

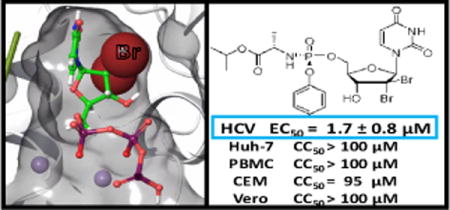
Abstract
Several β-D-2′-deoxy-2′-substituted nucleoside analogs have displayed potent and selective anti-HCV activities and some of them have reached human clinical trials. In that regard, we report herein the synthesis of a series of 2′-deoxy, 2′-dibromo substituted U, C, G and A nucleosides 10a–d and their corresponding phosphoramidate prodrugs 13a–d. The synthesized nucleosides 10a–d and prodrugs 13a–d were evaluated for their inhibitory activity against HCV as well as cellular toxicity. The results showed that the most potent compound was prodrug 13a, which exhibited micromolar inhibitory activity (EC50 = 1.5 ± 0.8 μM) with no observed toxicity. In addition, molecular modeling and free energy perturbation calculations for the 5′-triphosphate formed from 13a and related 2′-modified nucleotides are discussed.
Keywords: Synthesis, Nucleoside, Prodrug, Hepatitis C virus
Graphical abstract
Hepatitis C virus (HCV) is a blood-borne pathogen infecting an estimated 180 million subjects worldwide with 55–85% of infected individuals progressing to chronic HCV infection.1 More concerning is that approximately 30% of those chronically infected persons will develop liver cirrhosis and 1–7% of these will go on to develop hepatocellular carcinoma.2,3 Just a few years ago, the standard of care for HCV patients was pegylated interferon-α (PEG-IFN) and ribavirin (RBV) in combination with boceprevir, telaprevir, (two first-generation NS3/4A protease inhibitors) and simeprevir (a second-generation NS3/4A protease inhibitor), which demonstrated limited efficacy and often intolerable side effiects.4 Since their initial discovery, nucleoside analogs, as inhibitors of HCV NS5B polymerase, have been favored due to their high genetic barrier to drug resistance and pan-genotypic activity.5 To date, sofosbuvir is the only approved nucleoside analog for HCV treatment and remains the backbone of several combination therapies. Compared with previous treatments, sofosbuvir-based regimens provide a durable cure rate along with fewer side effects. However, the duration of approved sofosbuvir treatment regimens remains at 8–12 weeks.6 Therefore, there is still an unmet need to develop novel pan-genotypic and more efficacious nucleoside analogs, which could lead to new short to ultra-short combination therapies with improved safety profiles and higher barrier to resistance.7
Inspired by the success of 2′-halogenated nucleoside analogs such as sofosbuvir 18 or gemcitabine 2,9 several groups, including ours, studied extensively 2′-dihalogenated nucleosides as potential anti-HCV agents. Thus, Pinho et al. reported the discovery of a β-D-2′-deoxy-2′-dichlorouridine nucleotide prodrug 3 inhibiting HCV replication10 while we recently reported a new β-D-2′-Cl, 2′-F-uridine phosphoramidate nucleotide 4 as a non-toxic, pan-genotypic, potent and specific anti-HCV NS5B polymerase agent.11 As part of our continuing efforts in this area, we report herein the synthesis and antiviral evaluation of a series of 2′-dibromo nucleosides 10a–d and their corresponding phosphoramidate prodrugs 13a–d.
To determine if the HCV NS5 RdRp active site can accommodate 2′-dibromo substitutions, molecular modeling studies were initiated. Crystal structures of the HCV RdRP complexed to the RNA primer/template reveal dynamic rearrangement of residues contacting the 2′-OH versus 2′-F moieties12 and based on the β-D-2′-Me, 2′-F-uridine diphosphate HCV RdRp structure (PDBID 4WTG), we predicted that β-D-2′-di-Br-uridine triphosphate in the active site would incur only minimal steric clash (Figure 2). However, because reliable energy calculations require the use of methods that account for the flexibility in the polymerase active site we next performed alchemical free energy perturbation (FEP) calculations. FEP calculates relative binding free energy between two ligands by mutating one to the other over a series of molecular dynamics simulations in solution and bound to the protein.13 FEP calculations yield accurate ΔΔG as the change of binding energy between the first and second ligands.14 A negative ΔΔG value suggests a gain in binding energy. To evaluate this approach for 2′-modified HCV nucleotide analogs, FEP calculations were performed on known inhibitors relative to the UTP-bound complex (PDBID 4WTA) using Desmond MD with the OPLS-2005 force field on a GPU-enabled EXXACT MD server. It must be noted that the FEP ΔΔG results do not reflect the overall antiviral activity as many other pharmacologic steps are not included (cell penetration, prodrug processing, phosphorylation by host kinases and etc.), nor do these calculations necessarily recapture the enzymatic IC50 as they do not account for the chain-terminating chemical steps. However, these calculations predict how well the 5′-triphosphate analog binds to the active site; a critical step for antiviral activity. The calculations were performed in the physiologically relevant tri-anion and tetra-anion protonation states.15 The FEP ΔΔG for β-D-2′-Me, 2′-F-uridine triphosphate (the active form of sofosbuvir) was −2.52 ± 0.18 kcal/mol relative to UTP (Table 1), which agrees with the potent inhibitory activity of the drug. Two other 2′-halogenated analogs recently reported as HCV inhibitors, β-D-2′-Cl-2″-F and β-D-2′-Cl-2″-Cl-uridine, display negative FEP ΔΔG relative to UTP in both protonation states in agreement with their experimental antiviral activity. Interestingly, the FEP ΔΔG for the proposed 2′-dibromo UTP analog was large and negative for both tri- and tetra-anions (−2.87 ± 0.26 and −4.21 ± 0.18 kcal/mol, respectively) suggesting this analog can favorably bind to the active site relative to UTP thereby meriting synthesis and testing.
Figure 2.
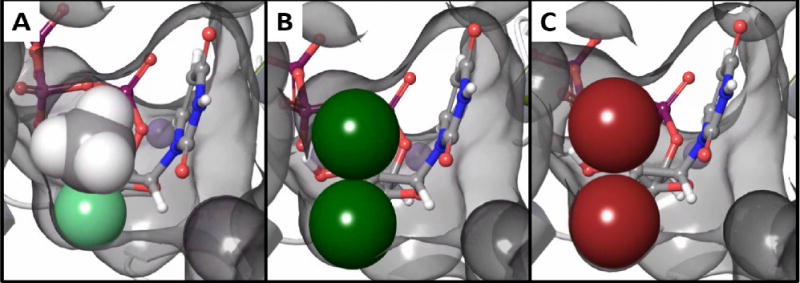
Models of 2′-modified uridine triphosphate analogs in the HCV NS5 active site (PDBID 4WTG) refined using Prime MMGBSA (5 Å cut-off). The active site surface is rendered in grey, and the Van der Waals radii of the 2′ substitutions are shown as spheres. A) β-D-2′-Me,2′-F-uridine 5′-triphosphate, the active form of sofosbuvir (Prime MMGBSA ΔGbind = −25.3 kcal/mol), B) β-D-2′-diCl-uridine 5′-triphosphate (−29.9 kcal/mol), and C) β-D-2′-diBr-uridine 5′-triphosphate (−30.7 kcal/mol).
Table 1.
Results of FEP calculations on 2′-modified nucleotide triphosphates in the HCV NS5 RdRp active site.
| 2′ Group |
2″ Group |
FEP ΔΔG (kcal/mol)
|
|||
|---|---|---|---|---|---|
| Tri-anion | Tetra-anion | ||||
| −CH3 | −F | −2.52 | ± 0.18 | 0.44 | ± 0.13 |
| −Cl | −F | −0.45 | ± 0.25 | −0.67 | ± 0.23 |
| −Cl | −Cl | −1.89 | ± 0.27 | −3.16 | ± 0.25 |
| −Br | −Br | −2.87 | ± 0.26 | −4.21 | ± 0.18 |
Synthesis of nucleosides 10a–d is described in Scheme 1. Oxidation of commercially available 2-deoxy-D-ribose 5 with Br2 followed by silylation with tert-butyldimethylchorosilane (TBDMSCl) yielded the di-silyl protected lactone 6 in 70% yield.16 Reaction of 6 with 2.2 equivalent of N-bromosuccinimide (NBS) in presence of LiHMDS gave the lactone 7 in 80% yield. Reduction of lactone 7 with lithium diisobutylaluminium hydride (DIBAL-H) provided the desired lactol intermediate, which was subjected to benzoylation with BzCl in presence of Et3N yielding benzoylate 8 in 81% yield. Initial glycosylation of 8 with silylated protected nucleobases (Uracil, N4-Bz-cytosine17, N6-diBoc-adenine or N4-diBoc-O6-Bn-Guanine18) to obtain nucleoside intermediates 9a–d was attempted using standard Vorbruggen conditions in the presence of TMSOTf. However, these coupling reactions, especially for C and G nucleosides (9b and 9d), needed long reaction times (> 24 h) and elevated temperatures (> 100 °C), which resulted in low yields probably due to the instability of these nucleosides to the reaction conditions. In order to improve the yields for formation of nucleosides 9a–d, a set of experiments were carried out under microwave irradiation (MW) using CH3CN as the solvent. After screening several temperatures and reaction times, it was found that the coupling reactions did not proceed at 80 °C, while a messy mixture was obtained when the temperature was increased to 140 °C. Best results were obtained when reactions were irradiated at 120 °C for 10 min with a maximum power of 200W. Under these conditions, compounds 9a–d were obtained as inseparable mixtures of α/β anomers, in yields ranging from 40% to 60%. It is noteworthy that the silylated purine nucleobases need to be Boc protected (N6-diBoc-adenine or N2-diBoc-O6-Bn-guanine) in order for the glycosyslation step to work however, these group fall off either during the reaction or during workup.
Scheme 1.
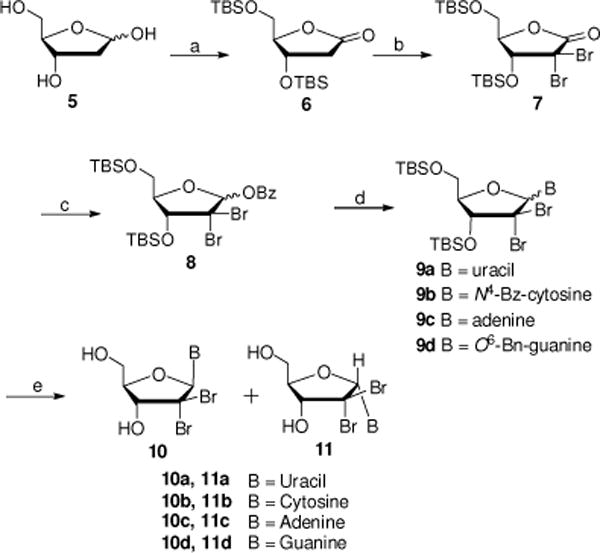
Reagents and conditions: (a) (i) Br2, H2O, rt, 5 d; (ii) TBDMCl, imidazole, DMF, rt, 24 h, 70% over two steps; (b) NBS, LiHMDS, THF, −78 °C to −10 °C, 4–6 h, 80%; (c) (i) DIBAL-H, toluene, −78 °C, 2 h, rt; (ii) BzCl, Et3N, DCM, 0 °C to rt, 12 h, 81%; (d) (i) nucleobase (Uracil, N4-Bz-cytosine, N6-diBoc-adenine or N4-diBoc-O6-Bn-Guanine), BSA, CH3CN, 60 °C, 30 min; (ii) TMSOTf, CH3CN, MW, 120 °C, 10 min; (e) for 9a and 9c: TBAF, THF, 0 °C, 1 h, 40–50% yield; for 9b: (i) NH3/MeOH, rt, 16–24 h; (ii) TBAF, THF, 0 °C, 1 h, 33% yield; for 9d: (i) TBAF, THF, 0 °C, 1 h; (ii) TFA, rt, 48–72 h, 33% yield.
Finally, deprotection of compounds 9a–d was performed and separation of each α and β isomers was achieved with the unprotected nucleoside analog using preparative reverse phase HPLC. Thus, treatment of compounds of 9a and 9c with TBAF in THF gave, after purification, β isomers 10a and 10c and α isomers 11a and 11c as anomer mixtures in 40–50% yield (10a:11a and 10c:11c ratio: 3:5 and 1:2 respectively). Debenzoylation of 9b in a saturated solution of NH3 in MeOH, followed by treatment with TBAF in THF yielded β isomer 10b and isomer 11b in 30–35% (ratio: 5:7). Desilylation of 9d with TBAF, followed by debenzylation in TFA afforded β isomer 10d and α isomer 11d in 30–35% (ratio = 1:1.2).
α and β anomers were identified using 1H 2D-NOESY experiments. For example, clear NOE enhancements were observed between H5 of the uracil nucleobase and H3′ of the sugar as well as between H1′ and H4′ for β-isomer 10a. While obvious NOE interactions between the H5 and H4′, H1′ and H3′ were observed for its α-isomer 11a (Figure 2).
In order to be a substrate for HCV NS5B, nucleosides analogs need to be activated to their triphosphate form. This multistep process is often limited by the first phosphorylation step which led to the development of nucleoside monophosphate prodrugs.19 These prodrugs allow for the intracellularly delivery of monophosphate species after enzymatic and/or chemical cleavage of different masking groups. Among all these prodrugs, the phosphoramidate/phosphonamidate approach is now, by far, the most popular and lead to the approval of compounds like sofosbuvir (HCV) and tenofovir alafenamide (HIV).20 As both of these compounds are Rp isomers, we have decided to prepare pure monophosphate prodrugs of nucleosides 10a–d using chiral pentafluorophenyl reagent 12 synthesized according to Ross et al’ procedure.21 Thus, reaction of nucleosides 10a–d with 12 in the presence of tert-butyl magnesium chloride provided the desired Rp prodrugs 13a–d in 18–25% yield.
Compounds 10a–d and their corresponding phosphoramidate prodrugs 13a–d were evaluated for inhibition of HCV genotype 1b RNA replication in Huh-7 cells using a subgenomic HCV replicon system.22 Cytotoxicity in Huh-7 cells was determined simultaneously by extraction and amplification of both HCV RNA and cellular ribosomal RNA (rRNA).23 In addition, cytotoxicity was determined in primary human peripheral blood mononuclear (PBM) cells, human lymphoblastoid CEM, and African Green monkey Vero cells.24,25 None of the nucleosides synthesized showed antiviral activity at concentration up to 10 μM (10b–d) or 33 μM (10a). However, while prodrugs 13b–d did not display any anti-HCV activity, uracil prodrug 13a was found weakly active (EC50 = 1.5 μM) and non-toxic versus all four tested cell lines. The discrepancy, in term of activity, between nucleoside 10a and its monophosphate prodrug 13a can most probably be attributed to a lack of phosphorylation by the cellular kinases.
In addition, all of the nucleosides 10a–d and prodrugs 13a–d were evaluated for inhibitory activities against West Nile, Dengue, Chikungunya, RSV, Influenza, Ebola, Norovirus and HIV, respectively. However, none of them exhibited obvious activities when tested up to 10 μM (Data not shown).
In summary, a series of β-D-2′-deoxy-2′-dibromo substituted U, C A and G nucleosides 10a–d and their corresponding phosphoramidate prodrugs 13a–d were synthesized and evaluated against HCV along with other viruses. Molecular modeling studies and FEP calculations indicated that the 2′-deoxy-2′-dibromo sugar ring modification binds favorably to the HCV RdRp active site. However, all four nucleosides 10a–d and three prodrugs 13b–d displayed no apparent inhibitory activities against HCV. Notably, the β-D-2′-Br2-uridine phosphoramidate prodrug analog 13a exhibited moderate inhibitory activity against HCV (EC50= 1.5 ± 0.8 μM). Since FEP calculations suggest that this agent may be a potent substrate to the HCV RdRp, the modest activity is likely due to other limiting factors unaccounted for by the model (such as prodrug processing or low host kinase phosphorylation). Despite the weak potency of prodrug 13a as HCV inhibitor, further modifications of these 2′-dihalogenonucleosides are currently being investigated and will be subject of future publications.
Supplementary Material
Figure 1.
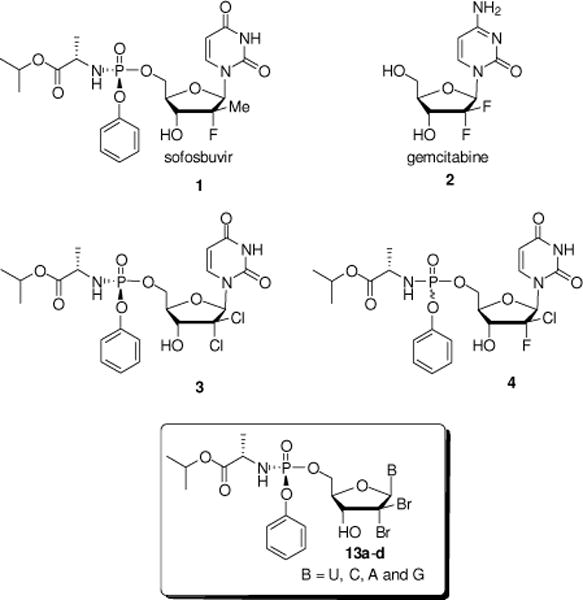
Selected β-D-2′-deoxy-2′-disubstituted nucleoside and nucleotide analogs and targeted 2′-dibromo nucleotide analogs 13a–d.
Figure 3.
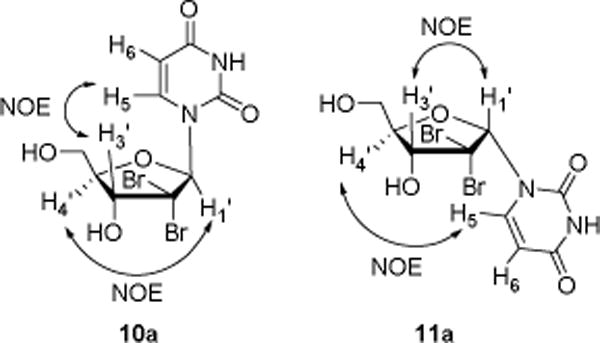
Anomer assignment for nucleosides 10a and 11a via NOE experiments.
Scheme 2.
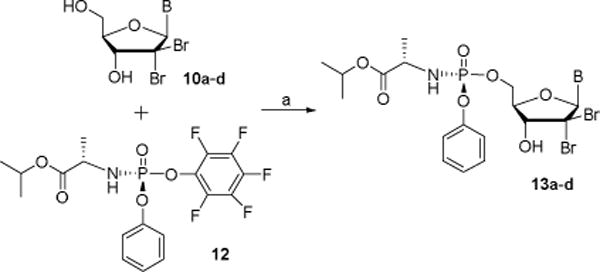
Synthesis of nucleoside phosphoramidates 13a–d. Reagents and conditions: (a) tBuMgCl, THF, 0 °C to rt, 24 h, 18–25%.
Table 2.
HCV genotype 1b replicon activity and cytotoxicity of nucleosides 10a–d and their phosphoramidate prodrugs 13a–d
| Compound | Anti HCV activity (μM) | Cytotoxicity, CC50 (μM) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| EC50 | EC90 | Huh-7 | PBM | CEM | Vero | |
| 10a | > 33 | > 33 | > 33 | > 100 | > 100 | > 100 |
| 10b | > 10 | > 10 | > 10 | > 100 | 82 | > 100 |
| 10c | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 |
| 10d | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 |
| 13a | 1.5 ± 0.8 | 2.7 ± 0.3 | > 10 | > 100 | 95 | > 100 |
| 13b | > 10 | > 10 | > 10 | > 100 | 73 | > 100 |
| 13c | > 10 | > 10 | > 10 | 57 | 33 | 60 |
| 13d | > 10 | > 10 | > 10 | > 100 | 34 | > 100 |
| 2′C-Me C | 1.7 ± 0.8 | 6.5 ± 1.9 | > 10 | > 100 | > 100 | > 100 |
| sofosbuvir | 0.2 ± 0.1 | 0.5 ± 0.3 | > 10 | > 100 | > 100 | > 100 |
All assays were performed in replicates. Only means + SD of three replicates are shown
Acknowledgments
This work was supported in part by NIH grant 5P30-AI-50409 (CFAR). Dr. Schinazi is the Chairman and a major shareholder of Cocrystal Pharma, Inc. Emory received no funding from Cocrystal Pharma, Inc. to perform this work and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at http://xxxx.
References and notes
- 1.Hepatitis C. Global Hepatitis Report. World Health Organization; Geneva: 2017. www.who.int. [Google Scholar]
- 2.Lavanchy D. Liver Int. 2009;29:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 3.Goossens N, Hoshida Y. Clin Mol Hepatol. 2015;21:105–114. doi: 10.3350/cmh.2015.21.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yau AH, Yoshida EM, Can J. Gastroenterol Hepatol. 2014;28:445–451. doi: 10.1155/2014/549624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coats SJ, Garnier-Amblard EC, Amblard F, Ehteshami M, Amiralaei S, Zhang H, Zhou L, Boucle SR, Lu X, Bondada L, Shelton JR, Li H, Liu P, Li C, Cho JH, Chavre SN, Zhou S, Mathew J, Schinazi RF. Antiviral Res. 2014;102:119–147. doi: 10.1016/j.antiviral.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curry MP, Tapper EB, Bacon B, Dieterich D, Flamm SL, Guest L, Kowdley KV, Lee Y, Milligan S, Tsai N, Younossi Z, Afdhal NH. Aliment Pharmacol Ther. 2017;46:540–548. doi: 10.1111/apt.14204. [DOI] [PubMed] [Google Scholar]
- 7.Lau G, Benhamou Y, Chen G, Li J, Shao Q, Ji D, Li F, Li B, Liu J, Hou J, Sun J, Wang C, Chen J, Wu V, Wong A, Wong C, Tsang S, Want Y, Bassit L, Tao S, Jiang Y, Hsiao H, Ke R, Perelson AS, Schinazi RF. Lancet Gastroenterol Hepatol. 2016;1:97–104. doi: 10.1016/S2468-1253(16)30015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentile I, Maraolo AE, Buonomo AR, Zappulo E, Borgia G, the discovery of sofosbuvir: a revolution for therapy of chronic hepatitis C Expert Opin Drug Discov. 2015;10:1363–1377. doi: 10.1517/17460441.2015.1094051. [DOI] [PubMed] [Google Scholar]
- 9.Shelton J, Lu X, Hollenbaugh J, Cho JH, Amblard F, Schinazi RF. Chem Rev. 2016:14379–14455. doi: 10.1021/acs.chemrev.6b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinho P, Kalayanov G, Westerlind H, Rosenquist Å, Wähling H, Sund C, Almeida M, Ayesa S, Tejbrant J, Targett-Adams P, Eneroth A, Lindqvist A. Bioorg Med Chem Lett. 2017;27:3468–3471. doi: 10.1016/j.bmcl.2017.05.075. [DOI] [PubMed] [Google Scholar]
- 11.Zhou S, Mahmoud S, Liu P, Zhou L, Ehteshami M, Bassit L, Tao S, Domaoal RA, Sari O, Schutter CD, Amiralaei S, Khalil A, Ollinger Russell O, McBrayer T, Whitaker T, Abou-Taleb N, Amblard F, Coats SJ, Schinazi RF. J Med Chem. 2017;60:5424–5437. doi: 10.1021/acs.jmedchem.7b00067. [DOI] [PubMed] [Google Scholar]
- 12.Appleby TC, Perry JK, Murakami E, Barauskas O, Feng J, Cho A, Fox D, Wetmore DR, McGrath ME, Ray AS, Sofia MJ. Science. 2015;347:771–775. doi: 10.1126/science.1259210. [DOI] [PubMed] [Google Scholar]
- 13.Christ CD, Mark AE, van Gunsteren WF. J Comput Chem. 2010;31:1569–1582. doi: 10.1002/jcc.21450. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL. J Am Chem Soc. 2015;137:2695–2703. doi: 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- 15.Corfu NA, Sigel H. Eur J Biochem. 1991;199:659–669. doi: 10.1111/j.1432-1033.1991.tb16168.x. [DOI] [PubMed] [Google Scholar]
- 16.Cen Y, Sauve AA. J Org Chem. 2009;74:5779–5789. doi: 10.1021/jo900637f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porcheddu A, Giacomelli G, Piredda I, Carta M, Nieddu G. Eur J Org Chem. 2008:5786–5797. [Google Scholar]
- 18.Kramer RA, Bleicher KH, Wennemers H. Helvetica Chimica Acta. 2012;95:2621–2634. [Google Scholar]
- 19.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Chem Rev. 2014;114:9154–9218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ray AS, Fordyce MW, Hitchcock MJM. Antiviral Research. 2016;125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Ross BS, Reddy PG, Zhang HR, Rachakonda S, Sofia M. J Org Chem. 2011;76:8311–8319. doi: 10.1021/jo201492m. [DOI] [PubMed] [Google Scholar]
- 22.Rondla R, Coats SJ, McBrayer TR, Grier J, Johns M, Tharnish PM, Whitaker T, Zhou L-H, Schinazi RF. Antivir Chem Chemother. 2009;20:99–106. doi: 10.3851/IMP1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2003;47:244–254. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie M-W, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061–1067. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.