

Abstract
Purpose
Neuropathic pain is a complex chronic condition occurring post-nervous system damage. The transcriptional reprogramming of injured dorsal root ganglia (DRGs) drives neuropathic pain. However, few comparative analyses using high-throughput platforms have investigated uninjured DRG in neuropathic pain, and potential interactions among differentially expressed genes (DEGs) and pathways were not taken into consideration. The aim of this study was to identify changes in genes and pathways associated with neuropathic pain in uninjured L4 DRG after L5 spinal nerve ligation (SNL) by using bioinformatic analysis.
Materials and methods
The microarray profile GSE24982 was downloaded from the Gene Expression Omnibus database to identify DEGs between DRGs in SNL and sham rats. The prioritization for these DEGs was performed using the Toppgene database followed by gene ontology and pathway enrichment analyses. The relationships among DEGs from the protein interactive perspective were analyzed using protein–protein interaction (PPI) network and module analysis. Real-time polymerase chain reaction (PCR) and Western blotting were used to confirm the expression of DEGs in the rodent neuropathic pain model.
Results
A total of 206 DEGs that might play a role in neuropathic pain were identified in L4 DRG, of which 75 were upregulated and 131 were downregulated. The upregulated DEGs were enriched in biological processes related to transcription regulation and molecular functions such as DNA binding, cell cycle, and the FoxO signaling pathway. Ctnnb1 protein had the highest connectivity degrees in the PPI network. The in vivo studies also validated that mRNA and protein levels of Ctnnb1 were upregulated in both L4 and L5 DRGs.
Conclusion
This study provides insight into the functional gene sets and pathways associated with neuropathic pain in L4 uninjured DRG after L5 SNL, which might promote our understanding of the molecular mechanisms underlying the development of neuropathic pain.
Keywords: spinal nerve ligation, neuropathic pain, uninjured afferent, bioinformatic analysis, microarray
Introduction
Neuropathic pain is a complex chronic condition usually resulting from damage or disease affecting the nervous system. Up to 10% of the general population suffers from neuropathic pain.1 Current treatments might relieve no more than 50% of pain intensity for only 30%–40% of patients in clinical practice.2,3 Therefore, identification of new and efficacious therapeutics toward neuropathic pain represents a major unmet medical need.
The transcriptional reprogramming of dorsal root ganglia (DRGs) following sensory nerve damage is considered to drive neuropathic pain.4 In the common neuropathic pain models that involve an injury to one or more peripheral nerves, the DRG in injured nerve exhibits abnormal gene expression and protein products that may play a role in neuropathic pain.5 However, accumulating evidence indicates that changes in multiple genes and cellular pathways in DRG in adjacent, uninjured areas may also contribute to the occurrence and development of neuropathic pain.6 For example, the expression of some G protein-coupled receptors and ion channel molecules was changed in uninjured L4 DRGs after L5 spinal nerve ligation (SNL).7,8 In addition, some targets such as glial cell-derived neurotrophic factor (GDNF) might exert their analgesic effects by acting on the central terminals of uninjured L4 DRG neurons instead of injured L5 DRG neurons in an L5 SNL model.9 Therefore, identifying the change in key transcripts and their protein products in uninjured DRG likely to initiate and sustain neuropathic pain might provide new insights into the underlying mechanism and treatment of this disease.
In recent decades, gene expression profiling for neuropathic pain has been performed using microarray technology, and hundreds of differentially expressed genes (DEGs) in different pathways, biological processes (BPs), or molecular functions (MFs) were identified.10 However, little comparative analyses by high-throughput platforms have been performed on uninjured DRGs in neuropathic pain, and potential interactions among DEGs and pathways were not taken into consideration. Notably, microarray technology combined with bioinformatic analysis allows comprehensive analysis of the expression changes of genes in neuropathic pain and other diseases such as cancer.10,11 We reanalyzed microarray data (GSE24982) submitted by von Schack et al12 from the Gene Expression Omnibus (GEO) database using bioinformatic technologies to identify the overall gene expression and pathway alterations in uninjured DRG and provide a more thorough insight into the pathological changes in neuropathic pain at the molecular level.
Materials and methods
Microarray data
We downloaded the microarray profile GSE24982 from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The microarray data were collected from the L4 DRGs in L5 SNL (N=5) or sham rats (N=5). The gene annotation platform was based on GPL1355 (Affymetrix Rat Genome 230 2.0 Array).
Microarray data preprocessing and DEG identification
Preprocessing for the cell intensity (CEL) files including conversion into expression measures and background correction, and quartile data normalization was performed using BRB-ArrayTools (version 4.5.1).13,14 The DEGs between SNL and sham samples were identified using the unpaired t-test. A fold change ≥2.0 and nominal significance level of 0.05 were applied in the BRB-ArrayTools to identify the DEGs between the SNL and sham rats.
Prioritization for the DEGs
To evaluate whether the DEGs obtained in the GEO dataset analysis might play a role in neuropathic pain, the prioritization for these DEGs was performed using the Toppgene database (http://toppgene.cchmc.org) with the threshold of P<0.05. Toppgene is an online tool used for identification and prioritization of novel disease candidate genes in the interactome.15 The set of training genes, which was obtained from the published literature to train the software, was mined and integrated from the online database GeneCards (http://genecards.org) by searching for the keywords “neuropathic pain.”16
Gene ontology and pathway enrichment analyses
To identify the prioritized DEGs obtained from the Toppgene database, the genes and their products were annotated using gene ontology (GO) annotation analysis, and the signaling pathways were enriched using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. GO term and KEGG pathway analysis were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/summary.jsp) online database.17,18 P<0.05 was considered statistically significant.
Construction of protein–protein interaction (PPI) network and module analysis
To evaluate the relationships among prioritized DEGs from the protein interactive perspective, we used the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; http://www.string-db.org) to construct and identify PPI information among these genes.19 There are 9,643,763 proteins from 2,031 organisms in the current version of the STRING database. A combination of more than 0.4 was considered the reliability threshold for PPI. The PPI network was then constructed and visualized using the Cytoscape software. The importance of a protein in the PPI network was determined by its connectivity degree, namely the number of the proteins it connected, according to a previous study.10
In addition, module analysis was performed using the plugin ClusterONE in Cytoscape with the threshold of P<0.001 to obtain subnetworks (modules). For genes in each module, protein domain enrichment analysis was performed using the InterPro database (http://www.ebi.ac.uk/interpro/) with the threshold of P<0.01.20
Expression confirmation of pain-related DEGs in uninjured L4 DRG
Rodent pain model
Healthy Sprague Dawley male rats weighting 200–250 g were provided by the Experiment Animal Center of Guangdong Province (SCXK Yue 2011-0015), housed at 23±2°C in separate cages with water, and fed ad libitum in a 12-hour reverse light cycle. All experimental protocols were performed in line with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals. The study was approved by the Animal Care Committee of Sun Yat-sen University. We made every possible effort to minimize unnecessary suffering of animals.
The rats were randomly assigned into a sham-operated group (sham) and SNL group (n=42 for each group). The SNL model was created in this study as described by Kim and Chung.21 In short, a midline incision was made on the back of the rat from the left L4 to S2 level. From the exposed L4 and L5 spinal nerves, the left para-spinal muscles and L5 transverse processes were carefully removed. Then, we ligated the L5 spinal nerve with a suture. For the sham rats, the same procedure was performed expect that the left L5 spinal nerve was only isolated without ligation.
Behavioral tests
As rats were more sensitive to mechanical stimulation than thermal stimulation after the ligation of the L5 spinal nerve,22 50% mechanical withdrawal thresholds (50% MWTs) were evaluated based on the up–down method for all rats before SNL surgery as basal responsiveness (day 0), and 1, 3, 5, 7, 10, and 14 days after SNL surgery.23 To acclimate to the testing environment, animals were placed on the test mesh floor at least 30 min prior to the behavioral test. The test was carried out between 9:00 and 13:00 hours. A series of von Frey hair monofilaments (Touch Test Sensory Evaluator; North Coast Medical, San Jose, CA, USA) were used on the webbing between the third and fourth digit of the rat hind paw. The evaluation of mechanical allodynia was carried out by an experimenter who was blinded to the treatment group.
Real-time polymerase chain reaction (RT-PCR)
L4 and L5 DRGs were harvested 0, 3, 7 and 14 days after SNL or sham surgery and immediately frozen on dry ice and stored at −80°C (n=6 at each time point). Total RNA extracted from snap-frozen L4 and L5 DRG tissues was isolated using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA). For the detection of RNA quality and concentration, we used the NanoDrop-1000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription was performed using ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan). Quantitative analysis of catenin beta 1 (Ctnnb1) mRNA was conducted with quantitative RT-PCR (qRT-PCR) using SYBR® Green Realtime PCR Master Mix (Toyobo) with Roche LightCycler 1.1. The sense and antisense oligonucleotide primers were as follows: Ctnnb1, F-CATGGGTGGAACACAGCA, R-CCCAGTGCACCCTTCAACC; Eif4a2, F-GCGGATTACAACAGAGAACATGG, R-CCTCGAAGAAGGGACTCCTTT; Achyl1, F-AATAGTGGGCTGTACGCACAT, R-GCAGCTACTTCATTCTGAGTTGA; Wt1, F-GAAATGGACAGAAGGGCAGA, R-GGGGTTGTGTGGTTCTCACT. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene. Data of transcripts were calculated relative to GAPDH using the 2−ΔΔCt method.24
Western blot
Western blot analysis was used to calculate the Ctnnb1 protein expression following the DRG protein extraction. The sample was solubilized in sodium dodecyl sulfate (SDS)-loading buffer (Bio-Rad Laboratories Inc., Hercules, CA, USA) by boiling. The samples were separated on a 10% polyacrylamide gel (Thermo Fisher Scientific) and SDS-polyacrylamide gel electrophoresis (PAGE; Bio-Rad Laboratories Inc.) and electro-transferred onto a polyvinylidene difluoride (Bio-Rad Laboratories Inc.) membrane. Subsequently, the membrane was blocked with 5% nonfat milk for 4 h at room temperature and was kept at 23±2°C, and then incubated with rabbit anti-Ctnnb1 polyclonal IgG (1:1000; Abcam, Cambridge, UK) and mouse anti-GAPDH monoclonal IgG (1:3000; Abcam) overnight at 4°C. All the blots were subsequently washed and incubated with the respective anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (1:3000; Abcam) or anti-mouse HRP-conjugated antibody (1:3000; Abcam). Images were acquired using the Tanon 5500 imaging system (Tanon, Shanghai, China). The images were scanned using the ImageJ scanning software, and the data are expressed as the values relative to the sham or control value.
Statistical analysis
All data were analyzed using SPSS 16.0 software (SPSS Inc., Chicago, IL, USA). Measurement data were expressed as mean ± standard deviation (SD). Data regarding the MWTs were analyzed statistically using the Mann–Whitney U test. Comparisons of mRNA and protein expressions among the groups were evaluated using one-way analysis of variance (ANOVA) followed by the Tukey’s test. P<0.05 was considered to be statistically significant.
Results
Screening and prioritization for DEGs
According to the criteria of fold change ≥2 and nominal significance level of 0.05, 430 DEGs were identified in L4 DRGs between SNL and sham rats, of which 173 were upregulated and 257 were downregulated.
According to the gene search results in the Genecard database, 470 DEGs were obtained and set as the training gene list, while the 430 DEGs from GSE24982 were set as the test gene set in the Toppgene database. With the criterion P<0.05, 206 candidate DEGs that might play a role in neuropathic pain were screened using the Toppgene database, of which 75 were upregulated and 131 were downregulated. The top 20 ranked genes are summarized in Table 1, and all 206 candidate genes in L4 DRGs are summarized in Table S1.
Table 1.
Results of the top 20 ranked DEGs from Toppgene
| Rank | Gene symbol | Gene name | Fold change (SNL versus sham) | Expression | Overall P-value |
|---|---|---|---|---|---|
| 1 | CAV1 | Caveolin 1 | 0.27 | Downregulated | 4.31E-05 |
| 2 | CTNNB1 | Catenin beta 1 | 4.41 | Upregulated | 8.17E-05 |
| 3 | IRS1 | Insulin receptor substrate 1 | 3.61 | Upregulated | 9.55E-05 |
| 4 | NRG1 | Neuregulin 1 | 0.28 | Downregulated | 1.07E-04 |
| 5 | STIM1 | Stromal interaction molecule 1 | 0.31 | Downregulated | 1.37E-04 |
| 6 | JAK1 | Janus kinase 1 | 0.22 | Downregulated | 2.61E-04 |
| 7 | VCL | Vinculin | 0.28 | Downregulated | 3.92E-04 |
| 8 | CDK6 | Cyclin-dependent kinase 6 | 3.90 | Upregulated | 4.24E-04 |
| 9 | IL6ST | Interleukin 6 signal transducer | 0.36 | Downregulated | 4.36E-04 |
| 10 | SYN1 | Synapsin 1 | 0.24 | Downregulated | 4.37E-04 |
| 11 | HTR3B | 5-Hydroxytryptamine receptor 3B | 0.29 | Downregulated | 4.53E-04 |
| 12 | MDM2 | MDM2 proto-oncogene | 4.31 | Upregulated | 5.01E-04 |
| 13 | STXBP1 | Syntaxin-binding protein 1 | 0.20 | Downregulated | 5.12E-04 |
| 14 | PRKAG2 | Protein kinase AMP-activated non-catalytic subunit gamma 2 | 4.32 | Upregulated | 5.16E-04 |
| 15 | SLC17A7 | Solute carrier family 17 member 7 | 0.30 | Downregulated | 5.91E-04 |
| 16 | Vamp2 | Vesicle-associated membrane protein 2 | 0.26 | Downregulated | 6.07E-04 |
| 17 | TPM1 | Tropomyosin 1 | 0.35 | Downregulated | 6.09E-04 |
| 18 | EEF1A2 | Eukaryotic translation elongation factor 1 alpha 2 | 0.38 | Downregulated | 6.54E-04 |
| 19 | TFRC | Transferrin receptor | 0.28 | Downregulated | 6.96E-04 |
| 20 | ATP2A1 | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 | 0.31 | Downregulated | 4.31E-04 |
Abbreviations: DEGs, differentially expressed genes; SNL, spinal nerve ligation.
GO annotation enrichment and KEGG pathway analysis
The GO annotation enrichment for the 206 prioritized DEGs is summarized in Table 2. The 75 upregulated prioritized genes were mostly enriched in BPs related to regulation of transcription and MF related to binding such as DNA binding. The 131 downregulated prioritized genes were mainly enriched in cell components (CCs) related to synapse and MF related to soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) binding as well as soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) activity.
Table 2.
Results of GO annotation enrichment analysis
| Expression | Category | GO term | Count of genes | P-value |
|---|---|---|---|---|
| Upregulated genes | Cluster 1 | |||
| BP | GO:0000122 – negative regulation of transcription from RNA polymerase II promoter | 12 | 1.7E-4 | |
| MF | GO:0043565 – sequence-specific DNA binding | 10 | 6.0E-4 | |
| BP | GO:0045944 – positive regulation of transcription from RNA polymerase II promoter | 13 | 6.8E-4 | |
| BP | GO:0045893 – positive regulation of transcription, DNA templated | 9 | 1.9E-3 | |
| MF | GO:0003682 – chromatin binding | 7 | 1.2E-2 | |
| MF | GO:0003700 – transcription factor activity, sequence-specific DNA binding | 9 | 1.3E-2 | |
| BP | GO:0006355 – regulation of transcription, DNA templated | 10 | 2.4E-2 | |
| BP | GO:0006351 – transcription, DNA templated | 8 | 3.7E-2 | |
| Downregulated genes | Cluster 1 | |||
| CC | GO:0030672 – synaptic vesicle membrane | 7 | 3.4E-6 | |
| CC | GO:0043229 – intracellular organelle | 4 | 1.1E-4 | |
| CC | GO:0008021 – synaptic vesicle | 6 | 2.1E-3 | |
| Cluster 2 | ||||
| MF | GO:0000149 – SNARE binding | 5 | 1.0E-3 | |
| MF | GO:0005484 – SNAP receptor activity | 3 | 3.0E-2 | |
| CC | GO:0031201 – SNARE complex | 3 | 4.7E-2 |
Abbreviations: BP, biological process; CC, cellular component; GO, gene ontology; MF, molecular function; SNAP, soluble N-ethylmaleimide-sensitive factor attachment protein; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptors.
According to the KEGG pathway enrichment analysis, as summarized in Table 3, the upregulated prioritized genes were mainly enriched in cell cycle and the FoxO signaling pathway, while the downregulated prioritized genes were mainly enriched in synaptic vesicle cycle pathway.
Table 3.
Results of KEGG pathway analysis
| Expression | Pathway name | Gene count | P-value | Genes |
|---|---|---|---|---|
| Upregulated genes | rno04110: cell cycle | 4 | 2.4E-2 | Mdm2, Ttk, Cdk6, Smc3 |
| rno04068: FoxO signaling pathway | 4 | 2.8E-2 | Klf2, Mdm2, Irs1, Prkag2 | |
| rno04610: complement and coagulation cascades | 3 | 4.8E-2 | Cd46, F5, C3 | |
| rno04520: adherens junction | 3 | 4.9E-2 | Lmo7, Ctnnb1, Tcf7l2, Vcan | |
| Downregulated genes | rno04721: synaptic vesicle cycle | 4 | 1.9E-2 | Rab3a, Slc17a7, Stxbp1, Vamp2 |
Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network and module screening
The constructed PPI network is shown in Figure 1. There were 138 nodes and 201 edges in the network. The top 10 proteins with relatively high connectivity degrees were as follows: Ctnnb1 (degree=16), ubiquitin-like 4A (Ubl4a, degree=15), transformed mouse 3T3 cell double minute 2 (Mdm2, degree=10), vesicle-associated membrane protein 2 (Vamp2, degree=9), Janus kinase 1 (Jak1, degree=9), insulin receptor substrate 1 (Irs1, degree=9), cyclin-dependent kinase 6 (Cdk6, degree=9), eukaryotic translation initiation factor 4A2 (Eif4a2, degree=8), vesicle-associated membrane protein 1 (Vamp1, degree=8), and ubiquitin-conjugating enzyme E2B (Ube2b, degree=8).
Figure 1.

PPI network of the screened DEGs.
Notes: The gray circles represent upregulated proteins, and the white circles represent downregulated proteins. The size of a protein is determined by the degree of its connection to other proteins, and the width of the edge connecting two proteins is determined by the combined score of two proteins.
Abbreviations: DEGs, differentially expressed genes; PPI, protein–protein interaction.
Based on the module analysis by ClusterONE, four protein modules, module 1 (P=1.687E-4), module 2 (P=5.285E-4), module 3 (P=5.650E-4), and module 4 (P=7.806E-4) were obtained as shown in Figure 2. The crucial nodes with high connectivity degrees in these four modules were Ctnnb1, Eif4a2, Ahcyl1, and Wt1, respectively. For protein domains in the PPI modules, P-loop containing nucleoside triphosphate hydrolase (IPR027417) was detected in module 3, while no domain was enriched for proteins in other modules.
Figure 2.

Protein modules in the PPI network.
Notes: The gray circles represent upregulated proteins, and the white circles represent downregulated proteins. The size of a protein is determined by the degree of its connection to other proteins, and the width of the edge connecting two proteins is determined by the combined score of two proteins. M1–4 represent modules 1–4, respectively.
Abbreviation: PPI, protein–protein interaction.
Behavioral tests for SNL model
The ligation of the L5 spinal nerve led to a significant increase in nociceptive response to mechanical stimuli in the hind paw ipsilateral to the injury. Mechanical allodynia was determined by 50% MWT, which was similar between SNL and sham rats prior to surgery and significantly increased in SNL rats compared to that in the sham rats after L5 SNL (Figure 3 and Table S2).
Figure 3.

Time-course curves of 50% MWT induced by L5 SNL.
Notes: About 50% MWT was measured before (day 0) and 1, 3, 5, 7, 10, and 14 days after surgery. *P<0.05, SNL rats versus sham rats (at each time point, n=6 per group).
Abbreviations: MWT, mechanical withdrawal threshold; SNL, spinal nerve ligation.
DEG expression in the ipsilateral L4 DRGs after L5 SNL
Because Ctnnb1, Eif4a2, Ahcyl1, and Wt1 were the proteins with highest connectivity degrees in the four module analysis by ClusterONE, we then detected the mRNA expressions of the four genes in both ipsilateral L4 and L5 DRGs at 0, 3, 7, and 14 days after L5 SNL. In comparison to the L4 DRG in sham rats, SNL led to a significant enhancement of Ctnnb1, Eif4a2, and Wt1, and a significant reduction in Ahcyl1 mRNA in L4 DRG for SNL rats at 3, 7, and 14 days after surgery. In addition, only the expression of Ctnnb1 statistically increased in both L4 and L5 DRGs after surgery (Figure 4 and Table S3).
Figure 4.

mRNA expression of Ctnnb1, Eif4a2, Ahcyl1, and Wt1 in the L4 and L5 DRGs.
Notes: The histograms show the mRNA expression of Ctnnb1 (A), Eif4a2 (B), Achyl1 (C), and Wt1 (D) in the ipsilateral L4 and L5 DRGs after SNL or sham surgery. Data were normalized with the values of L4 DRGs in the sham group at day 0. At each time point, mRNA bands from left to right represent sham L4 DRGs, sham L5 DRGs, SNL L4 DRGs, and SNL L5 DRGs. *P<0.05, L4 DRGs of SNL rats versus sham rats; #P<0.05, L5 DRGs of SNL rats versus sham rats (at each time point, n=6 per group).
Abbreviations: DRGs, dorsal root ganglia; SNL, spinal nerve ligation.
As Ctnnb1 was also the protein with highest connectivity degrees in the PPI network, the change in Ctnnb1 expression was also observed at protein level. SNL led to a significant increase in Ctnnb1 protein in both ipsilateral L4 and L5 DRGs, with the magnitude of increase being larger in L5 DRG at 3, 7, and 14 days after injury (Figure 5).
Figure 5.
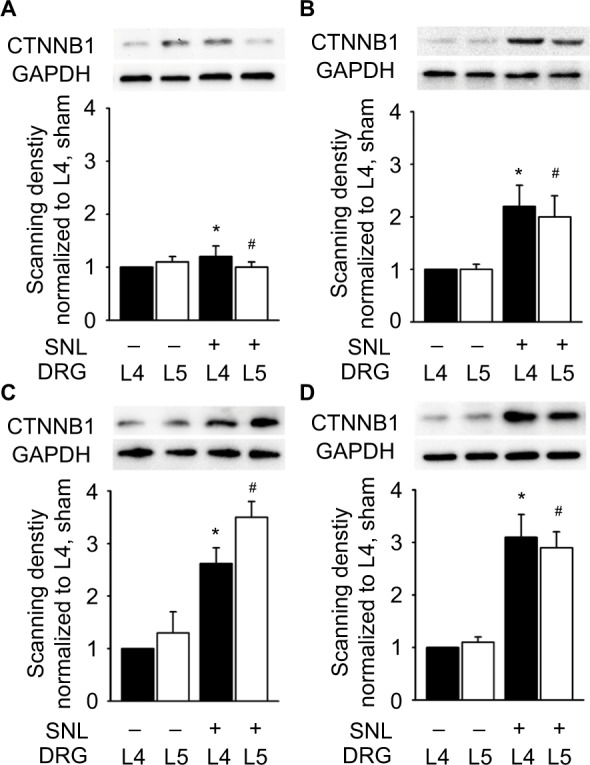
Protein expression of Ctnnb1 in the L4 and L5 DRGs.
Notes: Protein bands and histograms show a significant increase in Ctnnb1 protein in the ipsilateral L4 and L5 DRGs at 0 (A), 3 (B), 7 (C), and 14 (D) days after surgery. Data were normalized with the values of L4 DRGs in the sham group. At each time point, protein bands from left to right represent sham L4 DRGs, sham L5 DRGs, SNL L4 DRGs, and SNL L5 DRGs. *P<0.05, L4 DRGs of SNL rats versus sham rats; #P<0.05, L5 DRGs of SNL rats versus sham rats (at each time point, n=6 per group).
Abbreviations: DRG, dorsal root ganglia; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; SNL, spinal nerve ligation.
Discussion
Neuropathic pain is a complex chronic condition occurring after nervous system injury, but the exact mechanisms remain largely unknown. Accumulating evidence indicates that uninjured DRGs adjacent to the injured one also contribute to the occurrence and development of neuropathic pain. Mas-related G-protein-coupled receptor subtype C (MrgC) is changed in uninjured L4 DRGs after L5 SNL,7 and the expression of brain-derived neurotrophic factor (BDNF) increases in the uninjured DRGs in the L5 SNL model.25 In our study, we used the BRB-ArrayTools to analyze the microarray data and identify 206 candidate genes, including 75 upregulated and 131 downregulated genes. The GO term annotation and KEGG pathway enrichment analysis showed that these prioritized genes were related to SNARE-binding and synaptic vesicle cycle pathway. The primary role of SNARE proteins is to mediate vesicle fusion, including the docking of synaptic vesicles with the presynaptic membrane in neurons, and the disorder of synaptic vesicle fusion is related to the generation and maintenance of neuropathic pain.26 This DEG might also lead to an abnormal synaptic vesicle fusion in L4 DRG, which appears to play a role in neuropathic pain. In addition, the FoxO signaling pathway was also identified in L4 DRG. FoxO transcription factors belong to the large Forkhead family of proteins, a family of transcriptional regulators characterized by a conserved DNA-binding domain termed the “forkhead box”.27 The FoxO signaling pathway not only regulates gene transcription and expression but also plays an important role in cell proliferation, differentiation, and apoptosis in different tissues and cells in human beings and animals.28 Reduction and loss of FoxO transcription factors are related to the lumbar intervertebral disc and play an important role in low back pain.29 Our bioinformatic analysis indicated that the FoxO signaling pathway might be stimulated in the uninjured L4 DRG in L5 SNL model, which might provide new potential mechanism insights for SNL-mediated neuropathic pain.
PPI network and module screenings showed that Ctnnb1 had the highest connectivity degree (degree=16). In addition, Ctnnb1 was a crucial node with the highest connectivity degree in the module analysis. Ctnnb1 is the key downstream component of the canonical Wnt signaling pathway.30 The Wnt/β-catenin signaling pathway plays a significant role in neural development, axonal guidance, neuropathic pain remission, and neuronal survival.31 For example, Ctnnb1 protein is the central player in the pathological process of spinal cord injury (SCI), and it promotes the regeneration of axons and inhibits apoptosis and improves functional recovery after SCI. Ctnnb1 might translocate into the nucleus to bind with the TCF/LEF transcription factor, which might play a role in cell proliferation, cell apoptosis, or differentiation.32 Activation of Wnt/β-catenin signaling inhibits neural cell apoptosis after SCI.33 In addition, several studies reveal that activating Ctnnb1 protein and the Wnt/β-catenin signaling pathway induce axonal regeneration in the central nervous system.34,35 Suppression of the Wnt/β-catenin signaling pathway might induce apoptosis after SCI, and upregulation of Ctnnb1 leading to further activation of the Wnt/β-catenin signaling pathway might promote locomotor recovery after SCI.33,36
In neuropathic pain, Ctnnb1 and Wnt/β-catenin signaling pathways might contribute to the development of pain behavior. Zhang et al37 reported Ctnnb1 upregulation in the dorsal horn of the spinal cord after peripheral nerve injury (ligation of sciatic nerve), and they suggested that the upregulation of Ctnnb1 might be associated with N-methyl-D-aspartic acid receptor (NMDAR) activity and sprouting of Ab-fibers in the dorsal horn. Our results showed that the L5 SNL led to a significant enhancement of Ctnnb1 mRNA and protein expressions in L5 DRGs at 3, 7, and 14 days after injury, which is consistent with the bioinformatic analysis and the results obtained by Zhang et al.37 In addition, the expression of Ctnnb1 mRNA and protein was significantly increased in uninjured L4 DRGs, which suggested that Ctnnb1 might play a role in neuropathic pain in uninjured DRGs after L5 SNL. Moreover, the regulation of expression of some genes in uninjured DRG, such as overexpression of GDNF in L4 DRG after SNL, might exert analgesic effects in the neuropathic pain state by acting on the central terminals of uninjured DRG neurons and/or on the spinal cells targeted by the uninjured DRG neurons.9 Therefore, further molecular biological experiments are needed to confirm whether the Ctnnb1 in uninjured DRG after nerve injury might have therapeutic benefits and relieve symptoms of neuropathic pain.
In addition to Ctnnb1, we found that differential expressions of three crucial nodes with high connectivity degrees (Eif4a2, Ahcyl1, and Wt1) were unique in L4 DRGs, which were also confirmed by RT-PCR. Eif4a is an isoform in the Eif4a protein family, which exhibits RNA-dependent ATPase (adenosine triphosphatase) and bidirectional RNA helicase activity.38 Ahcyl1 is a member of Ahcyl family of proteins that influences the inositol 1,4,5-trisphosphate-induced Ca2+ signaling cascade essential for numerous cellular and physiological processes such as organ development, fertilization, and cell death.39 Wt1 regulates transcription, RNA metabolism, translation, and both oncogenic and tumor suppressor functions, and its overexpression has been reported in various tumors and is predictive of a poor prognosis for some cancers such as breast cancer.40 However, few studies demonstrate the roles of these three genes in either injured or uninjured nerve and nerve terminals in neuropathic pain. Further studies are needed to reveal the contribution of these genes to neuropathic pain.
Conclusion
The current study demonstrated that the DRGs adjacent to the injured one developed many abnormal gene expressions and signaling pathways, among which Ctnnb1 was the crucial protein with highest connectivity degrees. Further molecular biological experiments are required to confirm the function of the identified DEGs in uninjured DRG for neuropathic pain.
Acknowledgments
This Project was supported by the National Natural Science Foundation of China (number 81701104) and the Natural Science Foundation of Guangdong Province, China (number 2016A030310157).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers. 2017;3:17002. doi: 10.1038/nrdp.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backonja MM, Irving G, Argoff C. Rational multidrug therapy in the treatment of neuropathic pain. Curr Pain Headache Rep. 2006;10(1):34–38. doi: 10.1007/s11916-006-0007-1. [DOI] [PubMed] [Google Scholar]
- 3.Finnerup NB, Otto M, McQuay HJ, Jensen TS, Sindrup SH. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289–305. doi: 10.1016/j.pain.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Berger JV, Knaepen L, Janssen SP, et al. Cellular and molecular insights into neuropathy-induced pain hypersensitivity for mechanism-based treatment approaches. Brain Res Rev. 2011;67(1–2):282–310. doi: 10.1016/j.brainresrev.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Zang Y, Chen SX, Liao GJ, et al. Calpain-2 contributes to neuropathic pain following motor nerve injury via up-regulating interleukin-6 in DRG neurons. Brain Behav Immun. 2015;44:37–47. doi: 10.1016/j.bbi.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Djouhri L, Fang X, Koutsikou S, Lawson SN. Partial nerve injury induces electrophysiological changes in conducting (uninjured) nociceptive and nonnociceptive DRG neurons: possible relationships to aspects of peripheral neuropathic pain and paresthesias. Pain. 2012;153(9):1824–1836. doi: 10.1016/j.pain.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He SQ, Han L, Li Z, et al. Temporal changes in MrgC expression after spinal nerve injury. Neuroscience. 2014;261:43–51. doi: 10.1016/j.neuroscience.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith T, Al Otaibi M, Sathish J, Djouhri L. Increased expression of HCN2 channel protein in L4 dorsal root ganglion neurons following axotomy of L5- and inflammation of L4-spinal nerves in rats. Neuroscience. 2015;295:90–102. doi: 10.1016/j.neuroscience.2015.03.041. [DOI] [PubMed] [Google Scholar]
- 9.Takasu K, Sakai A, Hanawa H, Shimada T, Suzuki H. Overexpression of GDNF in the uninjured DRG exerts analgesic effects on neuropathic pain following segmental spinal nerve ligation in mice. J Pain. 2011;12(11):1130–1139. doi: 10.1016/j.jpain.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Ma SH, Tao R, Xia LJ, Liu L, Jiang YH. Gene expression profile changes in rat dorsal horn after sciatic nerve injury. Neurol Res. 2017;39(2):176–182. doi: 10.1080/01616412.2016.1273590. [DOI] [PubMed] [Google Scholar]
- 11.Yang YK, Lu XB, Wang YH, Yang MM, Jiang DM. Identification crucial genes in peripheral neuropathic pain induced by spared nerve injury. Eur Rev Med Pharmacol Sci. 2014;18(15):2152–2159. [PubMed] [Google Scholar]
- 12.von Schack D, Agostino MJ, Murray BS, et al. Dynamic changes in the microRNA expression profile reveal multiple regulatory mechanisms in the spinal nerve ligation model of neuropathic pain. PLoS One. 2011;6(3):e17670. doi: 10.1371/journal.pone.0017670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17(6):520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 14.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37(Web Server issue):W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:1.30.1–1.30.33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 17.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37(Database issue):D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50(3):355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 22.Engle MP, Merrill MA, Marquez De Prado B, Hammond DL. Spinal nerve ligation decreases γ-aminobutyric acidB receptors on specific populations of immunohistochemically identified neurons in L5 dorsal root ganglion of the rat. J Comp Neurol. 2012;520(8):1663–1677. doi: 10.1002/cne.23005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 24.Dixit M, Raghuvanshi A, Gupta CP, et al. Medicarpin, a natural pterocarpan, heals cortical bone defect by activation of notch and Wnt canonical signaling pathways. PLoS One. 2015;10(12):e0144541. doi: 10.1371/journal.pone.0144541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21(13):4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Steenwinckel J, Reaux-Le Goazigo A, Pommier B, et al. CCL2 released from neuronal synaptic vesicles in the spinal cord is a major mediator of local inflammation and pain after peripheral nerve injury. J Neurosci. 2011;31(15):5865–5875. doi: 10.1523/JNEUROSCI.5986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan X, Zhang Y, Kim HG, Liangpunsakul S, Dong XC. FOXO transcription factors protect against the diet-induced fatty liver disease. Sci Rep. 2017;7:44597. doi: 10.1038/srep44597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan CW, Jin X, Zhao Y, et al. AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 2017;36(8):995–1010. doi: 10.15252/embj.201695534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Garcia O, Matsuzaki T, Olmer M, Masuda K, Lotz MK. Age-related reduction in the expression of FOXO transcription factors and correlations with intervertebral disc degeneration. J Orthop Res. 2017 Apr 21; doi: 10.1002/jor.23583. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA, Song XJ. WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2013;123(5):2268–2286. doi: 10.1172/JCI65364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suh HI, Min J, Choi KH, Kim SW, Kim KS, Jeon SR. Axonal regeneration effects of Wnt3a-secreting fibroblast transplantation in spinal cord-injured rats. Acta Neurochir (Wien) 2011;153(5):1003–1010. doi: 10.1007/s00701-011-0945-1. [DOI] [PubMed] [Google Scholar]
- 32.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao K, Shen Z, Yuan Y, et al. Simvastatin inhibits neural cell apoptosis and promotes locomotor recovery via activation of Wnt/β-catenin signaling pathway after spinal cord injury. J Neurochem. 2016;138(1):139–149. doi: 10.1111/jnc.13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Wang X, Lu CC, et al. Repulsive Wnt signaling inhibits axon regeneration following central nervous system injury. J Neurosci. 2008;28:8376–8382. doi: 10.1523/JNEUROSCI.1939-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onishi K, Hollis E, Zou Y. Axon guidance and injury-lessons from Wnts and Wnt signaling. Curr Opin Neurobiol. 2014;27:232–240. doi: 10.1016/j.conb.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu GB, Niu FW, Zhang YC, et al. Methylprednisolone promotes recovery of neurological function after spinal cord injury: association with Wnt/β-catenin signaling pathway activation. Neural Regen Res. 2016;11(11):1816–1823. doi: 10.4103/1673-5374.194753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Chen G, Xue Q, Yu B. Early changes of beta-Catenins and Menins in spinal cord dorsal horn after peripheral nerve injury. Cell Mol Neurobiol. 2010;30(6):885–890. doi: 10.1007/s10571-010-9517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaoyan X, Juanjuan Y, Yalan T, Ping H, Jianzhong L, Qinian W. Downregulation of EIF4A2 in non-small-cell lung cancer associates with poor prognosis. Clin Lung Cancer. 2013;14(6):658–665. doi: 10.1016/j.cllc.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Jeong W, Kim J, Ahn SE, et al. AHCYL1 is mediated by estrogen-induced ERK1/2 MAPK cell signaling and microRNA regulation to effect functional aspects of the avian oviduct. PLoS One. 2012;7(11):e49204. doi: 10.1371/journal.pone.0049204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lv L, Chen G, Zhou J, Li J, Gong J. WT1-AS promotes cell apoptosis in hepatocellular carcinoma through down-regulating of WT1. J Exp Clin Cancer Res. 2015;34:119. doi: 10.1186/s13046-015-0233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]