

Summary
Cerebellar growth and foliation require the Hedgehog-driven proliferation of granule cell precursors (GCPs) in the external granule layer (EGL). However, that increased or extended GCP proliferation generally does not elicit ectopic folds suggests that additional determinants control cortical expansion and foliation during cerebellar development. Here, we find that genetic loss of the serine-threonine kinase Liver Kinase B1 (Lkb1) in GCPs increased cerebellar cortical size and foliation independent of changes in proliferation or Hedgehog signaling. This finding is unexpected given that Lkb1 has previously shown to be critical for Hedgehog pathway activation in cultured cells. Consistent with unchanged proliferation rate of GCPs, the cortical expansion of Lkb1 mutants is accompanied by thinning of the EGL. The plane of cell division, which has been implicated in diverse processes from epithelial surface expansions to gyrification of the human cortex, remains unchanged in the mutants when compared to wild-type controls. However, we find that Lkb1 mutants display delayed radial migration of post-mitotic GCPs that coincides with increased cortical size, suggesting that aberrant cell migration may contribute to the cortical expansion and increase foliation. Taken together, our results reveal an important role for Lkb1 in regulating cerebellar cortical size and foliation in a Hedgehog-independent manner.
Introduction
The cerebellum integrates sensory and motor information and has recently drawn attention for its extensive involvement in cognition, including emotional control1, learning2, memory3, and decision making4. Although the importance of the cerebellum during human brain evolution was initially dismissed based on the finding that it occupies a constant proportion of total brain volume5, subsequent analysis revealed that cerebellar surface area—a more accurate measure of processing capacity than volume—increases in an evolutionarily-dependent manner6. The capacity of the cerebellum to expand in surface area relative to its volume is facilitated by the presence of deep folds in the cerebellar surface known as fissures that separate the cerebellum into lobules (also known as folia). Like surface area, foliation complexity scales in an evolutionarily-dependent manner7. Despite the evolutionary import and functional significance of foliation, the cellular cues and genetic programs controlling the expansion and subsequent folding of the cerebellar cortex remain incompletely understood.
Cerebellar foliation occurs in two phases: an embryonic phase, which encompasses cardinal fissure formation, and a postnatal phase, during which time non-cardinal fissures form. Cardinal fissures form around embryonic day 17 (E17) in the mouse and divide the cerebellar surface into five cardinal lobes8. Cardinal fissure formation is at least partially genetically determined, as loss of the Engrailed homeobox genes En1/2 disrupts placement and depth of cardinal fissures9. By contrast, non-cardinal fissures are thought to form in response to mechanical forces; namely, the need to fit the expanding cortical surface within the confines of the skull7.
Expansion of the cerebellar cortex is driven in part by the proliferation of granule cell precursors (GCPs) in the external granule layer (EGL). Between late embryogenesis and the second postnatal week, GCPs in the EGL multiply in response to mitogenic Sonic Hedgehog (Shh) signaling before exiting the cell cycle and migrating radially along Bergmann glia to reach the internal granule layer (IGL), where they mature into granule cells10–12. The importance of the EGL in cortical expansion and foliation is evident from studies showing that reducing GCP proliferation, either using gamma irradiation7 or genetic ablation of Hedgehog (Hh) signaling13, leads to a small, hypoplastic cerebellum with fewer folds. However, mutations that increase or prolong GCP proliferation do not consistently increase foliation, even when hyperplasia is evident. For example, although Shh-P1 transgenic mice, in which Purkinje cell production of Shh is increased, have a larger cerebellum with 1–2 additional folia13, loss of the cell cycle inhibitor p27Kip1 extends GCP proliferation and increases cerebellar volume without the formation of additional folds14. Taken together, these studies suggest that GCP proliferation is necessary, but not sufficient, to induce cortical folding in the cerebellum. Thus, one intriguing question regarding cerebellar development is whether factors other than proliferation are important for cortical expansion and foliation.
With 14 known substrates, the tumor suppressor Liver Kinase B1 (Lkb1; also known as Stk11) controls diverse cellular activities, including cytoskeletal dynamics15,16, tight junction formation17, migration18, and proliferation19. In the developing neocortex, Lkb1 has been shown to regulate neuronal migration20,21, axon specification22,23, and terminal axon branching24. We initially became interested in Lkb1 following a genetic screen demonstrating that loss of Lkb1 reduced Hh pathway responsiveness in mouse embryonic fibroblasts25. To determine if Lkb1 is important for Hh signaling in vivo, we generated a mouse model of GCP-specific Lkb1 ablation. We find that GCP-specific loss of Lkb1 resulted in an expanded cerebellar cortex with increased foliation. In contrast to a recently published manuscript26, we find that increased foliation occurs without alteration of Hedgehog signaling or GCP proliferation. Neither the orientation of cell division nor mTOR signaling, a known downstream target of Lkb1, are responsible for changes in foliation. However, we find that post-mitotic GCPs display delayed radial migration in Lkb1 mutants, suggesting that aberrant migration may contribute to increasing in cortical size and foliation. Taken together, our study reveals that Lkb1 controls cerebellar cortical expansion and foliation without altering cell proliferation or perturbing Hedgehog pathway activity.
Results
Lkb1 is expressed in neural progenitors
In situ hybridization for Lkb1 at postnatal day 4 (P4) revealed that Lkb1 was highly expressed in the EGL, where Shh-responsive GCPs reside (Figure 1A–A′). Consistent with a role for Lkb1 in the EGL, Lkb1 immunostaining at P7 revealed Lkb1 protein was seen throughout EGL, consistent with a role of Lkb1 in granule cell function (Figure 1B–B′).
Figure 1. Lkb1 is expressed in the granule cell precursors.
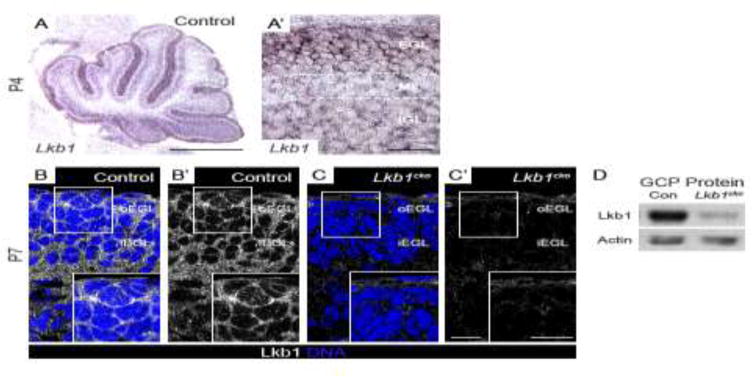
A–A′. Lkb1 in situ hybridization at postnatal day 4 (P4). Lkb1 is expressed in all cortical layers but is highest in the external granule layer (EGL). B–C. Immunohistochemistry for Lkb1 (white) and TO-PRO 3 (blue) at postnatal day 7 (P7). Lkb1 localizes to the cytoplasm and cell cortex of GCPs in the EGL of control cerebella (B–B′) but is absent in Lkb1cko cerebella (C–C′). D. Western blotting for Lkb1 reveals a significant reduction in Lkb1 protein levels in Lkb1cko GCPs compared to controls. Actin was used as loading control. Scalebars: A = 500 μm, A′ = 50 μm, C–D = 10 μm. Con = control, EGL = external granule layer, oEGL = outer EGL, iEGL = inner EGL, ML = molecular layer, IGL = internal granule layer. Full-length blots are shown in Supplementary Figure 9.
Loss of Lkb1 from GCPs increases cortical size and foliation
Mice null for Lkb1 die between E8 and E11 due to vascular defects38. In order to study the function of Lkb1 in the cerebellum, which develops postnatally, we generated Atoh1-cre; Lkb1fl/− mice (hereafter referred to as Lkb1cko). Expression of the Atoh1 transcription factor is restricted to cerebellar GCPs and deep cerebellar nuclei39–41. Lkb1 immunostaining (Figure 1C) and immunoblotting (Figure 1D) revealed a near-complete loss of Lkb1 protein in Lkb1cko cerebella. Consistent with previous reports of reduced Atoh1-cre activity in the posterior cerebellum42, Lkb1 protein levels in the EGL of lobes IX and X of Lkb1cko were higher than other regions (data not shown).
Lkb1−/− MEFs have reduced levels of Hh responsiveness25. If Lkb1 were responsible for Hh signaling in GCPs, we would expect Lkb1cko to be smaller than controls. However, consistent with the findings of others26, we noted that Lkb1cko cerebella were considerably more foliated than littermate controls at adult stages (Figure 2A–B). Given that folia pattern and number were similar among Atoh1-cre; Lkb1fl/+, Lkb1+/−, Lkb1fl/+, and Lkb1fl/− littermates, the term “control” collectively refers to littermates of any of these genotypes.
Figure 2. Granule cell precursor-specific loss of Lkb1 results in increased foliation and cortical expansion.
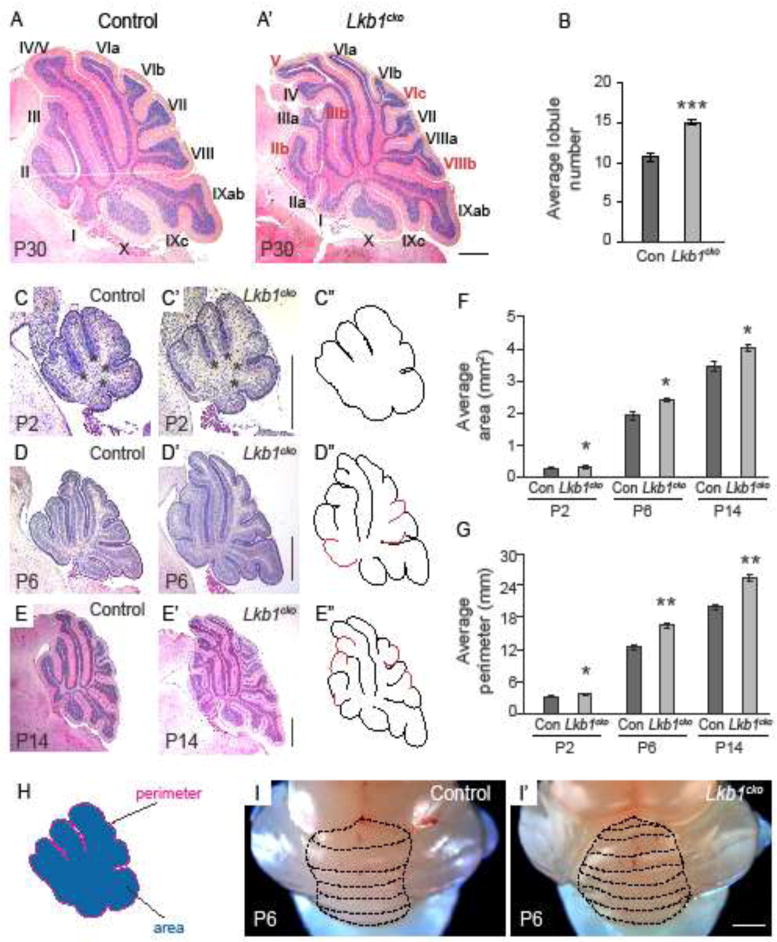
A–A′. Hematoxylin and eosin staining of control (A) and Lkb1cko (A′) cerebella at P30. Roman numerals denote lobule numbers. Red roman numerals indicate lobules present in Lkb1cko absent in control. B. Average lobule number of control and Lkb1cko cerebella. C–E. Hematoxylin and eosin staining of mid-vermal cerebellar cross sections at the indicated stages. Lobules present in Lkb1cko not present in the control are highlighted in red in C″–E″. Asterisks in C–C′ indicate cardinal fissures. F. Average cross sectional area of mid-vermal cerebellar sections at indicated stages. G. Average cross sectional perimeter of mid-vermal cerebellar cross sections at the indicated stages. H. Illustration showing how area and perimeter were determined. I–I′. Whole mount images of P6 control (I) and Lkb1cko (I′) cerebella. Dashed lines delineate folia. n=5 for all analyses. *, p<0.05, **, p<0.005, ***, p<0.0005, Student’s t-test. Scalebar 500 μm for all images. Con = control.
To determine when Lkb1cko first exhibited enhanced foliation, we collected cerebella sequentially during the first two postnatal weeks (Figure 2; Figure S1). The initial stages of cerebellar patterning, including cardinal fissure formation, were normal in Lkb1cko cerebella at P2 (Figure 2C–C″, asterisks denote principal fissures). However, Lkb1cko cerebella appeared visibly larger than controls at both P2 (Figure 2C–C′, F) and P4 (Figure S1). Indeed, mid-sagittal cross sectional area was larger in Lkb1cko cerebella relative to controls (0.31 +/− 0.029 mm2 in Lkb1cko vs. 0.27 +/− 0.027 mm2 in controls; Figure 2F). Additionally, mid-sagittal EGL perimeter was longer in P2 Lkb1cko cerebella (3.5 +/− 0.16 mm in Lkb1cko vs. 3.12 +/− 0.16 mm in controls; Figure 2G), suggesting that cerebellar surface area was increased. Thus, cortical expansion and increased cross sectional area preceded supernumerary folia in Lkb1cko. Of note, Lkb1cko cerebella also appeared similar to controls during late embryonic stages (Figure S1).
Lkb1cko first displayed increased foliation at P6, with multiple lobules not present in controls (Figure 2D–D″, H–H′). Additionally, mid-sagittal area (2.34 +/− 0.05 mm2 in Lkb1cko vs. 1.92 +/− 0.12 mm2 in controls) and perimeter (16.5 +/− 0.36 mm in Lkb1cko vs. 12.48 +/− 0.52 mm in controls) were larger in P6 Lkb1cko, indicative of increased cerebellar volume and surface area (Figure 2F–G). Consistent with increased volume, Lkb1cko cerebella were often adhered to the overlying skull, making them difficult to dissect. By P11, dramatic differences in the shape and pattern of Lkb1cko cerebella were apparent (Figure S1). At P14, when foliation patterns are established43, Lkb1cko were larger (area: 4.03 +/− 0.10 mm2 in Lkb1cko vs. 3.46 +/− 0.15 mm2 in controls; Figure 2F), had a longer perimeter (25.45 +/− 0.56 mm in Lkb1cko vs. 19.95 +/− 0.57 mm in controls; Figure 2G), and were considerably more foliated than controls (Figure 2E–E″, Figure S1). However, although P30 Lkb1cko cerebella were more foliated than littermate controls (Figure 2A–A′) and had a larger cross sectional perimeter (31.5 +/− 0.81 mm in Lkb1cko vs. 27.89 +/− 1.35 mm in controls; Figure S2), cross sectional area was not increased (Figure S1).
The central cerebellar vermis of most inbred mouse strains contains between 9 and 11 lobules and sublobules. Whereas the mixed strain control mice used in our studies had an average of 10.5 lobules, Lkb1cko had an average of 15 lobules, corresponding to a ~40% increase in foliation (Figure 2B). Additional lobules were consistently observed in lobes I, II, III, IVb and VIII (Figure 2A–A′, E–E′; Figure S1). Whereas lobes IV and V were usually fused in controls, lobes IV and V were distinct in the majority of Lkb1cko (Figure 2A–A′, E–E′, Figure S1). Other morphological changes were also evident in Lkb1cko: whereas the interface of lobes V and IV was normally straight in controls, this fissure often had an undulated, rippled appearance in Lkb1cko (Figure 2A′, E′ and Figure S1). Notably, throughout our analysis, lobes IX and X appeared normal in Lkb1cko, consistent with reduced recombination efficiency of Atoh1-cre in these regions42. Increased foliation patterns were also observed in the hemispheres of Lkb1cko (not shown).
Loss of Lkb1 does not increase Hedgehog signaling
Given that cardinal lobes formed normally in Lkb1cko (Figure 2C–C′), we hypothesized that Lkb1 controls the development of secondary and tertiary lobules, which are thought to form in response to expansion of the EGL7. A recent study has reported that loss of Lkb1 from granule cell precursors increased Hedgehog signaling and GCP proliferation26. To test this, we examined expression of the transcription factor Gli1, a transcriptional target of Hh signaling. Gli1 mRNA and protein levels are an established readout for pathway activity44. In situ hybridization for Gli1 revealed no discernible difference in Gli1 expression in Lkb1cko at P6 (Figure 3A–B) or at birth (P0; Figure S3). Additionally, Gli1 protein levels were unchanged in freshly isolated Lkb1cko GCPs (Figure 3C–D). Similarly, quantitative real-time PCR for Gli1 as well as the Hh target genes Ptch1 and Hhip1 indicated that the expression of Gli1, Ptch1, and Hhip1 was unchanged in Lkb1cko GCPs relative to controls (Figure 3E). Finally, no difference in Gli1 abundance was detectible in cultured GCPs derived from control and Lkb1cko and treated with the Shh agonist SAG (Figure 3F). Together, these data indicate that Lkb1 does not regulate cortical expansion or foliation by increasing Hedgehog pathway activity.
Figure 3. Hedgehog signaling is unchanged in Lkb1cko cerebella.
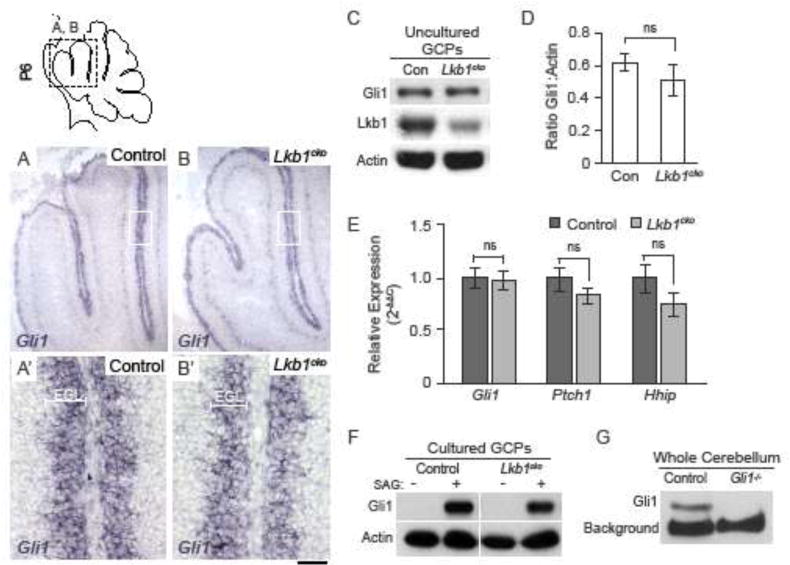
A–B. In situ hybridization for Hedgehog target gene Gli1 in postnatal day 6 control (A–A′) and Lkb1cko (B–B′) cerebella. C. Western blot for Gli1 in GCPs derived from control and Lkb1cko cerebella. Lkb1 and Actin were used to verify knockdown and loading, respectively. D. Densitometric measurements of Gli1 protein levels relative to Actin. E. Quantitative real-time PCR for Gli1, Ptch1, and Hhip1 in freshly isolated control and Lkb1cko GCPs. n=3, no significant difference, Student’s t-test. F. Western blot for Gli1 in cultured GCPs derived from control and Lkb1cko cerebella in the presence (+) or absence (−) of 20nm SAG. G. Western blot for Gli1 in whole cerebella derived from control and Gli1−/− mutants. Scalebar 50 μm. Con = control, EGL = external granule layer. Full-length blots are shown in Supplementary Figure 9.
Lkb1−/− MEFs have a shorter primary cilium, a microtubule-based organelle important for Hedgehog signaling25. However, loss of Lkb1 did not alter primary cilia length in GCPs (Figure S3). Thus, Lkb1 does not maintain Hh signaling or cilia length in GCPs.
Loss of Lkb1 does not increase GCP proliferation
Our data suggest that Lkb1 functions outside of the Hh pathway in GCPs. Although Hh signaling is essential for GCP proliferation, it remains possible that cortical expansion and increased foliation in Lkb1cko could result from Hh-dependent proliferation. Indeed, a recent study26 demonstrated that loss of Lkb1 from GCPs increased GCP proliferation.
To systematically and globally measure proliferation in the developing cerebellum, we generated an algorithm to automatically quantify cells in the EGL based on size and brightness threshold using the cell counting software CellProfiler (Figure S4). Short-term (1 hour) labeling with the thymidine analog BrdU was used to measure proliferation in control and Lkb1cko cerebella at P2, when changes in foliation were not yet evident, and P6, around the onset of altered foliation in Lkb1cko. However, the proportion of BrdU+ GCPs was unchanged in Lkb1cko compared to controls at both P2 and P6 (Figure 4A–C). Given that EGL perimeter length was longer in Lkb1cko (Figure 2), we speculated that the total number of mitotic cells might be increased even if the proportion of dividing cells was not. However, the total number of mitotic (phospho-histone H3+) EGL GCPs per mid-vermal section was unchanged in Lkb1cko at both P2 and P6 (Figure 4D). Since previous studies26 demonstrated increased proliferation of Lkb1-deficient GCPs at P4 we also quantified the number of mitotic cells per unit area in the EGL at P4 and found no difference between Lkb1cko and control (Figure 4E). Additionally, no difference was observed when comparing the number of Brdu+ cells per unit area of the EGL (Figure 4F) or when we limited our analysis to the proliferating (Ki67+) fraction of GCPs at P6 (Figure S5). Similar to GCPs, we observed no change in the number of Purkinje cells in the P8 Lkb1cko cerebellum (Figure S5), nor did we observe any alteration in the number of Pax2+ interneuron precursors in the molecular layer at P8 (Figure S5).
Figure 4. Neither proliferation nor cell cycle kinetics are altered in Lkb1cko cerebella.
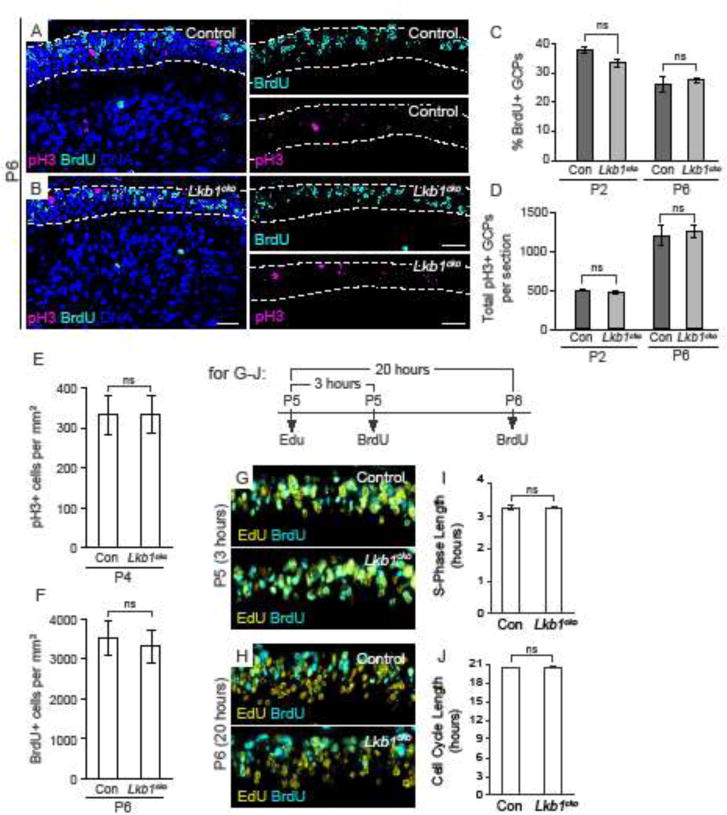
A–B. Immunostaining of P6 control (A) and Lkb1cko (B) for phospho-histone H3 (pH3) and BrdU one hour after BrdU injection. Dashed lines delimit the EGL. DNA was counterstained with TO-PRO 3. C. Quantification of the percentage of BrdU+ cells in the EGL of control and Lkb1cko cerebella at P2 and P6. D. Quantification of the total number of pH3+ cells per mid-vermal cross section of control and Lkb1cko cerebella at the indicated stages. E. Number of pH3+ cells per mm2 at P4. F. Number of BrdU+ cells per mm2 at P6. G. EdU/BrdU double labeling at P5 (3 hour timepoint). H. EdU/BrdU double labeling at P6 (20 hour timepoint). I. S-phase length of control and Lkb1cko GCPs. J. Cell cycle length of control and Lkb1cko GCPs. n=3, no significant difference, Student’s t-test. Scalebar 20 μm. Con = control.
Lkb1−/− MEFs divide more quickly than do controls45. Sequential Edu-BrdU labeling was used to determine if cell cycle kinetics were altered in Lkb1cko GCPs36,37. However, neither S-phase length nor cell cycle length were altered in Lkb1cko cerebella (Figure 3G–J). Together, these data suggested cortical expansion and increased foliation in Lkb1cko were not due to increased GCP proliferation.
As a final means of assuring that proliferation was not increased in Lkb1cko, we examined cerebella at P30. If loss of Lkb1 increased GCP proliferation, we would expect cerebellar size and IGL area to be increased in Lkb1cko. We noted that at this stage, although the perimeter remained larger in Lkb1cko (27.889 +/− 1.35 mm in controls vs. 31.51 +/− 0.81 mm in Lkb1cko) neither cross-sectional area nor IGL area were increased relatively to controls (cross-sectional area: 7.52 +/− 0.35 mm2 in controls vs. 7.08 +/− 0.34 mm2 in Lkb1cko; Figure S2).
Lkb1cko cerebella have a thinner outer EGL
Given that Lkb1cko were larger at P6 (Figure 2) but did not harbor an increased number of proliferating GCPs (Figure 4, Figure S5), we wondered if the oEGL was thinner Lkb1cko cerebella. In other words, if GCP proliferation was equivalent in control and Lkb1cko mice, but GCPs were distributed over a larger area in Lkb1cko, we would expect the oEGL to be thinner in Lkb1cko. Indeed, Ki67 staining of P7 sections revealed that many regions of the oEGL appeared thinner in Lkb1cko compared to controls (Figure 5). The average number of Ki67+ GCPs per mm of EGL was significantly reduced in Lkb1cko (538.7 +/− 15 cells/mm in control vs. 436.9 cells/mm in Lkb1cko; Figure 5C). Similarly, the average thickness of oEGL is significantly thinner in Lkb1cko cerebella (13.7 μm2 +/− 0.9 μm2 in control vs 11.14 +/− 0.6 μm2 in Lkb1cko), whereas the thickness of iEGL remains comparable between Lkb1cko cerebella and controls (6.9 +/− 0.6 μm2 vs 7.4 +/− 0.7 μm2; Figure 5D–F). Thus, loss of Lkb1 from GCPs leads to a thinner oEGL. Of note, when we quantified the total oEGL area, we saw no difference between controls and Lkb1cko (Figure S6), in keeping with our data showing there was no difference in proliferation. Similarly, we observed no difference in iEGL area between Lkb1cko and controls (Figure S6). In addition to thinner EGL, cell size may also contribute to surface area expansion. We, therefore, estimated average cell size of GCPs by measuring the area of Ki67+ and Ki27kip1+ cells and found no difference between controls and Lkb1cko cerebella (Figure S6D).
Figure 5. Loss of Lkb1 from granule cell precursors results in a thinner outer EGL.
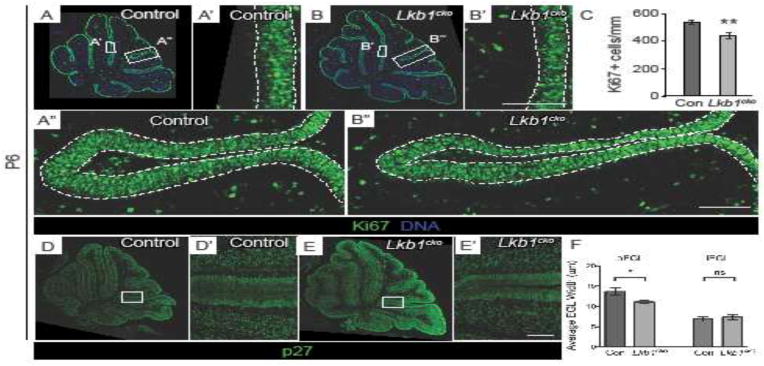
A–B. Representative Ki67-stained P6 control (A) and Lkb1cko (B) cerebella. Dashed lines in A′–A″ and B′–B″ delimit outer EGL, where proliferative cells reside. Lkb1cko have a visibly thinner layer of proliferative (Ki67+) GCPs than do controls (compare A′ to B′, A″ to B″). C. Quantification of oEGL thickness using Ki67+ cells per mm EGL. n=3, p<0.01, Student’s t-test. D–E. Representative p27-stained P6 control (D) and Lkb1cko (E) cerebella. Dashed lines in D′ and E′ delimit inner EGL, where differentiating cells reside. F. Quantification of average thickness of oEGL (p<0.05) and iEGL (p=0.6), Student’s t-test. Scalebar 50 μm. Con = control.
The plane of cell division regulates surface area expansion and organ shape in a number of tissues, including the lung and epidermis46–48. To determine if cortical expansion and increased foliation in Lkb1cko were due to perturbations in spindle orientation, control and Lkb1cko cerebella at P2, P4, and P6 were labeled with phospho-histone H3 and Aurora B kinase to label mitotic DNA and spindle-associated microtubules, respectively (Figure S8). These markers, together with a nuclear dye, were used to quantify the proportion of anaphase GCPs dividing vertical (60–90°), horizontal (0–30°), or tangential (30–60°) to the cerebellar surface (Figure 6Y) across the entire EGL (Figure S7). Although there was a slight decrease in the number of vertical divisions at P6 in Lkb1cko, we found that the majority of GCPs divide horizontal to cerebellar surface at regardless of genotype. Together, these data indicate that Lkb1 does not regulate the plane of GCP division.
Figure 6. Lkb1cko cerebella have defects in granule cell migration.
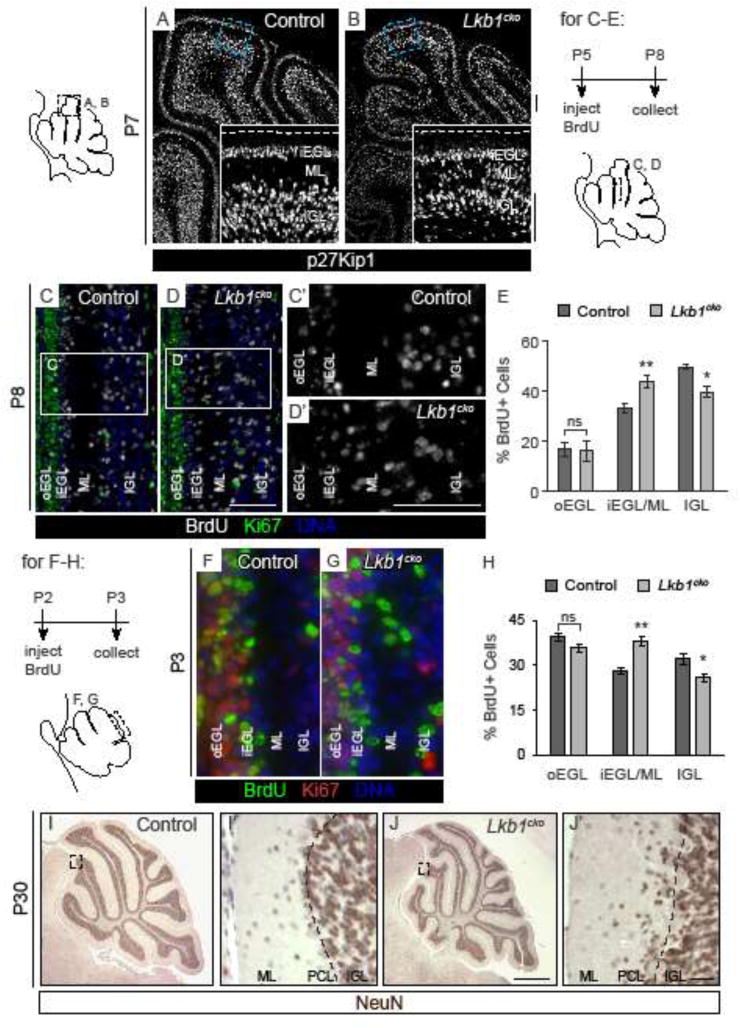
A–B. p27Kip1 immunostaining labels post-mitotic GCPs in P7 control (A) and Lkb1cko (B) cerebella. Dashed line in inset denotes cerebellar surface. C–D. BrdU/Ki67 co-staining of P8 control (C) and Lkb1cko (D) cerebella three days after BrdU injection. C′–D′. Enlarged images of boxed regions in C and D. E. Quantification of the proportion of BrdU+ cells in each of the specified regions three days after BrdU pulse. n=3 controls, n=5 Lkb1cko. *, p<0.05, ** p<0.005. Student’s t-test. F–G. P3 cerebella injected with BrdU at P2. H. Quantification of the proportion of BrdU+ cells in each of the specified regions one day after BrdU pulse. n=3, *, p<0.05, ** p<0.01. Student’s t-test. I–J. Representative staining for Neuron-specific nuclear protein (NeuN), a marker of mature granule cells, in P30 control (I) and Lkb1cko (J) cerebella. I′ and J′ are enlargements of the boxed regions in I and J. Dashed lines in I′ and J′ corresponds to Purkinje cell layer (PCL). Note that a number of granule cells fail to migrate past the Purkinje cell layer in Lkb1cko. All scalebars 50 μm except I–J, in which scalebar is 500 μm. oEGL = outer external granule layer, iEGL = inner external granule layer, ML = molecular layer, IGL = internal granule layer. Full-length blots are shown in Supplementary Figure 9.
Lkb1cko cerebella have impaired radial migration
Our data so far indicated that increased cortical size and foliation in Lkb1cko cerebella is not due to changes in Hedgehog signaling, proliferation, cell cycle kinetics or the plane of GCP division. Given the role of Lkb1 in neuronal migration in forebrain neurons20,21, and the recently documented role of Lkb1 in GCP migration26, we examined whether Lkb1 regulated migration in the cerebellum.
Differentiating granule cells migrate radially through the molecular layer along Bergmann glia to reach internal granule layer (IGL). Staining for p27Kip1, a marker of post-mitotic GCPs and some interneurons, at P7 revealed increased numbers of p27Kip1+ cells in the molecular layer of Lkb1cko (Figure 6A–B). Long-term BrdU labeling was employed to track the location of GCPs over time. Mice were injected with BrdU at P5 and collected at P8. Whereas BrdU-labeled cells formed tight bands corresponding to the EGL and IGL in controls, BrdU-labeled cells in Lkb1cko were distributed between all cortical layers (Figure 6C–D). Automated quantification using the counting software Cell Profiler revealed that Lkb1cko had significantly more BrdU+ cells undergoing migration and significantly fewer BrdU+ cells in the IGL (Figure 6E), consistent with defective radial migration. Similarly, injection of BrdU at P2 and collection at P3 revealed an increased number of post-mitotic BrdU+ cells (e.g. BrdU+Ki67−) cells in the molecular layer of Lkb1cko mice compared to controls (Figure 6F–H), indicating that Lkb1-deficient GCPs exhibit defects in migration as early as P2. We also examined the capacity of GCP to re-enter the cell cycle after 24 hours of BrdU chase by measuring the proportion of Ki67+ BrdU+/Ki67+ cells and found no difference in cell cycle re-entry between mutants and controls (Figure S6D), ruling out the possibility that Lkb1 also promotes neurogenic proliferation of GCPs. Finally, staining of adult P30 cerebella for the granule cell-specific marker Neuron-specific nuclear protein (NeuN) revealed that, in contrast to controls, many more NeuN+ granule cells failed to reach the IGL in Lkb1cko (Figure 6I–J).
Lkb1 regulates GCP polarity and migration independent of mTOR signaling
Under conditions of energetic stress, Lkb1 activates the catalytic α subunits of AMPK via phosphorylation, and activated AMPK inhibits processes that expend energy, including fatty acid synthesis and mTORC1 signaling49. AMPK inhibits mTOR signaling through phosphorylation of the TSC1/TSC2 complex. Accordingly, loss of Lkb1, AMPK, TSC1, or TSC2 leads to hyperactivation of mTORC1 signaling50. Aberrant mTOR activity disrupts cerebellar development, as mice harboring a dominant-negative TSC2 allele exhibit ectopic granule cells and increased GCP proliferation51.
Immunostaining and Western blotting for the mTOR target phosphorylated-S6 ribosomal protein (p-S6)52, revealed elevated p-S6 levels in Lkb1cko GCPs (Figure 7A–C). Increased mTORC1 signaling in Lkb1cko likely resulted from reduced AMPK activity, as both AMPK phosphorylation and phosphorylation of the AMPK substrate acetyl co-A carboxylase (ACC) were reduced in Lkb1cko GCPs (Figure 7D). Together, these data suggest that loss of Lkb1 reduced AMPK activity and increased mTOR signaling in GCPs.
Figure 7. Increased foliation and altered migration in Lkb1cko cerebella is mTOR-independent.
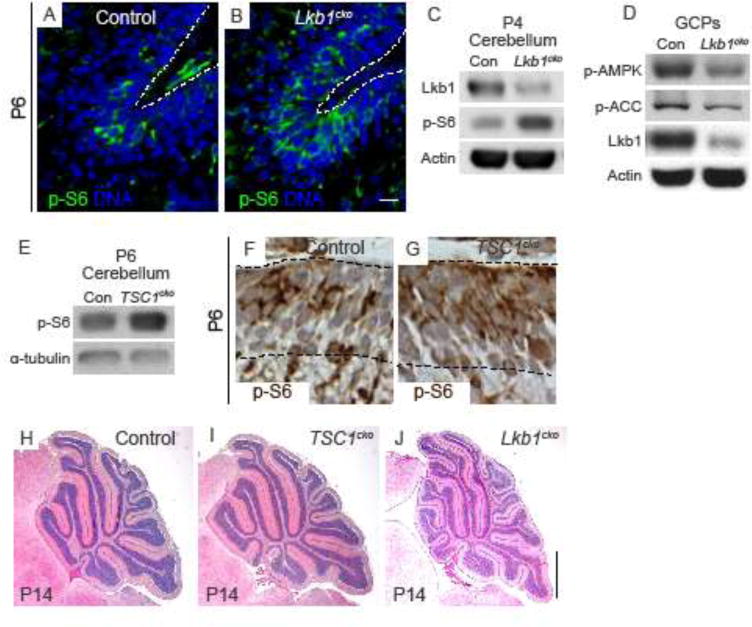
A–B. Representative staining for p-S6 in P6 control (A) and Lkb1cko (B) cerebella. Scalebar 10 μm. C. Western blotting (cropped) of P4 cerebella for p-S6. Lkb1 and Actin were used to verify knockdown and loading, respectively. D. Western blot (cropped) of control and Lkb1cko GCPs for p-AMPK (Thr172) and p-ACC. Lkb1 and Actin were used to verify knockdown and loading, respectively. E. Western blot (cropped) for p-S6 in control and TSC1cko cerebella. F–G. Immunohistochemistry for p-S6 in Control (F) and TSC1cko (G) cerebella. H–J. H&E staining of P14 control (H), TSC1cko (I), and Lkb1cko (J) cerebella. Scalebar 500 μm. Con = control.
To determine if the defects in foliation and migration in Lkb1cko resulted from mTOR pathway upregulation, we generated Atoh1-cre; TSC1flox/− mice (TSC1cko mice). Western blotting and immunostaining for p-S6 revealed increased mTOR signaling in TSC1cko (Figure 7E–G). However, foliation patterns were normal in TSC1cko mutants at P14 (Figure 7H–I, Figure S9). Notably, in contrast TSC2 dominant-negative mice51, TSC1cko cerebella did not harbor ectopic granule cell clusters (Figure S9). Together, these data suggested that mTORC1 signaling was not responsible for increased foliation in Lkb1cko.
Discussion
The determinants of cortical folding are poorly understood. Our data indicate that loss of Lkb1 from granule cell precursors increases cerebellar surface area and foliation in the absence of changes in proliferation, Hedgehog signaling, and cell polarity. Although mTOR signaling is increased in cells lacking Lkb1, we find that genetic loss of TSC1, a downstream target of Lkb1, does not lead to increased foliation, suggesting that increased mTOR signaling is not responsible for increased foliation in Lkb1 mutants. Finally, in accordance with the work of others26, we find that Lkb1-deficient GCPs display impaired radial migration. Impaired migration coincides with increased cortical area and foliation, suggesting that impaired migration may be responsible for hyperfoliation in Lkb1 mutants.
Previous studies have demonstrated the loss of Lkb1 decreases25 or increases26 Hedgehog signaling in MEFs and GCPs, respectively. Notably, we find that GCP loss of Lkb1 does not affect Hedgehog signaling activity in vivo or in freshly isolated GCPs. This finding is supported by in situ hybridization, quantitative real time PCR, and western blotting for Hedgehog target genes. Additionally, we find that deletion of Lkb1 does not alter GCP proliferation. This finding is significant because existing mouse models of increased foliation demonstrate increased numbers of proliferating GCPs13,53. By contrast, we find no difference in the proportion of proliferating cells in the EGL of Lkb1cko, nor do we observe any difference in cell cycle length. Importantly, we measured proliferation in a non-biased manner by quantifying the proportion of proliferating GCPs in the EGL by automated cell counting. That GCP proliferation is unaffected by Lkb1 deletion is further supported by the finding that neither cerebellar size nor IGL area are increased at adult (e.g. P30) stages of cerebellar development.
We find that Lkb1-deficient GCPs have impaired radial migration as early as post-natal day 2. This coincides with the increased surface area in Lkb1cko, thus it is possible that impaired migration may be responsible for increased cerebellar surface area and foliation in Lkb1cko. Indeed, in hypothyroid rats, which have increased foliation, radial migration is impaired although GCP proliferation is not54. Moreover, the dramatic increase in foliation seen in the human cerebellum has been credited to a prolonged period of proliferation and migration of granule cell precursors8. Whereas all GCPs have migrated to the IGL by postnatal day 21 in the mouse, the window of GCP proliferation and migration extends an entire year in humans. Interestingly, most mutations that impair radial migration in mice do not lead to an increase in cerebellar folding. More often than not, mutations disrupting GCP migration lead to a reduction in foliation, often due to secondary effects in glial morphology, Purkinje cell development, and/or GCP proliferation55–58. As such, they Lkb1cko provide a novel tool in which to examine how migration and foliation are coupled during cerebellar development.
Putting this work in the context of existing literature, two important questions arise. First, how is it that we find no change in Hedgehog signaling in Lkb1-deficient GCPs, whereas others see decreased25 or increased26 pathway activity? Moreover, why do we see no change in proliferation, whereas Men et al. see an increase in a similar model? Thought we cannot be certain, these differences may attributable to differences in cell type. Our analysis focused on cerebellar granule cell precursors, whereas Jacob et al. performed their analysis on MEFs and Men et al. considered the entire cerebellum. Another potential difference is that we used an enhancer-driven Atoh1-cre instead of knock-in Atoh1-cre line, although we do not think that this could account for the different conclusions regarding proliferation and Hedgehog signaling. Second, how is it that Lkb1cko have a transient increase in cerebellar surface area during development in the absence of changes in proliferation? We believe defective GCP may explain this phenomenon; Lkb1-deficient GCPs are preferentially located nearer to the cerebellar surface, perhaps causes the volume to expand despite similar cell numbers compared to controls. This belief is supported by the fact that as GCPs migrate inward in Lkb1cko, cerebellar size normalizes and ultimately approximates the size of controls.
Cortical folding, whether gyrification in the neocortex or foliation in the cerebellum, is a complex process involving cell proliferation, migration, differentiation and neuronal connectivity59. In the gyrencephalic neocortex of humans and some mammals, cortical folding has been attributed to outer radial glial (oRG), a population of radial glia that are largely absent in mice and other smooth-brained (lissencephalic) species60. Interestingly, recent studies in mice suggest that cortex folding does not require progenitor cell proliferation but is associated with clustering of neurons along the tangential axis due to defective cell migration61.
In the cerebellum, cortical folding has been attributed to the postnatal expansion the EGL, and differences in the degree of cerebellar foliation have historically been credited to differences in GCP number or to a protracted period of GCP proliferation and maturation7,8. However, not all mutations that increase GCP proliferation or number are sufficient to increase foliation, even when the overall size and surface area of the cerebellum are larger14,62. The nonlinear relationship between GCP number and foliation might be rooted in the need to maintain an optimal EGL thickness in order for folding to occur. Indeed, many mutations that increase GCP proliferation lead to EGL hyperplasia as well as loss of foliation14,58,63,64. Perhaps the most dramatic example of EGL hyperplasia is seen in mouse models of medulloblastoma, in which foliation is lost or completely absent63,64. A thicker EGL may therefore inhibit folding by increasing surface tension and the force required to deform the cerebellar surface, the first described step in fissure formation43. By contrast, when Lkb1 is deleted from GCPs, proliferation is unaffected but the EGL becomes thinner and more receptive to folding.
Materials and Methods
Mice
All animal experiments were approved by the VUMC Animal Care Committee and performed using young neonatal and adult animals (ages P0-P30), according to regulation of the NIH and VUMC Division of Animal Care. Lkb1fl/fl mice27, Sox2-cre mice28 were obtained from Jackson laboratories. TSC1fl/fl mice29 were kindly donated from Kevin Ess (Vanderbilt University). Atoh1-cre mice30 were kindly donated from David Rowitch (UCSF). Lkb1+/− and TSC1+/− mice were generated by crossing fl/fl animals to Sox2-cre females. BrdU (Roche) was dissolved in PBS to a final concentration of 10 mg/ml and administered by intraperitoneal injection.
GCP Isolation
GCPs were isolated as previously described31. Briefly, cerebella were isolated from P4-P6 mice in Hanks buffered saline solution (HBSS) (Gibco) supplemented with glucose. Meninges were removed and cerebella were treated with Trypsin-EDTA. Cerebella were dissociated, large cells were allowed to settle, and GCP-containing supernatants were moved to a fresh tube. For western blotting, cells were spun down and resuspended in RIPA buffer. For RNA extraction, cells were resuspended according to QIAGEN protocols. GCPs cultures were prepared by pre-plating GCPs onto poly-L-lysine-coated plates for 20 minutes, and GCP-containing supernatants were plated onto poly-ornithine-coated plates in the presence of 20 nm SAG or vehicle control.
Western blotting
Whole cerebella or isolated GCPs were homogenized in RIPA buffer containing protease inhibitors (Roche). Protein concentration was measured using the BCA method, and 20–50 μg protein was separated by SDS-PAGE before being transferred onto nitrocellulose membranes.
RNA Isolation and Reverse Transcription
Total RNA was purified from freshly isolated GCPs using RNAeasy mini kit (QIAGEN) and cell homogenization performed using QIAshredder columns (QIAGEN). cDNAs were synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosciences). Quantitative real-time PCR was performed as previously described32. The ΔΔCT method was used to compare expression levels using a Student’s t-test.
Tissue Processing, Immunohistochemistry, and In Situ Hybridization
Tissue was collected and processed as described previously33. To collect mid-sagittal sections, we sectioned only through the vermis, and relied on architecture of nearby brain regions to serve as landmarks, such as the midbrain, brainstem, and choroid plexus. Paraffin sections underwent antigen-retrieval using Citrate Buffer pH=6.0. For γ-tubulin staining, frozen sections were dried, post-fixed, washed in PBS and submerged in ice cold acetone before blocking. In situ hybridizations were performed as described previously34.
Quantification
Statistical analysis
T-tests were performed in Excel. Paired t-tests were used to account for litter-dependent variability in cerebellar size and perimeter. Unpaired t-tests were used for all other analyses. All t-tests were 2-tailed with the exception of P3 BrdU cells in the molecular layer, which was 1-tailed. A 1-tailed t-test was used because of existing evidence regarding defective migration in Lkb1cko.
Area, perimeter, and lobule number
For vermal area and perimeter, at least 3 and up to 10 mid-vermal cross sections were measured and averaged for each mouse, such that each mouse was assigned a single value for area and perimeter. These values were gathered for n=5 control and Lkb1cko mice, and t-tests were performed in Excel. For lobule number, lobules were defined as in35, by the separation of individual lobules by molecular layer as well as the presence of white matter. Lobule counts were obtained for n=3 or n=5 animals.
Automated Cell Counting
Using CellProfiler (Broad Institute, MIT), a pipeline was constructed to count all BrdU and Ki67 double positive nuclei in a number of high-power fields of view. The first module served to split the original RGB image files into the red (Ki67) and green (BrdU) color components and convert them into individual grayscale images. The next two modules were designed to identify and count the nuclei positive for BrdU and Ki67 in the separated images, respectively. The diameters of accepted nuclei were required to fall in a range between 3 and 18 microns (randomly measured nuclei averaged to 6 – 8 microns in diameter). For each grayscale image, a minimum threshold was applied (pixel intensities < 38 on an 8-bit scale were removed). A RelateObjects module grouped nuclei together that were positive for both BrdU and Ki67. Finally, the FilterObjects module served to filter out the nuclei that were BrdU positive but were negative for Ki67 staining. CellProfiler then displayed a program-altered final image of nuclei being bordered by colored lines to clearly indicate which cells were counted; this allows for strict quality control of accurate quantification.
Proliferation
For P2 and P6 BrdU, sections were stained and a total of 5 regions (excluding lobes IX and X in which recombination was inefficient) from each of three sections were imaged using an Olympus fluorescent microscope at 40× magnification. These images were cropped so as to only contain the EGL; all non-EGL areas were edited out first in Photoshop. The automated cell counting software Cell Profiler was used to count the total number of Dapi+ cells as well as BrdU+ cells per region. Outliers were excluded, and regions were averaged to yield a single value corresponding to the proliferative index of each mouse. This was done for n=3 mice of each genotype, and t-tests were performed in Excel. For pH3+ counts of the entire cerebellum, the total number of pH3+ cells in the EGL was determined at 20× magnification using a hand tally counter. For each animal, at least 3 and up to 6 sections were analyzed. These numbers were averaged such that each mouse was assigned a single value.
S-phase and cell cycle length
The length of S-phase and the cell cycle were determined as described previously36. Briefly, P5 control and Lkb1cko pups were injected with EdU (4 mg/kg). After 3 or 20 hours, pups were injected with BrdU (5 mg/kg) and collected after 45 minutes, processed for paraffin sectioning, sectioned, and stained with BrdU and EdU. Slides were scanned through the Vanderbilt DHSR. Three sections were analyzed for each animal at each stage using Cell Profiler. Outliers were excluded. The following equations were used to determine S-phase (Ts) and cell cycle length (Tc), as described36,37:
Students t-tests were used to compare S-phase length and cell cycle length for n=3 animals of each genotype.
Cilia length
40× images were taken on an LSM 510. 4 regions from lobes I–XIII were analyzed. Images were cropped so as to only include the EGL, and Cell Profiler was used to measure cilia length. An average cilia length was obtained for each animal.
EGL thickness
Ki67 and P27-stained cerebella were scanned through the DHSR. At least 3 sections for each mouse were cropped so as to omit any non-EGL cells (eg, cells of the white matter). The total number of Ki67+ cells per section was determined. EGL length was determined by measuring the perimeter of each section in ImageJ, similar to above measurements of perimeter and area. The number of Ki67+ cells was divided by perimeter length to give the average Ki67 cells per section for each of three sections. The average thickness of oEGL and iEGL was determined by measuring the width of Ki67+ and p27+ cells, respectively, in 30 non-overlapping regions per lobule along the entire cerebellar axis using Image J. These numbers were averaged for each mouse, yielding a single value corresponding to each animal.
Cell size
Cellular area was measured for a minimum of 20 cells per field from both oEGL and iEGL using ImageJ for n=3 controls and n=3 Lkb1cko.
P8 Migration analysis
Animals were injected with BrdU at P5 (2 injections 1 hour apart) and collected 3 days later at P8. Paraffin sections were co-stained with BrdU and Ki67 and scanned through the Vanderbilt DHSR. Cell Profiler was used to determine the number of cells in each of three regions: the Ki67+ outer EGL, the nuclei-dense IGL, and the region between the oEGL and IGL (iEGL/ML) within the region as shown in Figure 7. The proportion of cells in each region was determined for n=3 controls and n=5 Lkb1cko, and these values were compared using a Student’s unpaired t-test in Excel.
P3 Migration analysis
Animals were injected with BrdU at P2 and collected 1 day later at P3. Paraffin sections were stained with BrdU and Ki67 and scanned through the Vanderbilt DHSR. CellProfiler was used to determine the number of cells in each of three regions as described above for P8. The proportion of cells in each region was determined (n=3), and these values were compared using a Student’s unpaired t-test in Excel.
Orientation of cell division
Sections were stained with Aurora B and/or pH3 for n=3 mice of each genotype (Lkb1cko or littermate controls). At least 3 and up to 5 stained sections from each mouse were imaged at 20× magnification in non-overlapping fields over the entire cerebellum (approximately 12–15 images per section). Angle measurements were taken using the angle tool in ImageJ. Between 20 and 40 cells were measured for each section depending on stage. Only cells in anaphase or later were measured. The proportion of GCPs dividing parallel (0–30 degrees), perpendicular (60–90 degrees) or tangential (30–60 degrees) was determined for each section. These proportions were averaged such that each mouse was assigned a single set of numbers corresponding to the proportion of GCPs dividing in each orientation.
Microscopy
Bright-field images were collected on an Olympus BX51 upright microscope or a Leica M165 FC stereoscope. Fluorescent images were taken on a Zeiss LSM510, Leica TSC SP5 Confocal, or Olympus fluorescent microscope with an Optigrid system (Qioptiq Imaging). For automated cell counting of entire postnatal cerebella, slides were scanned on an automated scanning microscope system (Ariol SL-50) through the Vanderbilt Digital Histology Shared Resource (DHSR). The Ariol system is based on the Leica DM6000 B microscope.
Antibodies
The following antibodies were used for immunohistochemistry: p27Kip1 (BD Biosciences, 1:300), Tag1 (Hybridoma Bank, 1:10), γ-tubulin (Sigma, 1:300), BrdU (Hybridoma Bank, 1:100), Ki67 (Thermo Scientific, 1:200), phosphohistone H3 (Upstate, 1:300), p-S6 (Cell Signaling, 1:200), Aurora B (BD Biosciences, 1:300), ARL13B (a kind gift from Jonathan Eggenschwiler, 1:5000), Keratin (Sigma, 1:250), N-Cadherin (Hybridoma Bank, 1:200), Lkb1 (Santa Cruz, 1:200). EdU was purchased from Molecular Probes.
For Western: p-S6 (Cell Signaling, 1:1000), p-ACC (Cell Signaling, 1:1000), ACC (Cell Signaling, 1:1000), S6 (Cell Signaling, 1:1000), Lkb1 (Sigma, 1:3000), α-tubulin (Hybridoma Bank, 1:10,000), β-Actin (Thermo Scientific, 1:5000), Gli1 (Cell Signaling, 1:1500), p-AMPK (Cell Signaling, 1:1000).
Supplementary Material
Highlights.
Lkb1 regulates cerebellar cortical size and foliation.
Cerebellar foliation is independent of mTOR signaling.
Loss of Lkb1 does not alter Hh signaling or proliferation.
Radial migration of post-mitotic GCP depends on the Lkb1 function.
Acknowledgments
We are grateful to Kevin Ess and Eric Armour for providing the TSC1flox/flox mice and to David Rowitch for providing the Atoh1-cre mice. We are also grateful to Yongliang Huo for his assistance with Lkb1 immunohistochemistry. We thank Joe Roland of the Digital Histology Shared Resource for his assistance with Cell Profiler, and Sean Schaffer at the Vanderbilt University Cell Imaging Shared Resource for use of the confocal microscope. We are grateful to Laura Lee and Ian Macara for their careful reading and assistance preparing the manuscript. This work was supported by American Heart Association (AHA) pre-doctoral fellowship (K.E.R.), Vanderbilt-Ingram Cancer Center Support Grant P30 CA068485 (C.C.), and National Institutes of Health NS 097898 (C.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
K.R. and C.C. designed and wrote the manuscript. K.R., P.K., E.B., J.F., and F.C. performed experiments. K.R., Y.L. and C.C. analyzed data.
Additional Information
The authors declare no competing financial interests.
References
- 1.Tavano A, Borgatti R. Evidence for a link among cognition, language and emotion in cerebellar malformations. Cortex. 2010;46:907–918. doi: 10.1016/j.cortex.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Bellebaum C, Daum I. Mechanisms of cerebellar involvement in associative learning. Cortex. 2011;47:128–136. doi: 10.1016/j.cortex.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Rochefort C, et al. Cerebellum shapes hippocampal spatial code. Science. 2011;334:385–389. doi: 10.1126/science.1207403. [DOI] [PubMed] [Google Scholar]
- 4.Ito M. Control of mental activities by internal models in the cerebellum. Nat Rev Neurosci. 2008;9:304–313. doi: 10.1038/nrn2332. [DOI] [PubMed] [Google Scholar]
- 5.Clark DA, Mitra PP, Wang SS. Scalable architecture in mammalian brains. Nature. 2001;411:189–193. doi: 10.1038/35075564. [DOI] [PubMed] [Google Scholar]
- 6.Sultan F. Analysis of mammalian brain architecture. Nature. 2002;415:133–134. doi: 10.1038/415133b. [DOI] [PubMed] [Google Scholar]
- 7.Altman J, Bayer S. Development of the cerebellar system: in relation to its evolution, structure, and functions. CRC Press Inc; New York: 1997. [Google Scholar]
- 8.Sillitoe RV, Joyner AL. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, et al. The Engrailed homeobox genes determine the different foliation patterns in the vermis and hemispheres of the mammalian cerebellum. Development. 2010;137:519–529. doi: 10.1242/dev.027045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 11.Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 12.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 13.Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 14.Miyazawa K, et al. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci. 2000;20:5756–5763. doi: 10.1523/JNEUROSCI.20-15-05756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baas AF, et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 2004;116:457–466. doi: 10.1016/s0092-8674(04)00114-x. [DOI] [PubMed] [Google Scholar]
- 16.Xu X, Omelchenko T, Hall A. LKB1 tumor suppressor protein regulates actin filament assembly through Rho and its exchange factor Dbl independently of kinase activity. BMC Cell Biol. 2010;11:77. doi: 10.1186/1471-2121-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng B, Cantley LC. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:819–822. doi: 10.1073/pnas.0610157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcus AI, Zhou W. LKB1 regulated pathways in lung cancer invasion and metastasis. J Thorac Oncol. 2010;5:1883–1886. doi: 10.1097/JTO.0b013e3181fbc28a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boudeau J, Sapkota G, Alessi DR. LKB1, a protein kinase regulating cell proliferation and polarity. FEBS Lett. 2003;546:159–165. doi: 10.1016/s0014-5793(03)00642-2. [DOI] [PubMed] [Google Scholar]
- 20.Asada N, Sanada K. LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J Neurosci. 2010;30:8852–8865. doi: 10.1523/JNEUROSCI.6140-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asada N, Sanada K, Fukada Y. LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J Neurosci. 2007;27:11769–11775. doi: 10.1523/JNEUROSCI.1938-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Barnes AP, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 24.Courchet J, et al. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacob LS, et al. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4:ra4. doi: 10.1126/scisignal.2001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Men Y, et al. LKB1 Regulates Cerebellar Development by Controlling Sonic Hedgehog-mediated Granule Cell Precursor Proliferation and Granule Cell Migration. Sci Rep. 2015;5:16232. doi: 10.1038/srep16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi S, Lewis P, Pevny L, McMahon AP. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev. 2002;119(Suppl 1):S97–S101. doi: 10.1016/s0925-4773(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 29.Uhlmann EJ, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 30.Schuller U, et al. Forkhead transcription factor FoxM1 regulates mitotic entry and prevents spindle defects in cerebellar granule neuron precursors. Mol Cell Biol. 2007;27:8259–8270. doi: 10.1128/MCB.00707-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parathath SR, Mainwaring LA, Fernandez LA, Campbell DO, Kenney AM. Insulin receptor substrate 1 is an effector of sonic hedgehog mitogenic signaling in cerebellar neural precursors. Development. 2008;135:3291–3300. doi: 10.1242/dev.022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regard JB, et al. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat Med. 2013;19:1505–1512. doi: 10.1038/nm.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleming JT, et al. The Purkinje neuron acts as a central regulator of spatially and functionally distinct cerebellar precursors. Dev Cell. 2013;27:278–292. doi: 10.1016/j.devcel.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Zhang H, Litingtung Y, Chiang C. Cholesterol modification restricts the spread of Shh gradient in the limb bud. Proc Natl Acad Sci U S A. 2006;103:6548–6553. doi: 10.1073/pnas.0600124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lancaster MA, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011;17:726–731. doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandt MD, Hubner M, Storch A. Brief report: Adult hippocampal precursor cells shorten S-phase and total cell cycle length during neuronal differentiation. Stem Cells. 2012;30:2843–2847. doi: 10.1002/stem.1244. [DOI] [PubMed] [Google Scholar]
- 37.Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kaern M, Cheng HY. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 2013;5:961–973. doi: 10.1016/j.celrep.2013.10.037. [DOI] [PubMed] [Google Scholar]
- 38.Wang VY, Zoghbi HY. Genetic regulation of cerebellar development. Nat Rev Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- 39.Ben-Arie N, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 40.Wang VY, Rose MF, Zoghbi HY. Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron. 2005;48:31–43. doi: 10.1016/j.neuron.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 41.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48:17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 42.Pan N, Jahan I, Lee JE, Fritzsch B. Defects in the cerebella of conditional Neurod1 null mice correlate with effective Tg(Atoh1-cre) recombination and granule cell requirements for Neurod1 for differentiation. Cell Tissue Res. 2009;337:407–428. doi: 10.1007/s00441-009-0826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sudarov A, Joyner AL. Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev. 2007;2:26. doi: 10.1186/1749-8104-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryan KE, Chiang C. Hedgehog secretion and signal transduction in vertebrates. J Biol Chem. 2012;287:17905–17913. doi: 10.1074/jbc.R112.356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bardeesy N, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 46.Ochoa-Espinosa A, Affolter M. Branching morphogenesis: from cells to organs and back. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray S, Lechler T. Regulation of asymmetric cell division in the epidermis. Cell Div. 2011;6:12. doi: 10.1186/1747-1028-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poulson ND, Lechler T. Robust control of mitotic spindle orientation in the developing epidermis. J Cell Biol. 2010;191:915–922. doi: 10.1083/jcb.201008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhatia B, et al. Tuberous sclerosis complex suppression in cerebellar development and medulloblastoma: separate regulation of mammalian target of rapamycin activity and p27 Kip1 localization. Cancer Res. 2009;69:7224–7234. doi: 10.1158/0008-5472.CAN-09-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–380. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wefers AK, Lindner S, Schulte JH, Schuller U. Overexpression of Lin28b in Neural Stem Cells is Insufficient for Brain Tumor Formation, but Induces Pathological Lobulation of the Developing Cerebellum. Cerebellum. 2016 doi: 10.1007/s12311-016-0774-0. [DOI] [PubMed] [Google Scholar]
- 54.Hosaka YZ, Neki Y, Hasebe M, Shinozaki A, Uehara M. Formation of excess sublobules in the cerebellum of hypothyroid rats. Ann Anat. 2012;194:329–333. doi: 10.1016/j.aanat.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Kullmann JA, et al. Profilin1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 2012;13:75–82. doi: 10.1038/embor.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kokubo M, et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J Neurosci. 2009;29:8901–8913. doi: 10.1523/JNEUROSCI.0040-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, et al. Nuclear factor I coordinates multiple phases of cerebellar granule cell development via regulation of cell adhesion molecules. J Neurosci. 2007;27:6115–6127. doi: 10.1523/JNEUROSCI.0180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 59.Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci. 2014;15:217–232. doi: 10.1038/nrn3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borrell V, Gotz M. Role of radial glial cells in cerebral cortex folding. Curr Opin Neurobiol. 2014;27C:39–46. doi: 10.1016/j.conb.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Del Toro D, et al. Regulation of Cerebral Cortex Folding by Controlling Neuronal Migration via FLRT Adhesion Molecules. Cell. 2017;169:621–635 e616. doi: 10.1016/j.cell.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 62.Tanori M, et al. Developmental and oncogenic effects of insulin-like growth factor-I in Ptc1+/− mouse cerebellum. Mol Cancer. 2010;9:53. doi: 10.1186/1476-4598-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng FY, et al. Widespread contribution of Gdf7 lineage to cerebellar cell types and implications for hedgehog-driven medulloblastoma formation. PLoS One. 2012;7:e35541. doi: 10.1371/journal.pone.0035541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dey J, et al. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol. 2012;32:4104–4115. doi: 10.1128/MCB.00862-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.