fluorescence anisotropy. Affinity constants (Kd) were calculated to be 3.3 ± 1.4 and 4.6 ± 2.8, respectively. (C) Representative two-dimensional class averages of full-length Chp2. (C) Example raw negative stain electron micrograph of Chp2.