

Abstract
We explore the impact of a host genetic factor on heterosexual HIV epidemics by using a deterministic mathematical model. A protective allele unequally distributed across populations is exemplified in our models by the 32-bp deletion in the host-cell chemokine receptor CCR5, CCR5Δ32. Individuals homozygous for CCR5Δ32 are protected against HIV infection whereas those heterozygous for CCR5Δ32 have lower pre-AIDS viral loads and delayed progression to AIDS. CCR5Δ32 may limit HIV spread by decreasing the probability of both risk of infection and infectiousness. In this work, we characterize epidemic HIV within three dynamic subpopulations: CCR5/CCR5 (homozygous, wild type), CCR5/CCR5Δ32 (heterozygous), and CCR5Δ32/CCR5Δ32 (homozygous, mutant). Our results indicate that prevalence of HIV/AIDS is greater in populations lacking the CCR5Δ32 alleles (homozygous wild types only) as compared with populations that include people heterozygous or homozygous for CCR5Δ32. Also, we show that HIV can provide selective pressure for CCR5Δ32, increasing the frequency of this allele.
Nineteen million people have died of AIDS since the discovery of HIV in the 1980s. In 1999 alone, 5.4 million people were newly infected with HIV (ref. 1 and http://www.unaids.org/epidemicupdate/report/Epireport.html). (For brevity, HIV-1 is referred to as HIV in this paper.) Sub-Saharan Africa has been hardest hit, with more than 20% of the general population HIV-positive in some countries (2, 3). In comparison, heterosexual epidemics in developed, market-economy countries have not reached such severe levels. Factors contributing to the severity of the epidemic in economically developing countries abound, including economic, health, and social differences such as high levels of sexually transmitted diseases and a lack of prevention programs. However, the staggering rate at which the epidemic has spread in sub-Saharan Africa has not been adequately explained. The rate and severity of this epidemic also could indicate a greater underlying susceptibility to HIV attributable not only to sexually transmitted disease, economics, etc., but also to other more ubiquitous factors such as host genetics (4, 5).
To exemplify the contribution of such a host genetic factor to HIV prevalence trends, we consider a well-characterized 32-bp deletion in the host-cell chemokine receptor CCR5, CCR5Δ32. When HIV binds to host cells, it uses the CD4 receptor on the surface of host immune cells together with a coreceptor, mainly the CCR5 and CXCR4 chemokine receptors (6). Homozygous mutations for this 32-bp deletion offer almost complete protection from HIV infection, and heterozygous mutations are associated with lower pre-AIDS viral loads and delayed progression to AIDS (7–14). CCR5Δ32 generally is found in populations of European descent, with allelic frequencies ranging from 0 to 0.29 (13). African and Asian populations studied outside the United States or Europe appear to lack the CCR5Δ32 allele, with an allelic frequency of almost zero (5, 13). Thus, to understand the effects of a protective allele, we use a mathematical model to track prevalence of HIV in populations with or without CCR5Δ32 heterozygous and homozygous people and also to follow the CCR5Δ32 allelic frequency.
We hypothesize that CCR5Δ32 limits epidemic HIV by decreasing infection rates, and we evaluate the relative contributions to this by the probability of infection and duration of infectivity. To capture HIV infection as a chronic infectious disease together with vertical transmission occurring in untreated mothers, we model a dynamic population (i.e., populations that vary in growth rates because of fluctuations in birth or death rates) based on realistic demographic characteristics (18). This scenario also allows tracking of the allelic frequencies over time. This work considers how a specific host genetic factor affecting HIV infectivity and viremia at the individual level might influence the epidemic in a dynamic population and how HIV exerts selective pressure, altering the frequency of this mutant allele.
CCR5 is a host-cell chemokine receptor, which is also used as a coreceptor by R5 strains of HIV that are generally acquired during sexual transmission (6, 19–25). As infection progresses to AIDS the virus expands its repertoire of potential coreceptors to include other CC-family and CXC-family receptors in roughly 50% of patients (19, 26, 27). CCR5Δ32 was identified in HIV-resistant people (28). Benefits to individuals from the mutation in this allele are as follows. Persons homozygous for the CCR5Δ32 mutation are almost nonexistent in HIV-infected populations (11, 12) (see ref. 13 for review). Persons heterozygous for the mutant allele (CCR5 W/Δ32) tend to have lower pre-AIDS viral loads. Aside from the beneficial effects that lower viral loads may have for individuals, there is also an altruistic effect, as transmission rates are reduced for individuals with low viral loads (as compared with, for example, AZT and other studies; ref. 29). Finally, individuals heterozygous for the mutant allele (CCR5 W/Δ32) also have a slower progression to AIDS than those homozygous for the wild-type allele (CCR5 W/W) (7–10), remaining in the population 2 years longer, on average. Interestingly, the dearth of information on HIV disease progression in people homozygous for the CCR5Δ32 allele (CCR5 Δ32/Δ32) stems from the rarity of HIV infection in this group (4, 12, 28). However, in case reports of HIV-infected CCR5 Δ32/Δ32 homozygotes, a rapid decline in CD4+ T cells and a high viremia are observed, likely because of initial infection with a more aggressive viral strain (such as X4 or R5X4) (30).
The Model
Because we are most concerned with understanding the severity of the epidemic in developing countries where the majority of infection is heterosexual, we consider a purely heterosexual model. To model the effects of the allele in the population, we examine the rate of HIV spread by using an enhanced susceptible-infected-AIDS model of epidemic HIV (for review see ref. 31). Our model compares two population scenarios: a CCR5 wild-type population and one with CCR5Δ32 heterozygotes and homozygotes in addition to the wild type. To model the scenario where there are only wild-type individuals present in the population (i.e., CCR5 W/W), we track the sexually active susceptibles at time t [Si,j(t)], where i = 1 refers to genotype (CCR5 W/W only in this case) and j is either the male or female subpopulation. We also track those who are HIV-positive at time t not yet having AIDS in Ii,j,k(t) where k refers to stage of HIV infection [primary (A) or asymptomatic (B)]. The total number of individuals with AIDS at time t are tracked in A(t). The source population are children, χi,j(t), who mature into the sexually active population at time t (Fig. 1, Table 1). We compare the model of a population lacking the CCR5Δ32 allele to a demographically similar population with a high frequency of the allele. When genetic heterogeneity is included, male and female subpopulations are each further divided into three distinct genotypic groups, yielding six susceptible subpopulations, [Si,j(t), where i ranges from 1 to 3, where 1 = CCR5W/W; 2 = CCR5 W/Δ32; 3 = CCR5 Δ32/Δ32]. The infected classes, Ii,j,k(t), also increase in number to account for these new genotype compartments. In both settings we assume there is no treatment available and no knowledge of HIV status by people in the early acute and middle asymptomatic stages (both conditions exist in much of sub-Saharan Africa). In addition, we assume that sexual mixing in the population occurs randomly with respect to genotype and HIV disease status, all HIV-infected people eventually progress to AIDS, and no barrier contraceptives are used. These last assumptions reflect both economic and social conditions.
Figure 1.

A schematic representation of the basic compartmental HIV epidemic model. The criss-cross lines indicate the sexual mixing between different compartments. Each of these interactions has a positive probability of taking place; they also incorporate individual rates of transmission indicated as λ, but in full notation is λî,ĵ,k̂→i,j, where i,j,k is the phenotype of the infected partner and î,ĵ is the phenotype of the susceptible partner. Also shown are the different rates of disease progression, γi,j,k, that vary according to genotype, gender, and stage. Thus, the interactions between different genotypes, genders, and stages are associated with a unique probability of HIV infection. M, male; F, female.
Table 1.
Children's genotype
| Parents | Mother | |||
|---|---|---|---|---|
| Father | W/W | W/Δ32 | Δ32/Δ32 | |
| W/W | χ1,j | χ1,j, χ2,j | χ2,j | |
| W/Δ32 | χ1,j, χ2,j | χ1,j, χ2,j, χ3,j | χ2,j, χ3,j | |
| Δ32/Δ32 | χ2,j | χ2,j, χ3,j | χ3,j | |
χ1,j = wild-type children; (W/W); χ2,j = heterozygous children (W/Δ32); χ3,j = homozygous children (Δ32/Δ32) of gender j. Children's genotypes are determined by using Mendelian inheritance patterns.
Parameter Estimates for the Model.
Estimates for rates that govern the interactions depicted in Fig. 1 were derived from the extensive literature on HIV. Our parameters and their estimates are summarized in Tables 2–4. The general form of the equations describing the rates of transition between population classes as depicted in Fig. 1 are summarized as follows:
 |
1 |
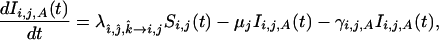 |
2 |
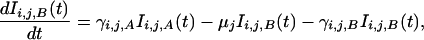 |
3 |
 |
4 |
where, in addition to previously defined populations and rates (with i equals genotype, j equals gender, and k equals stage of infection, either A or B), μj, represents the non-AIDS (natural) death rate for males and females respectively, and μA is estimated by the average (μF + μM/2). This approximation allows us to simplify the model (only one AIDS compartment) without compromising the results, as most people with AIDS die of AIDS (δAIDS) and very few of other causes (μA). These estimates include values that affect infectivity (λî,ĵ,k̂→i,j), transmission (βî,ĵ,k̂→i,j), and disease progression (γi,j,k) where the î,ĵ,k̂ notation represents the genotype, gender, and stage of infection of the infected partner, and j ≠ ĵ.
Table 2.
Transmission probabilities
| HIV-infected partner (î, ĵ, k̂) | Susceptible partner (i, j)
|
|||
|---|---|---|---|---|
| (ĵ to j) | W/W | W/Δ32 | Δ32/Δ32 | |
| Acute/primary | ||||
| W/W or Δ32/Δ32 | M to F | 0.040 | 0.040 | 0.00040 |
| F to M | 0.020 | 0.020 | 0.00020 | |
| W/Δ32 | M to F | 0.030 | 0.030 | 0.00030 |
| F to M | 0.015 | 0.015 | 0.00015 | |
| Asymptomatic | ||||
| W/W or Δ32/Δ32 | M to F | 0.0010 | 0.0010 | 10 × 10−6 |
| F to M | 0.0005 | 0.0005 | 5 × 10−6 | |
| W/Δ32 | M to F | 0.0005 | 0.0005 | 5 × 10−6 |
| F to M | 0.00025 | 0.00025 | 2.5 × 10−6 | |
Listed are the different transmission probabilities (βî,ĵ,k̂→i,j) for random sexual mixing between persons where i, j, k is the phenotype of the infected partner and i, j is the phenotype of the susceptible partner. M, male; F, female.
Table 4.
Parameter values
| Parameter | Definition | Value |
|---|---|---|
| μF, μM | All-cause mortality for adult females (males) | 0.015 (0.016) per year |
| μχ | All-cause childhood mortality (<15 years of age) | 0.01 per year |
| Br | Birthrate | 0.25 per woman per year |
| SAF | Percent females acquiring new partners (sexual activity) | 10% |
| SAM | Percent males acquiring new partners (sexual activity) | 25% |
mF(σ ) ) |
Mean (variance) no. of new partners for females | 1.8 (1.2) per year |
σ
|
Variance in no. of new partners for males | 5.5 per year |
| 1 − pv | Probability of vertical transmission | 0.30 per birth |
| Ii,j,k(0) | Initial total population HIV-positive | 0.50% |
| χi,j(0) | Initial total children in population (<15 years of age) | 45% |
| W/W (0) | Initial total wild types (W/W) in population | 80% |
| W/Δ32(0) | Initial total heterozygotes (W/Δ32) in population | 19% |
| Δ32/Δ32(0) | Initial total homozygotes (Δ32/Δ32) in population | 1% |
| rM(rF) | Initial percent males (females) in total population | 49% (51%) |
| φF, φM | Number of sexual contacts a female (male) has | 30 (24) per partner |
| ɛi,j,k | % effect of mutation on transmission rates (see Table 2) | 0 < ɛi,j,k < 1 |
| δ | Death rate for AIDS population | 1.0 per year |
| q | Allelic frequency of Δ32 allele | 0.105573 |
The effects of the CCR5 W/Δ32 and CCR5 Δ32/Δ32 genotypes are included in our model through both the per-capita probabilities of infection, λî,ĵ,k̂→i,j, and the progression rates, γi,j,k. The infectivity coefficients, λî,ĵ,k̂→i,j, are calculated for each population subgroup based on the following: likelihood of HIV transmission in a sexual encounter between a susceptible and an infected (βî,j,k̂→i,j) person; formation of new partnerships (cj); number of contacts in a given partnership (φj); and probability of encountering an infected individual (Iî,ĵ,k̂/Nĵ). The formula representing this probability of infection is
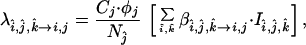 |
where j ≠ ĵ is either male or female. Nĵ represents the total population of gender ĵ (this does not include those with AIDS in the simulations).
The average rate of partner acquisition, cj, includes the mean plus the variance to mean ratio of the relevant distribution of partner-change rates to capture the small number of high-risk people: cj = mj + (σ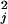 /mj) where the mean (mj) and variance (σ
/mj) where the mean (mj) and variance (σ ) are annual figures for new partnerships only (32). These means are estimated from Ugandan data for the number of heterosexual partners in the past year (33) and the number of nonregular heterosexual partners (i.e., spouses or long-term partners) in the past year (34). In these sexual activity surveys, men invariably have more new partnerships; thus, we assumed that they would have fewer average contacts per partnership than women (a higher rate of new partner acquisition means fewer sexual contacts with a given partner; ref. 35). To incorporate this assumption in our model, the male contacts/partnership, φM, was reduced by 20%. In a given population, the numbers of heterosexual interactions must equate between males and females. The balancing equation applied here is SAF⋅mF⋅NF = SAM⋅mM⋅NM, where SAj are the percent sexually active and Nj are the total in the populations for gender j. To specify changes in partner acquisition, we apply a male flexibility mechanism, holding the female rate of acquisition constant and allowing the male rates to vary (36, 37).
) are annual figures for new partnerships only (32). These means are estimated from Ugandan data for the number of heterosexual partners in the past year (33) and the number of nonregular heterosexual partners (i.e., spouses or long-term partners) in the past year (34). In these sexual activity surveys, men invariably have more new partnerships; thus, we assumed that they would have fewer average contacts per partnership than women (a higher rate of new partner acquisition means fewer sexual contacts with a given partner; ref. 35). To incorporate this assumption in our model, the male contacts/partnership, φM, was reduced by 20%. In a given population, the numbers of heterosexual interactions must equate between males and females. The balancing equation applied here is SAF⋅mF⋅NF = SAM⋅mM⋅NM, where SAj are the percent sexually active and Nj are the total in the populations for gender j. To specify changes in partner acquisition, we apply a male flexibility mechanism, holding the female rate of acquisition constant and allowing the male rates to vary (36, 37).
Transmission probabilities.
The effect of a genetic factor in a model of HIV transmission can be included by reducing the transmission coefficient. The probabilities of transmission per contact with an infected partner, βî,ĵ,k̂→i,j, have been estimated in the literature (see ref. 38 for estimates in minimally treated groups). We want to capture a decreased risk in transmission based on genotype (ref. 39, Table 2). No studies have directly evaluated differences in infectivity between HIV-infected CCR5 W/Δ32 heterozygotes and HIV-infected CCR5 wild types. Thus, we base estimates for reduced transmission on studies of groups with various HIV serum viral loads (40), HTLV-I/II viral loads (41), and a study of the effect of AZT treatment on transmission (29). We decrease transmission probabilities for infecting CCR5Δ32/Δ32 persons by 100-fold to reflect the rarity of infections in these persons. However, we assume that infected CCR5Δ32/Δ32 homozygotes can infect susceptibles at a rate similar to CCR5W/W homozygotes, as the former generally have high viremias (ref. 30, Table 2). We also assume that male-to-female transmission is twice as efficient as female-to-male transmission (up to a 9-fold difference has been reported; ref. 42) (ref. 43, Table 2).
Given the assumption of no treatment, the high burden of disease in people with AIDS is assumed to greatly limit their sexual activity. Our initial model excludes people with AIDS from the sexually active groups. Subsequently, we allow persons with AIDS to be sexually active, fixing their transmission rates (βAIDS) to be the same across all CCR5 genotypes, and lower than transmission rates for primary-stage infection (as the viral burden on average is not as high as during the acute phase), and larger than transmission rates for asymptomatic-stage infection (as the viral burden characteristically increases during the end stage of disease).
Disease progression.
We assume three stages of HIV infection: primary (acute, stage A), asymptomatic HIV (stage B), and AIDS. The rates of transition through the first two stages are denoted by γi,j,k, where i represents genotype, j is male/female, and k represents either stage A or stage B. Transition rates through each of these stages are assumed to be inversely proportional to the duration of that stage; however, other distributions are possible (31, 44, 45). Although viral loads generally peak in the first 2 months of infection, steady-state viral loads are established several months beyond this (46). For group A, the primary HIV-infecteds, duration is assumed to be 3.5 months. Based on results from European cohort studies (7–10), the beneficial effects of the CCR5 W/Δ32 genotype are observed mainly in the asymptomatic years of HIV infection; ≈7 years after seroconversion survival rates appear to be quite similar between heterozygous and homozygous individuals. We also assume that CCR5Δ32/Δ32-infected individuals and wild-type individuals progress similarly, and that men and women progress through each disease stage at the same rate. Given these observations, and that survival after infection may be shorter in untreated populations, we choose the duration time in stage B to be 6 years for wild-type individuals and 8 years for heterozygous individuals. Transition through AIDS, δAIDS, is inversely proportional to the duration of AIDS. We estimate this value to be 1 year for the time from onset of AIDS to death. The progression rates are summarized in Table 3.
Table 3.
Progression rates
| Genotype | Disease stage | Males/females |
|---|---|---|
| W/W | A | 3.5 |
| B | 0.16667 | |
| W/Δ32 | A | 3.5 |
| B | 0.125 | |
| Δ32/Δ32 | A | 3.5 |
| B | 0.16667 |
Shown are the rates of progression, γi,j,k reflecting the different rates at which persons progress through different stages of disease by genotype, gender, and disease stage.
Demographic Setting.
Demographic parameters are based on data from Malawi, Zimbabwe, and Botswana (3, 47). Estimated birth and child mortality rates are used to calculate the annual numbers of children (χi,j) maturing into the potentially sexually active, susceptible group at the age of 15 years (3). For example, in the case where the mother is CCR5 wild type and the father is CCR5 wild type or heterozygous, the number of CCR5 W/W children is calculated as follows [suppressing (t) notation]: χ1,j =
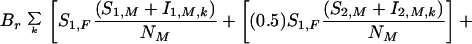 |
 |
where the probability of HIV vertical transmission, 1 − pv, and the birthrate, Br, are both included in the equations together with the Mendelian inheritance values as presented in Table 1. The generalized version of this equation (i.e., χi,j) can account for six categories of children (including gender and genotype). We assume that all children of all genotypes are at risk, although we can relax this condition if data become available to support vertical protection (e.g., ref. 48). All infected children are assumed to die before age 15. Before entering the susceptible group at age 15, there is additional loss because of mortality from all non-AIDS causes occurring less than 15 years of age at a rate of μχ × χi,j (where μχ is the mortality under 15 years of age). Children then enter the population as susceptibles at an annual rate, σj × χi,j/15, where σj distributes the children 51% females and 49% males. All parameters and their values are summarized in Table 4.
Prevalence of HIV
Demographics and Model Validation.
The model was validated by using parameters estimated from available demographic data. Simulations were run in the absence of HIV infection to compare the model with known population growth rates. Infection was subsequently introduced with an initial low HIV prevalence of 0.5% to capture early epidemic behavior.
In deciding on our initial values for parameters during infection, we use Joint United Nations Programme on HIV/AIDS national prevalence data for Malawi, Zimbabwe, and Botswana. Nationwide seroprevalence of HIV in these countries varies from ≈11% to over 20% (3), although there may be considerable variation within given subpopulations (2, 49).
In the absence of HIV infection, the annual percent population growth rate in the model is ≈2.5%, predicting the present-day values for an average of sub-Saharan African cities (data not shown). To validate the model with HIV infection, we compare our simulation of the HIV epidemic to existing prevalence data for Kenya and Mozambique (http://www.who.int/emc-hiv/fact-sheets/pdfs/kenya.pdf and ref. 51). Prevalence data collected from these countries follow similar trajectories to those predicted by our model (Fig. 2).
Figure 2.

Model simulation of HIV infection in a population lacking the protective CCR5Δ32 allele compared with national data from Kenya (healthy adults) and Mozambique (blood donors, ref. 17). The simulated population incorporates parameter estimates from sub-Saharan African demographics. Note the two outlier points from the Mozambique data were likely caused by underreporting in the early stages of the epidemic.
Effects of the Allele on Prevalence.
After validating the model in the wild type-only population, both CCR5Δ32 heterozygous and homozygous people are included. Parameter values for HIV transmission, duration of illness, and numbers of contacts per partner are assumed to be the same within both settings. We then calculate HIV/AIDS prevalence among adults for total HIV/AIDS cases.
Although CCR5Δ32/Δ32 homozygosity is rarely seen in HIV-positive populations (prevalence ranges between 0 and 0.004%), 1–20% of people in HIV-negative populations of European descent are homozygous. Thus, to evaluate the potential impact of CCR5Δ32, we estimate there are 19% CCR5 W/Δ32 heterozygous and 1% CCR5 Δ32/Δ32 homozygous people in our population. These values are in Hardy-Weinberg equilibrium with an allelic frequency of the mutation as 0.105573.
Fig. 3 shows the prevalence of HIV in two populations: one lacking the mutant CCR5 allele and another carrying that allele. In the population lacking the protective mutation, prevalence increases logarithmically for the first 35 years of the epidemic, reaching 18% before leveling off.
Figure 3.

Prevalence of HIV/AIDS in the adult population as predicted by the model. The top curve (○) indicates prevalence in a population lacking the protective allele. We compare that to a population with 19% heterozygous and 1% homozygous for the allele (implying an allelic frequency of 0.105573. Confidence interval bands (light gray) are shown around the median simulation (⧫) providing a range of uncertainty in evaluating parameters for the effect of the mutation on the infectivity and the duration of asymptomatic HIV for heterozygotes.
In contrast, when a proportion of the population carries the CCR5Δ32 allele, the epidemic increases more slowly, but still logarithmically, for the first 50 years, and HIV/AIDS prevalence reaches ≈12% (Fig. 3). Prevalence begins to decline slowly after 70 years.
In the above simulations we assume that people with AIDS are not sexually active. However, when these individuals are included in the sexually active population the severity of the epidemic increases considerably (data not shown). Consistent with our initial simulations, prevalences are still relatively lower in the presence of the CCR5 mutation.
Because some parameters (e.g., rate constants) are difficult to estimate based on available data, we implement an uncertainty analysis to assess the variability in the model outcomes caused by any inaccuracies in estimates of the parameter values with regard to the effect of the allelic mutation. For these analyses we use Latin hypercube sampling, as described in refs. 52–56, Our uncertainty and sensitivity analyses focus on infectivity vs. duration of infectiousness. To this end, we assess the effects on the dynamics of the epidemic for a range of values of the parameters governing transmission and progression rates: βî,ĵ,k̂→i,j and γi,j,k. All other parameters are held constant. These results are presented as an interval band about the average simulation for the population carrying the CCR5Δ32 allele (Fig. 3). Although there is variability in the model outcomes, the analysis indicates that the overall model predictions are consistent for a wide range of transmission and progression rates. Further, most of the variation observed in the outcome is because of the transmission rates for both heterosexual males and females in the primary stage of infection (β2,M,A→i,F, β2,F,A→i,M). As mentioned above, we assume lower viral loads correlate with reduced infectivity; thus, the reduction in viral load in heterozygotes has a major influence on disease spread.
HIV Induces Selective Pressure on Genotype Frequency
To observe changes in the frequency of the CCR5Δ32 allele in a setting with HIV infection as compared with the Hardy-Weinberg equilibrium in the absence of HIV, we follow changes in the total number of CCR5Δ32 heterozygotes and homozygotes over 1,000 years (Fig. 4). We initially perform simulations in the absence of HIV infection as a negative control to show there is not significant selection of the allele in the absence of infection. To determine how long it would take for the allelic frequency to reach present-day levels (e.g., q = 0.105573), we initiate this simulation for 1,000 years with a very small allelic frequency (q = 0.00105). In the absence of HIV, the allelic frequency is maintained in equilibrium as shown by the constant proportions of CCR5Δ32 heterozygotes and homozygotes (Fig. 4, solid lines). The selection for CCR5Δ32 in the presence of HIV is seen in comparison (Fig. 4, dashed lines). We expand the time frame of this simulation to 2,000 years to view the point at which the frequency reaches present levels (where q ∼0.105573 at year = 1200). Note that the allelic frequency increases for ≈1,600 years before leveling off.
Figure 4.

Effects of HIV-1 on selection of the CCR5Δ32 allele. The Hardy-Weinberg equilibrium level is represented in the no-infection simulation (solid lines) for each population. Divergence from the original Hardy-Weinberg equilibrium is shown to occur in the simulations that include HIV infection (dashed lines). Fraction of the total subpopulations are presented: (A) wild types (W/W), (B) heterozygotes (W/Δ32), and (C) homozygotes (Δ32/Δ32). Note that we initiate this simulation with a much lower allelic frequency (0.00105) than used in the rest of the study to better exemplify the actual selective effect over a 1,000-year time scale. (D) The allelic selection effect over a 2,000-year time scale.
Discussion
This study illustrates how populations can differ in susceptibility to epidemic HIV/AIDS depending on a ubiquitous attribute such as a prevailing genotype. We have examined heterosexual HIV epidemics by using mathematical models to assess HIV transmission in dynamic populations either with or without CCR5Δ32 heterozygous and homozygous persons. The most susceptible population lacks the protective mutation in CCR5. In less susceptible populations, the majority of persons carrying the CCR5Δ32 allele are heterozygotes. We explore the hypothesis that lower viral loads (CCR5Δ32 heterozygotes) or resistance to infection (CCR5Δ32 homozygotes) observed in persons with this coreceptor mutation ultimately can influence HIV epidemic trends. Two contrasting influences of the protective CCR5 allele are conceivable: it may limit the epidemic by decreasing the probability of infection because of lower viral loads in infected heterozygotes, or it may exacerbate the epidemic by extending the time that infectious individuals remain in the sexually active population. Our results strongly suggest the former. Thus, the absence of this allele in Africa could explain the severity of HIV disease as compared with populations where the allele is present.
We also observed that HIV can provide selective pressure for the CCR5Δ32 allele within a population, increasing the allelic frequency. Other influences may have additionally selected for this allele. Infectious diseases such as plague and small pox have been postulated to select for CCR5Δ32 (57, 58). For plague, relatively high levels of CCR5Δ32 are believed to have arisen within ≈4,000 years, accounting for the prevalence of the mutation only in populations of European descent. Smallpox virus uses the CC-coreceptor, indicating that direct selection for mutations in CCR5 may have offered resistance to smallpox. Given the differences in the epidemic rates of plague (59), smallpox, and HIV, it is difficult to directly compare our results to these findings. However, our model suggests that the CCR5Δ32 mutation could have reached its present allelic frequency in Northern Europe within this time frame if selected for by a disease with virulence patterns similar to HIV. Our results further support the idea that HIV has been only recently introduced as a pathogen into African populations, as the frequency of the protective allele is almost zero, and our model predicts that selection of the mutant allele in this population by HIV alone takes at least 1,000 years. This prediction is distinct from the frequency of the CCR5Δ32 allele in European populations, where pathogens that may have influenced its frequency (e.g., Yersinia pestis) have been present for much longer.
Two mathematical models have considered the role of parasite and host genetic heterogeneity with regard to susceptibility to another pathogen, namely malaria (60, 61). In each it was determined that heterogeneity of host resistance facilitates the maintenance of diversity in parasite virulence. Given our underlying interest in the coevolution of pathogen and host, we focus on changes in a host protective mutation, holding the virulence of the pathogen constant over time.
Even within our focus on host protective mutations, numerous genetic factors, beneficial or detrimental, could potentially influence epidemics. Other genetically determined host factors affecting HIV susceptibility and disease progression include a CCR5 A/A to G/G promoter polymorphism (62), a CCR2 point mutation (11, 63), and a mutation in the CXCR4 ligand (64). The CCR2b mutation, CCR264I, is found in linkage with at least one CCR5 promoter polymorphism (65) and is prevalent in populations where CCR5Δ32 is nonexistent, such as sub-Saharan Africa (63). However, as none of these mutations have been consistently shown to be as protective as the CCR5Δ32 allele, we simplified our model to incorporate only the effect of CCR5Δ32. Subsequent models could be constructed from our model to account for the complexity of multiple protective alleles. It is interesting to note that our model predicts that even if CCR264I is present at high frequencies in Africa, its protective effects may not augment the lack of a protective allele such as CCR5Δ32.
Although our models demonstrate that genetic factors can contribute to the high prevalence of HIV in sub-Saharan Africa, demographic factors are also clearly important in this region. Our models explicitly incorporated such factors, for example, lack of treatment availability. Additional factors were implicitly controlled for by varying only the presence of the CCR5Δ32 allele. More complex models eventually could include interactions with infectious diseases that serve as cofactors in HIV transmission. The role of high sexually transmitted disease prevalences in HIV infection has long been discussed, especially in relation to core populations (15, 50, 66). Malaria, too, might influence HIV transmission, as it is associated with transient increases in semen HIV viral loads and thus could increase the susceptibility of the population to epidemic HIV (16).
In assessing the HIV/AIDS epidemic, considerable attention has been paid to the influence of core groups in driving sexually transmitted disease epidemics. Our results also highlight how characteristics more uniformly distributed in a population can affect susceptibility. We observed that the genotypic profile of a population affects its susceptibility to epidemic HIV/AIDS. Additional studies are needed to better characterize the influence of these genetic determinants on HIV transmission, as they may be crucial in estimating the severity of the epidemic in some populations. This information can influence the design of treatment strategies as well as point to the urgency for education and prevention programs.
Acknowledgments
We thank Mark Krosky, Katia Koelle, and Kevin Chung for programming and technical assistance. We also thank Drs. V. J. DiRita, P. Kazanjian, and S. M. Blower for helpful comments and discussions. We thank the reviewers for extremely insightful comments.
References
- 1.Weiss H A, Hawkes S. Leprosy Rev. 2001;72:92–98. doi: 10.5935/0305-7518.20010016. [DOI] [PubMed] [Google Scholar]
- 2.Taha T E, Dallabetta G A, Hoover D R, Chiphangwi J D, Mtimavalye L A R. AIDS. 1998;12:197–203. doi: 10.1097/00002030-199802000-00010. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization/UNAIDS. AIDS Epidemic Update. Geneva: World Health Organization; 1998. pp. 1–17. [Google Scholar]
- 4.D'Souza M P, Harden V A. Nat Med. 1996;2:1293–1300. doi: 10.1038/nm1296-1293. [DOI] [PubMed] [Google Scholar]
- 5.Martinson J J, Chapman N H, Rees D C, Liu Y T, Clegg J B. Nat Genet. 1997;16:100–103. doi: 10.1038/ng0597-100. [DOI] [PubMed] [Google Scholar]
- 6.Roos M T L, Lange J M A, deGoede R E Y, Miedema P T, Tersmette F, Coutinho M, Schellekens R A. J Infect Dis. 1992;165:427–432. doi: 10.1093/infdis/165.3.427. [DOI] [PubMed] [Google Scholar]
- 7.Garred P, Eugen-Olsen J, Iversen A K N, Benfield T L, Svejgaard A, Hofmann B. Lancet. 1997;349:1884. doi: 10.1016/s0140-6736(05)63874-3. [DOI] [PubMed] [Google Scholar]
- 8.Katzenstein T L, Eugen-Olsen J, Hofman B, Benfield T, Pedersen C, Iversen A K, Sorensen A M, Garred P, Koppelhus U, Svejgaard A, Gerstoft J. J Acquired Immune Defic Syndr Hum Retrovirol. 1997;16:10–14. doi: 10.1097/00042560-199709010-00002. [DOI] [PubMed] [Google Scholar]
- 9.deRoda H, Meyer K, Katzenstain W, Dean M. Science. 1996;273:1856–1862. [Google Scholar]
- 10.Meyer L, Magierowska M, Hubert J B, Rouzioux C, Deveau C, Sanson F, Debre P, Delfraissy J F, Theodorou I. AIDS. 1997;11:F73–F78. doi: 10.1097/00002030-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Smith M W, Dean M, Carrington M, Winkler C, Huttley D A, Lomb G A, Goedert J J, O'Brien T R, Jacobson L P, Kaslow R, et al. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 12.Samson M, Libert F, Doranz B J, Rucker J, Liesnard C, Farber C M, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, et al. Nature (London) 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 13.McNicholl J M, Smith D K, Qari S H, Hodge T. Emerging Infect Dis. 1997;3:261–271. doi: 10.3201/eid0303.970302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michael N L, Chang G, Louie L G, Mascola J R, Dondero D, Birx D L, Sheppard H W. Nat Med. 1997;3:338–340. doi: 10.1038/nm0397-338. [DOI] [PubMed] [Google Scholar]
- 15.Mayaud P, Mosha F, Todd J, Balira R, Mgara J, West B, Rusizoka M, Mwijarubi E, Gabone R, Gavyole A, et al. AIDS. 1997;11:1873–1880. doi: 10.1097/00002030-199715000-00013. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman I F, Jere C S, Taylor T E, Munthali P, Dyer J R. AIDS. 1998;13:487–494. doi: 10.1097/00002030-199903110-00007. [DOI] [PubMed] [Google Scholar]
- 17.U.S. Bureau of the Census. HIV/AIDS Surveillance Database. International Programs Center, Washington, DC: Population Division; 1999. [Google Scholar]
- 18.Anderson R M, May R M, McLean A R. Nature (London) 1988;332:228–234. doi: 10.1038/332228a0. [DOI] [PubMed] [Google Scholar]
- 19.Berger E A, Doms R W, Fenyo E M, Korber B T, Littman D R, Moore J P, Sattentau Q J, Schuitemaker H, Sodroski J, Weiss R A. Nature (London) 1998;391:240. doi: 10.1038/34571. [DOI] [PubMed] [Google Scholar]
- 20.Alkhatib G, Broder C C, Berger E A. J Virol. 1996;70:5487–5494. doi: 10.1128/jvi.70.8.5487-5494.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRosa G, Newman W, et al. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 22.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, et al. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 23.Doranz B J, Rucker J, Yi Y, Smyth R J, Samsom M, Peiper M, Parmentier S C, Collman R G, Doms R W. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 24.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 25.Zhu T, Mo H, Wang N, Nam D S, Cao Y, Koup R A, Ho D D. Science. 1993;261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 26.Bjorndal A, Deng H, Jansson M, Fiore J R, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman D R, Fenyo E M. J Virol. 1997;71:7478–7487. doi: 10.1128/jvi.71.10.7478-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conner R I, Sheridan K E, Ceradinin D, Choe S, Landau N R. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 29.Mussico M, Lazzarin A, Nicolosi A, Gasparini M, Costigliola P, Arici C, Saracco A. Arch Intern Med (Moscow) 1994;154:1971–1976. [PubMed] [Google Scholar]
- 30.Michael N L, Nelson J A, KewalRamani V N, Chang G, O'Brien S J, Mascola J R, Volsky B, Louder M, White G C, Littman D R, et al. J Virol. 1998;72:6040–6047. doi: 10.1128/jvi.72.7.6040-6047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hethcote H W, Yorke J A. Gonorrhea Transmission Dynamics and Control. Berlin: Springer; 1984. [Google Scholar]
- 32.Anderson R M, May R M. Nature (London) 1988;333:514–522. doi: 10.1038/333514a0. [DOI] [PubMed] [Google Scholar]
- 33.Asiimwe-Okiror G, Opio A A, Musinguzi J, Madraa E, Tembo G, Carael M. AIDS. 1997;11:1757–1763. doi: 10.1097/00002030-199714000-00013. [DOI] [PubMed] [Google Scholar]
- 34.Carael M, Cleland J, Deheneffe J C, Ferry B, Ingham R. AIDS. 1995;9:1171–1175. doi: 10.1097/00002030-199510000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Blower S M, Boe C. J AIDS. 1993;6:1347–1352. [PubMed] [Google Scholar]
- 36.Kirschner D. J Appl Math. 1996;56:143–166. [Google Scholar]
- 37.Le Pont F, Blower S. J AIDS. 1991;4:987–999. [PubMed] [Google Scholar]
- 38.Kim M Y, Lagakos S W. Ann Epidemiol. 1990;1:117–128. doi: 10.1016/1047-2797(90)90003-b. [DOI] [PubMed] [Google Scholar]
- 39.Anderson R M, May R M. Infectious Disease of Humans: Dynamics and Control. Oxford: Oxford Univ. Press; 1992. [Google Scholar]
- 40.Ragni M V, Faruki H, Kingsley L A. J Acquired Immune Defic Syndr. 1998;17:42–45. doi: 10.1097/00042560-199801010-00006. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan J E, Khabbaz R F, Murphy E L, Hermansen S, Roberts C, Lal R, Heneine W, Wright D, Matijas L, Thomson R, et al. J Acquired Immune Defic Syndr Hum Retrovirol. 1996;12:193–201. doi: 10.1097/00042560-199606010-00014. [DOI] [PubMed] [Google Scholar]
- 42.Padian N S, Shiboski S C, Glass S O, Vittinghoff E. Am J Edu. 1997;146:350–357. doi: 10.1093/oxfordjournals.aje.a009276. [DOI] [PubMed] [Google Scholar]
- 43.Leynaert B, Downs A M, de Vincenzi I. Am J Edu. 1998;148:88–96. doi: 10.1093/oxfordjournals.aje.a009564. [DOI] [PubMed] [Google Scholar]
- 44.Garnett G P, Anderson R M. J Acquired Immune Defic Syndr. 1995;9:500–513. [PubMed] [Google Scholar]
- 45.Stigum H, Magnus P, Harris J R, Samualson S O, Bakketeig L S. Am J Edu. 1997;145:636–643. doi: 10.1093/oxfordjournals.aje.a009161. [DOI] [PubMed] [Google Scholar]
- 46.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 47.World Resources Institute. World Resources (1998–1999) Oxford: Oxford Univ. Press; 1999. [Google Scholar]
- 48.Kostrikis L G, Neumann A U, Thomson B, Korber B T, McHardy P, Karanicolas R, Deutsch L, Huang Y, Lew J F, McIntosh K, et al. J Virol. 1999;73:10264–10271. doi: 10.1128/jvi.73.12.10264-10271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Low-Beer D, Stoneburner R L, Mukulu A. Nat Med. 1997;3:553–557. doi: 10.1038/nm0597-553. [DOI] [PubMed] [Google Scholar]
- 50.Grosskurth H, Mosha F, Todd J, Senkoro K, Newell J, Klokke A, Changalucha J, West B, Mayaud P, Gavyole A. AIDS. 1995;9:927–934. doi: 10.1097/00002030-199508000-00015. [DOI] [PubMed] [Google Scholar]
- 51.Melo J, Beby-Defaux A, Faria C, Guiraud G, Folgosa E, Barreto A, Agius G. J AIDS. 2000;23:203–204. doi: 10.1097/00126334-200002010-00013. [DOI] [PubMed] [Google Scholar]
- 52.Iman R L, Helton J C, Campbell J E. J Quality Technol. 1981;13:174–183. [Google Scholar]
- 53.Iman R L, Helton J C, Campbell J E. J Quality Technol. 1981;13:232–240. [Google Scholar]
- 54.Blower S M, Dowlatabadi H. Int Stat Rev. 1994;62:229–243. [Google Scholar]
- 55.Porco T C, Blower S M. Theor Popul Biol. 1998;54:117–132. doi: 10.1006/tpbi.1998.1366. [DOI] [PubMed] [Google Scholar]
- 56.Blower S M, Porco T C, Darby G. Nat Med. 1998;4:673–678. doi: 10.1038/nm0698-673. [DOI] [PubMed] [Google Scholar]
- 57.Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, Czeizel A, Claustres M, de la Rua C, Ferrari M, et al. Hum Mol Genet. 1998;7:399–406. doi: 10.1093/hmg/7.3.399. [DOI] [PubMed] [Google Scholar]
- 58.Lalani A S, Masters J, Zeng W, Barrett J, Pannu R, Everett H, Arendt C W, McFadden G. Science. 1999;286:1968–1971. doi: 10.1126/science.286.5446.1968. [DOI] [PubMed] [Google Scholar]
- 59.Kermack W O, McKendrick A G. Proc R Soc London. 1927;261:700–721. [Google Scholar]
- 60.Gupta S, Hill A V S. Proc R Soc London Ser B. 1995;260:271–277. [Google Scholar]
- 61.Ruwende C, Khoo S C, Snow R W, Yates S N R, Kwiatkowski D, Gupta S, Warn P, Allsopp C E, Gilbert S C, Peschu N. Nature (London) 1995;376:246–249. doi: 10.1038/376246a0. [DOI] [PubMed] [Google Scholar]
- 62.McDermott D H, Zimmerman P A, Guignard F, Kleeberger C A, Leitman S F, Murphy P M. Lancet. 1998;352:866–870. doi: 10.1016/s0140-6736(98)04158-0. [DOI] [PubMed] [Google Scholar]
- 63.Kostrikis L G, Huang Y, Moore J P, Wolinsky S M, Zhang L, Guo Y, Deutsch L, Phair J, Neumann A U, Ho D D. Nat Med. 1998;4:350–353. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- 64.Winkler C, Modi W, Smith M W, Nelson G W, Wu X, Carrington M, Dean M, Honjo T, Tashiro K, Yabe D, et al. Science. 1998;279:389–393. doi: 10.1126/science.279.5349.389. [DOI] [PubMed] [Google Scholar]
- 65.Martinson J J, Hong L, Karanicolas R, Moore J P, Kostrikis L G. AIDS. 2000;14:483–489. doi: 10.1097/00002030-200003310-00003. [DOI] [PubMed] [Google Scholar]
- 66.Vernazza P L, Eron J J, Fiscus S A, Cohen M S. AIDS. 1999;13:155–166. doi: 10.1097/00002030-199902040-00003. [DOI] [PubMed] [Google Scholar]