

Abstract
Estrogens are potent regulators of vascular tone, yet underlying receptor- and ligand-specific signaling pathways remain poorly characterized. The primary physiological estrogen 17β-estradiol (E2), a non-selective agonist of classical nuclear estrogen receptors (ERα and ERβ) as well as the G protein-coupled estrogen receptor (GPER), stimulates formation of the vasodilator nitric oxide (NO) in endothelial cells. Here, we studied the contribution of GPER signaling in E2-dependent activation of endothelial NO formation and subsequent vasodilation. Employing E2 and the GPER-selective agonist G-1, we investigated eNOS phosphorylation and NO formation in human endothelial cells, and endothelium-dependent vasodilation in the aortae of wild-type and Gper-deficient mice. Both E2 and G-1 induced phosphorylation of eNOS at the activation site Ser1177 to similar extents. Endothelial NO production to E2 was comparable to that of G-1, and was substantially reduced after pharmacological inhibition of GPER. Similarly, the clinically used ER-targeting drugs 4OH-tamoxifen, raloxifene, and ICI182,780 (faslodex, fulvestrant™) induced NO formation in part via GPER. We identified c-Src, EGFR, PI3K and ERK signaling pathways to be involved in GPER-dependent NO formation. In line with activation of NO formation in cells, E2 and G-1 induced equally potent vasodilation in the aorta of wild-type mice. Gper deletion completely abrogated the vasodilator response to G-1, while reducing the response to E2 by ~50%. These findings indicate that a substantial portion of E2-induced endothelium-dependent vasodilation and NO formation is mediated by GPER. Thus, selective targeting of vascular GPER may be a suitable approach to activate the endothelial NO pathway, possibly leading to reduced vascular tone and inhibition of atherosclerotic vascular disease.
Keywords: eNOS, estrogen, endothelium, GPER, GPR30, NO, SERD, SERM, vascular, vasodilation
INTRODUCTION
Epidemiological studies show a lower incidence of coronary artery disease, hypertension and stroke in premenopausal women compared to age-matched men; however, these sex differences lessen with the onset of menopause [1, 2]. Such observations suggest that the loss of ovarian sex hormones coincides with a loss of protection against vascular diseases, implicating a beneficial functional role for natural estrogen (predominantly 17β-estradiol, E2) in arterial health [3–5]. In particular, endothelial cells, which form the luminal cell monolayer of the vascular wall, have emerged as critical mediators of estrogen’s salutary effects in the cardiovascular system [3, 6, 7].
Endothelial cells release multiple vasoactive substances including the endothelium-derived relaxing factor (EDRF) [8], which was identified as nitric oxide (NO) [9, 10]. The enzyme that catalyzes NO formation (utilizing L-arginine and molecular oxygen as substrates) is NO synthase (NOS) [11, 12]. Three NOS isoforms exist, with one isoform (NOS III) being predominantly expressed in endothelial cells and hence referred to as endothelial NOS (eNOS) [11, 12]. eNOS-derived NO is a potent vasodilator, but also conveys vasoprotection through multiple mechanisms such as inhibition of leukocyte adhesion and migration, platelet aggregation and thrombosis, as well as mitigating proliferation and migration of the underlying vascular smooth muscle cells [12]. E2 has been shown to stimulate NO formation in cultured human endothelial cells [13]. In particular, E2 binding to estrogen receptors (ER) triggers signaling cascades that include activation of the kinases c-Src [14], ERK [15, 16], PI3K [17], and Akt [18], the latter eliciting phosphorylation of the eNOS activation residue Ser1177 [19–22]. Studies in murine aortae have confirmed the involvement of these pathways in endothelium-dependent, NO-mediated vasodilation induced by E2 [19, 23, 24]. Different ER subtypes (the holoreceptor ERα its truncated isoform ER46, and possibly ERβ) localized to the plasma membrane have been associated with these actions [25–27].
In 1997, an orphan G protein-coupled receptor termed GPR30 was cloned from human endothelial cells exposed to fluid shear stress [28], a prototypic physiologic stimulus of eNOS activation [29]. Following the demonstration of binding and signaling in response to E2 [17, 30], GPR30 was renamed G protein-coupled estrogen receptor (GPER) [31]. Experiments utilizing GPER-deficient (Gper−/−) mice [32] and GPER-selective ligands, the agonist G-1 [33, 34] and antagonists G15 and G36 [35, 36], have established the vascular actions of this third estrogen receptor in vascular biology [37–45] as well as many other aspects of physiology [46–51]. We and others have previously shown that G-1 induces eNOS activation in human endothelial cells [52–54], as well as endothelium-dependent, NO-mediated vasodilation of multiple human and rodent arteries [3, 53, 55, 56]. These findings established GPER as an ER mediating eNOS activation; however, the extent to which GPER contributes to the overall E2-dependent response and the activation of eNOS by multiple ER-targeting drugs remain unresolved. Hence, we set out to determine the role of GPER in eNOS phosphorylation and the mechanisms of NO production in human endothelial cells, as well as vasodilation in the aorta of wild-type and Gper−/− mice. To this end, we utilized the GPER-selective agonist G-1 [33] and the non-selective ER agonist E2, as well as the selective ER modulators (SERMs) 4OH-tamoxifen and raloxifene and the selective ER downregulator (SERD) ICI182,780 (faslodex, fulvestrant™), which have been shown to act as GPER agonists in other systems [57].
METHODS
Human endothelial cells
Telomerase-immortalized human umbilical vein endothelial (TIVE) cells were a generous gift from Rolf Renne, PhD (University of Florida, Gainesville, FL), and their derivation has been described previously [58]. Sex of the cells was determined by fluorescence in situ hybridization (FISH) analysis (TriCore Reference Laboratories, Albuquerque, NM). Primary single donor human umbilical vein endothelial cells (HUVEC) were obtained from Lonza (cat # C2517AS). TIVE and HUVEC cells were cultured in M199 basal media supplemented with 20% FBS, 100 μg/mL bovine neural-derived endothelial growth factor and antibiotics (50 U/mL penicillin, 50 μg/mL streptomycin). All tissue culture vessels were coated for 30–60 min at room temperature with 0.1% sterile gelatin in sterile milliQ-filtered water prior to seeding cells. The expression pattern of endothelial cell-specific markers remains unchanged from passages 2 to 12 in this cell line [58], and TIVE cells were used up to passage 9 for experiments. HUVEC were used below passage 4.
Quantitative real-time polymerase chain reaction (qPCR)
Gene expression of GPER and eNOS in TIVE cells was analyzed at passages 3, 6, 9 and 12 and in HUVEC at passage 3. Total cellular RNA was extracted using Trizol Reagent and eluted using Qiagen RNeasy Mini kit (Qiagen, Valencia, CA) according the manufacturer’s instructions. RNA (400 ng) was reverse transcribed using the First Strand cDNA synthesis kit (Applied Biosystems, Carlsbad, CA). SYBR green-based detection of amplified gene-specific cDNA fragments was performed on a 7500 FAST real-time PCR system (Applied Biosystems). Primer sequences are provided in Table 1. GAPDH served as a house-keeping control.
Table 1.
Sets of primers used for amplification of gene-specific cDNA fragments by qPCR.
| Gene (Accession number) |
Forward Primer | Reverse Primer |
|---|---|---|
| Human GPER (NM_001098201.1) | 5′-GTA CCC AGA AGT GAG CAG CT-3′ | 5′-GTG CAT CCG TGG AGG CGA GG-3′ |
| Human eNOS (NM_008713) | 5′-AGA GCC TGC AAT TAC TAC CA-3′ | 5′-GTG GAT TTG CTG CTC TGT AG-3′ |
| Human ERα (NM_000125.3) | 5′TGA TTG GTC TCG TCT GGC G-3′ | 5′-CAT GCC CTC TAC ACA TTT TCC C-3′ |
| Human GAPDH (NM_00804) | 5′-TTC ACC ACC ATG GAG AAG GC-3′ | 5′-GGC ATG GAC TGT GGT CAT GA-3′ |
Fluorescence microscopy
Cells were seeded onto gelatin-coated coverslips (approximately 30,000 cells per coverslip in a 24 well cell culture plate) for 48 hours and fixed in PBS containing 4% paraformaldehyde for 15 min at room temperature. For examining GPER localization, cells were treated with either permeabilizing (PBS containing 3% BSA and 0.1% Triton X-100) or non-permeabilizing (PBS containing 3% BSA) blocking buffer for 1 hour at room temperature, and incubated with a rabbit anti-mouse GPER antibody targeting a sequence within the second extracellular loop (acetyl-FADVREVQWLEVTLGFIC, 1:10,000) [18] or negative control pre-immune rabbit serum overnight at 4°C. Slides were then washed 3 times with permeabilizing (0.1% Triton X-100) or non-permeabilizing PBS, incubated with goat anti-rabbit IgG conjugated to Alexa Fluor 488 (1:500) for 1 h at room temperature, washed three times with PBS, and mounted in Vectashield supplemented with DAPI (200 ng/mL). To evaluate ERα and GPER staining in TIVE and HUVEC. cells were permeabilized and incubated with 1:50 mouse IgG antibody targeting ERα (Santa Cruz sc8002 clone F10) overnight at 4°C, washed three times with PBS-T and incubated with 1:500 rabbit anti-mouse Alexa Fluor 488. Cells were co-stained using a GPER antibody targeting the N-terminus [17] at 1:10,000 and 1:500 anti-rabbit Alexa Fluor 568. DAPI was used as a nuclear counterstain described above. Fluorescence signals were visualized using a Zeiss LSM510 Meta or Zeiss 720 confocal fluorescent microscope.
Western blotting
TIVE cells (200,000 cells per well) were seeded and grown to 70–80% confluence in 6-well plates and serum starved overnight. The next morning, cells were treated with the non-selective estrogen receptor agonist E2 (100 nM), the GPER-selective agonist G-1 (1–100 nM) or vehicle (DMSO 0.01 %) for up to 15 min [52], briefly washed with ice cold PBS and lysed on ice with 50 μL of ice cold NP-40 lysis buffer supplemented with 1% SDS, 5 μM NaVO4 and 5 μM NaF to preserve phosphorylated proteins. Cell debris was pelleted at 12,000 × g at 4 °C and the supernatant of soluble proteins was collected, aliquoted and stored at −80°C. Protein concentrations were determined by Coomassie (Bradford) assay (Pierce, Rockfield, IL). For each sample, 20 μg of total protein was resolved by 10% SDS PAGE and blotted to PVDF membrane. Blots were blocked for 1 hour at room temperature in TBS-T 0.01% supplemented with 3% newborn calf serum and incubated overnight at 4°C with mouse anti-human pSer1177-eNOS (1:500, antibody 612392, BD Biosciences, Sparks, MD) or mouse anti-human β-actin (1:10,000, antibody MAB1501, Millipore, Darmstadt, Germany) antibodies. Blots were then washed, incubated with secondary HRP-conjugated antibodies (1:5000) for 1 hour at room temperature, developed with PicoWest Chemiluminescence detection (ThermoScientific) for one minute at room temperature. Blots were imaged on Kodak X ray film and quantified using ImageJ densitometry analysis software (National Institutes of Health).
Detection of NO
NO was determined by detection of the stable NO metabolites NO2/NO3 (Nitric Oxide Detection Kit, ab65328, Abcam, Boston, MA). TIVE cells (300,000) were seeded onto 60 mm dishes. At 70% confluence, cells were serum starved in HEPES-buffered physiologic saline solution (HEPES-PSS, composition in mM: 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 0.026 EDTA, 10 glucose, and 10 HEPES; pH 7.4) for three hours prior to experiments. Cells were treated with either the non-selective ER agonist E2 (100 nM), the GPER-selective agonist G-1 (100 nM), the SERMs raloxifene (100 nM) and 4OH-tamoxifen (100 nM), the SERD ICI182,780 (100 nM), acetylcholine (100 nM), or vehicle (DMSO 0.01%) for 15 min in HEPES-PSS at 37°C. TIVE cells were lysed on ice with 80 μL Griess assay sample buffer. Per the manufacturer’s recommendation, we further lysed each sample by pulling it through a 27.5 gauge needle, briefly pelleted cell debris (3 min in at 13,000 rpm at 4°C) and collected the supernatants for Griess assay. Samples were either stored overnight at −80°C or immediately loaded into a 96-well plate, treated with nitrate reductase and nitrate reductase cofactor, and incubated at room temperature for 90 min to convert nitrates. Sequential addition of Griess reagents 1 and 2 was used to develop azo purple for 5 min at room temperature. Spectrophotometric detection of azo purple absorbance at 540 nm was performed on a Synergy H1 multimode microplate reader (BioTek, Winooski, VT) at room temperature. Three readings per plate were taken. Background signal was subtracted and net azo signal was normalized to total protein (determined by Coomassie blue readings at 533 nm) for each replicate. Samples were analyzed in triplicate for each experiment, and at least three experiments were performed for each condition. As indicated, selected experiments were repeated in the presence of the GPER-selective inhibitor G36 (1 μM), the EGFR inhibitor AG1478 (1 μM), the c-Src inhibitor PP2 (1 μM), the PI3K inhibitor LY294002 (1 μM), or the ERK inhibitor PD98059 (1 μM) for 30 min at 37°C prior to stimulation with E2 or G-1.
Animals
Gper−/− mice (Procter & Gamble, Cincinnati, OH, provided by Jan S. Rosenbaum) were generated and backcrossed onto the C57BL/6J background as described [52]. Male Gper−/− and wild-type mice (Harlan Laboratories, Indianapolis, IN) were bred and housed at the University of New Mexico Animal Resources Facility under controlled temperature of 22–23°C on a 12 hour light-dark cycle. Mice were given ad libitum access to water and a rodent diet devoid of alfalfa or soybean meal to minimize the presence of natural phytoestrogens (Teklad 2020SX, Harlan Laboratories, Madison, WI). All procedures were approved by the University of New Mexico Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Vascular function studies
At 3–5 months of age, Gper−/− and wild-type mice were sacrificed by intraperitoneal injection of 2 mg/g sodium pentobarbital. The thoracic aorta was immediately excised, transferred to ice-cold PSS (in mM: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose; pH 7.4), and cleaned of fat and connective tissue to exclude perivascular adipose-dependent contractile effects [59]. Vessels were cut into 3 mm long rings, and carefully mounted onto two 200 μm pins of a Mulvany-Halpern myograph (620M Multi Wire Myograph System, Danish Myo Technology, Aarhus, Denmark) to record isometric tension using a PowerLab 8/35 data acquisition system and LabChart Pro software (AD Instruments, Colorado Springs, CO). Vascular function experiments were performed as previously described [59]. Briefly, functional integrity of the vascular smooth muscle was assessed by repeatedly exposing vessels to KCl (PSS with equimolar substitution of 60 mM potassium for sodium). The presence of an intact endothelium was confirmed by precontracting vascular rings with phenylephrine (1 μM), followed by exposure to acetylcholine (1 μM) to induce endothelium-dependent, NO-mediated relaxation. A relaxation response of >80% was considered to represent an intact and functional endothelium. Rings were then precontracted with prostaglandin F2α to 30–40% of the KCl-induced contraction, and the vasorelaxation response to E2 (3 μM) or G-1 (3 μM) was recorded for 50 min as described [38]. Ethanol at a final concentration of 0.1% served as solvent control. Experiments were conducted in the presence of meclofenamate (pretreated with 1 μM for 30 min) to exclude effects of endothelial vasoconstrictor prostanoids [38]. Relaxation is expressed as the percentage of precontraction to prostaglandin F2α.
Materials
Prostaglandin F2α, meclofenamate, AG1478, PP2, LY294002, and PD98059 were from Cayman Chemical (Ann Arbor, MI). M199 medium was from Gibco (ThermoFisher). G-1 and G36 were synthesized as described [33, 36] and provided by Jeffrey Arterburn (New Mexico State University, Las Cruces, NM). All other drugs were from Sigma-Aldrich (St. Louis, MO).
Statistical analyses
When comparing multiple groups, data were analyzed using one-way or two-way analysis of variance (ANOVA) with repeated measures as appropriate followed by Bonferroni’s post-hoc test. When comparing two groups, the unpaired Student’s t-test was used. All analyses were performed using GraphPad Prism version 5.0 for Macintosh (GraphPad Software, San Diego, CA). Values are expressed as mean of independent experiments, and error bars represent SEM. n equals the number of animals or independent experiments. A p value <0.05 was considered statistically significant.
RESULTS
Expression and intracellular localization of GPER in human endothelial cells
To study the role of GPER in stimulated endothelial NO release, we employed a well-characterized hTERT-immortalized human endothelial cell line, TIVE cells [58]. The cells were derived from male fetal umbilical cord as demonstrated by FISH analysis for X and Y chromosome centromeres (Figure 1A). As GPER was originally cloned from endothelial cells [28], we confirmed expression of its mRNA, which was unaffected by limited serial passaging (Figure 1B). However, since eNOS gene expression decreased between passages 9 and 12 in TIVE cells (Figure 1B), all experiments were performed in cells passaged less than 9 times. RNA expression of ERα, eNOS, and GPER in TIVE cells was found to be similar to primary HUVECs (Figure 1C). GPER protein staining employing an antibody that recognizes a putative “extracellular loop” [18] detected GPER in TIVE cells only under permeabilizing (and not non-permeabilizing) conditions, indicating that the majority of the protein is localized intracellularly in TIVE cells (Figure 1D, E). Comparing GPER staining to ERα staining in both TIVE cells and HUVECs, the majority of ERα localized to the nucleus in both cell types, whereas GPER was localized to membranes in the cytosol, with strong perinuclear localization (Figure 1F, G). We therefore conclude that GPER is predominantly localized to intracellular membranes in human endothelial cells under steady-state conditions, similar to previous observations in other cell types [17, 52], and that GPER and ERα are each similarly expressed in HUVEC and TIVE cells.
Figure 1. GPER is expressed on intracellular membranes in human endothelial cells.
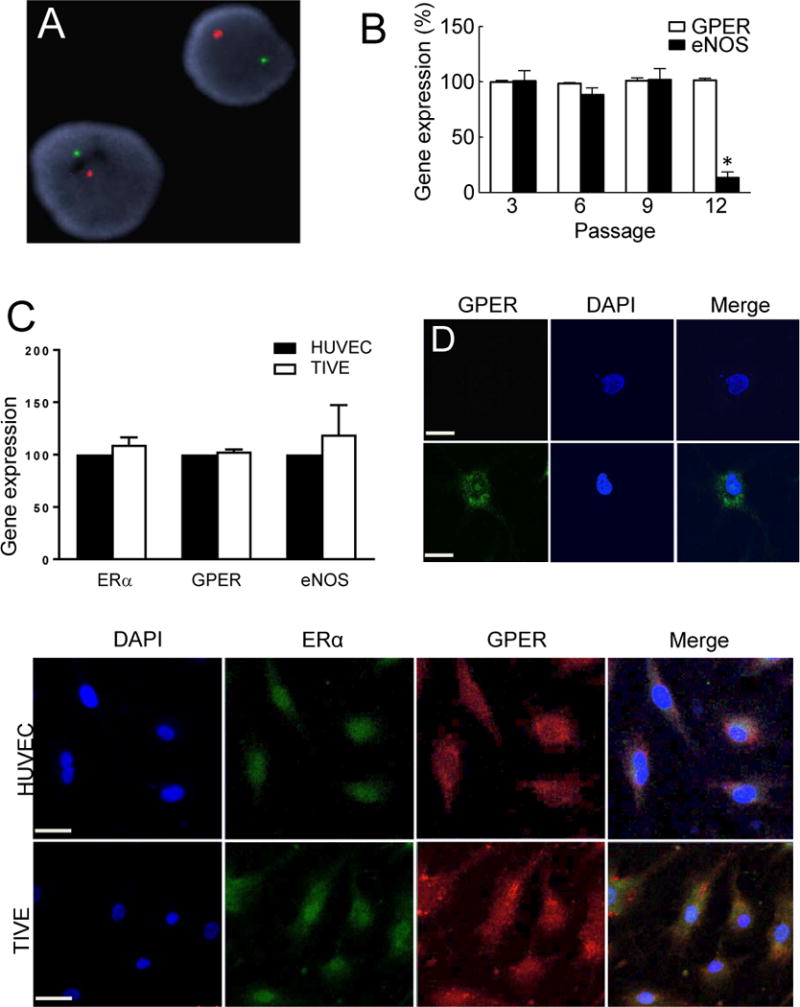
The donor sex of TIVE cells was determined to be male by FISH analysis for X (red) and Y (green) chromosomes (A). GPER (white bars) and eNOS (black bars) expression in TIVE cells was determined by qPCR at the indicated passages (B). Gene expression of ERα and GPER was similar between TIVE and HUVEC at passage 3 (C). TIVE cells were stained by immunofluorescence for GPER (green) under non-permeabilizing (D) or permeabilizing (E) conditions demonstrating intracellular expression (staining only under permeabilizing conditions) of GPER. Cells were counterstained with nuclear DAPI (blue). Immunofluorescence of ERα (green) and GPER (red) indicate predominantly cytosolic localization for GPER and predominantly nuclear localization for ERα in both HUVEC (F) and TIVE (G) cells. Data were analyzed by one-way ANOVA with repeated measures followed by Bonferroni’s post-hoc test (B) or Student’s t-test (C) and graphed as mean±s.e.m.; *P<0.01 vs. passage 3.
GPER mediates phosphorylation of eNOS at Ser1177
We next examined the effects of the non-selective ER agonist E2 and the GPER-selective agonist G-1 [33] on eNOS activation by determining the phosphorylation status of the critical activation residue Ser1177 in human endothelial cells. We observed a G-1-mediated, dose-dependent increase in eNOS phosphorylation with a 3-fold increase in activation at 100 nM (p<0.05 vs. vehicle, n=4–7), which was similar to eNOS phosphorylation stimulated by 100 nM E2 (Figure 2).
Figure 2. GPER stimulates eNOS phosphorylation.
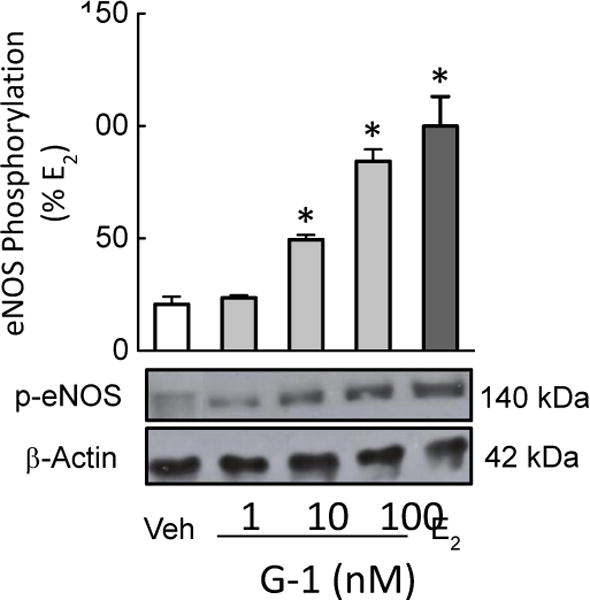
Endothelial cells were treated with the GPER-selective agonist G-1 (1, 10, and 100 nM) or E2 (100 nM) and blotted for eNOS phosphorylation at activation residue Ser1177. Data (n=4–7) were analyzed by one-way ANOVA followed by Bonferroni’s post-hoc test and graphed as mean±s.e.m.; *P<0.05 vs. vehicle (Veh, DMSO 0.01%).
E2 and G-1 mediate NO formation via GPER
We next sought to determine whether phosphorylation of eNOS by E2 and G-1 translated into increased NO production. Relative to basal NO, non-selective ER activation by E2 rapidly stimulated NO formation by 5-fold (p<0.01 vs. vehicle, n=4–6, Figure 3), with selective activation of GPER by G-1 yielding approximately 73% of the E2-induced response (p<0.01 vs. vehicle, n=4–6, Figure 3). Pretreating cells with the GPER-selective antagonist G36 [36] reduced NO production in response to E2 by 58%, and completely blocked the G-1-induced response (both p<0.01, n=4, Figure 3). By comparison, activation of the NO pathway by the muscarinic M3 receptor agonist acetylcholine showed no difference in NO formation compared with E2 (124±13% vs. 100±11%, n=6–8, p=n.s.), and was unaffected by GPER inhibition (n=5, Figure 3). These findings indicate that GPER alone can mediate a substantial portion of the E2-induced eNOS activation response and NO formation in human endothelial cells.
Figure 3. Selective and non-selective GPER activation mediates NO formation.
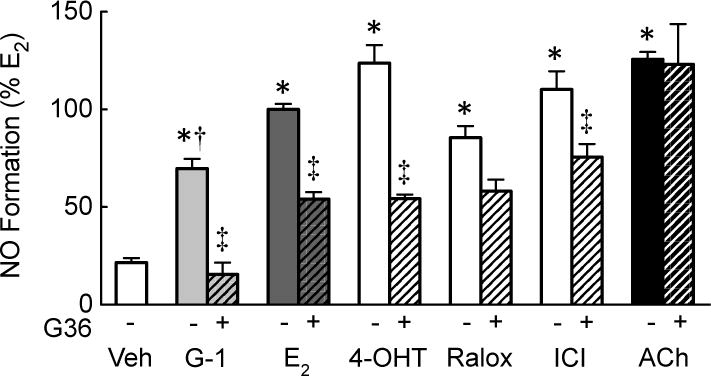
Endothelial cells were treated with vehicle (Veh, DMSO 0.01%) or the GPER-selective antagonist G36 (1 μM) prior to stimulation with the GPER-selective agonist G-1, the non-selective ER agonist E2, the SERMs 4OH-tamoxifen (4-OHT) and raloxifene (Ralox), or the SERD ICI182,780 (ICI, 100 nM each). For comparison, the response to the muscarinic M3 receptor agonist acetylcholine (ACh, 100 nM) is shown. NO formation was determined through the detection of stable NO metabolites NO2−/NO3−. Data (n=3–8) were analyzed by two-way ANOVA followed by Bonferroni’s post-hoc test and graphed as mean±s.e.m.; *P<0.01 vs. vehicle, †P<0.05 vs. E2, ‡P<0.05 vs. without G36.
GPER-dependent NO formation is induced by selective estrogen receptors modulators and downregulators
SERMs such as 4OH-tamoxifen and raloxifene as well as SERDs such as ICI182,780 (faslodex, fulvestrant™) are classically thought to target ERα/ERβ, yet more recent data identified these compounds as GPER agonists [15, 57]. SERMs/SERDs are used in the treatment of estrogen-sensitive cancer and osteoporosis, inducing both pro-estrogenic and anti-estrogenic effects depending on the tissue and cell type [60]. We hypothesized that SERMs/SERDs might also induce NO production through GPER activation in endothelial cells. 4OH-tamoxifen, raloxifene and ICI182,780 stimulated NO production to a similar extent compared to E2 (Figure 3). Acute inhibition of GPER using G36 reduced NO production to 4OH-tamoxifen by 56% (n=3, p<0.05), to raloxifene by 31% (n=3, p=0.086), and to ICI 182,780 by 33% (n=3, p<0.05, Figure 3). These data establish that GPER contributes to endothelial NO formation induced by SERMs and SERDs and extend our knowledge regarding the pharmacological properties of these clinically approved drugs.
GPER induces NO production in endothelial cells through c-Src, EGFR, PI3K and ERK pathways
We next sought to determine the signaling pathway(s) involved in GPER-mediated NO formation in human endothelial cells using inhibitors targeting multiple pathways previously associated with GPER signaling, including c-Src, trans-activation of the epidermal growth factor receptor (EGFR), PI3K and ERK1/2 [40]. c-Src has been shown to directly phosphorylate eNOS at the activation residue Tyr83 [22, 61]. In endothelial cells, inhibition of c-Src by PP2 reduced NO production in response to the GPER-selective agonist G-1 by 63%, and to the non-selective ER agonist E2 by 54% (both n=4–6, p<0.01, Figure 4A). Similarly, inhibition of EGFR with AG1478 diminished NO production in response to G-1 by 62%, and to E2 by 84% (both n=3–4, p<0.01, Figure 4B). EGFR transactivation initiates a number of downstream secondary messengers such as PI3K/Akt, which directly mediates eNOS phosphorylation at ser1177 [21]. Indeed, PI3K inhibition, by pretreatment of endothelial cells with LY294002, reduced NO production in response to G-1 by 46%, and to E2 by 54% (n=4, p<0.05, Figure 4C). An alternative signaling target of GPER is the kinase ERK1/2, shown to activate eNOS at phosphorylation sites Thr602 and Thr604 [62]. Inhibition of ERK1/2, by pretreatment with PD98059, reduced NO production in response to G-1 by 59%, and to E2 by 48% (both n=4, p<0.05, Figure 4D), identifying ERK as an additional component of GPER-dependent NO formation in human endothelial cells. Taken together, these data indicate that both non-selective ER (E2) and selective GPER (G-1) activation of NO formation is mediated by c-Src, EGFR, PI3K and ERK pathways.
Figure 4. GPER contributes to NO production via multiple signaling pathways.
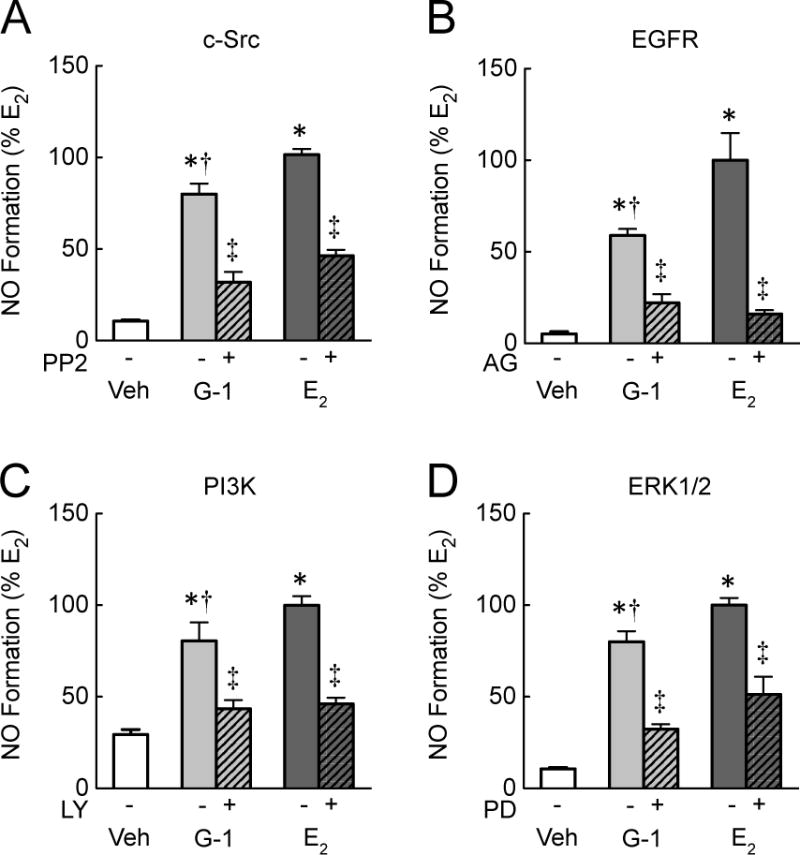
NO formation in endothelial cells was induced by the GPER-selective agonist G-1 or the non-selective ER agonist E2 (100 nM each). Cells were pretreated with multiple inhibitors of GPER signaling components that are known upstream activators of eNOS: PP2 for c-Src (A), AG1478 (AG) for EGFR (B), LY294002 (LY) for PI3K (C), and PD98059 (PD) for ERK1/2 (D). Data (n=4–6) were analyzed by two-way ANOVA followed by Bonferroni’s post-hoc test and graphed as mean±s.e.m.; *P<0.01 vs. vehicle (Veh, DMSO 0.01%), †P<0.05 vs. E2, ‡P<0.05 vs. without inhibitor.
GPER contributes to endothelium-dependent arterial dilatation by E2
Given the similar levels of E2- and G-1-induced activation of eNOS in endothelial cell, we next studied the contribution of GPER to E2- and G-1-induced, endothelium-dependent vasodilation in the aorta of wild-type and Gper−/− mice. This vascular bed displays dilation to E2 largely mediated by endothelial NO production [63–66]. In WT mice, both the non-selective ER agonist E2 and the GPER-selective agonist G-1 induced a time-dependent vasodilation (60±10% and 76±7%, n=3–8, both p<0.001 vs. solvent, Figure 5A). Interestingly, E2 induced a rapid relaxation within 10 minutes that was more potent compared to G-1 (31±8% vs. 8±5% at 10 min, p<0.05), whereas the sustained vasodilator response to E2 and G-1 was similar. Deletion of Gper reduced E2-induced vasodilation by 48% (from 60±10% to 31±6%, p<0.001, Figure 5A), indicating that a substantial portion of the overall vasodilator response is mediated by GPER. As expected, the GPER-selective agonist G-1 did not induce vasodilation in aortae from Gper−/− mice, demonstrating the specificity of G-1 towards GPER (Figure 5A). Furthermore, endothelium-dependent, NO-mediated vasodilation induced by acetylcholine was similar in WT and Gper−/− mice, excluding inherent differences in endothelial function between the two genotypes (92±2% vs. 90±3%, n=4–8, p=n.s., Figure 5B). Together, these findings indicate that E2 induces potent, endothelium-dependent vasodilation that is to a significant extent mediated by GPER.
Figure 5. GPER partially mediates endothelium-dependent vasodilation to E2.
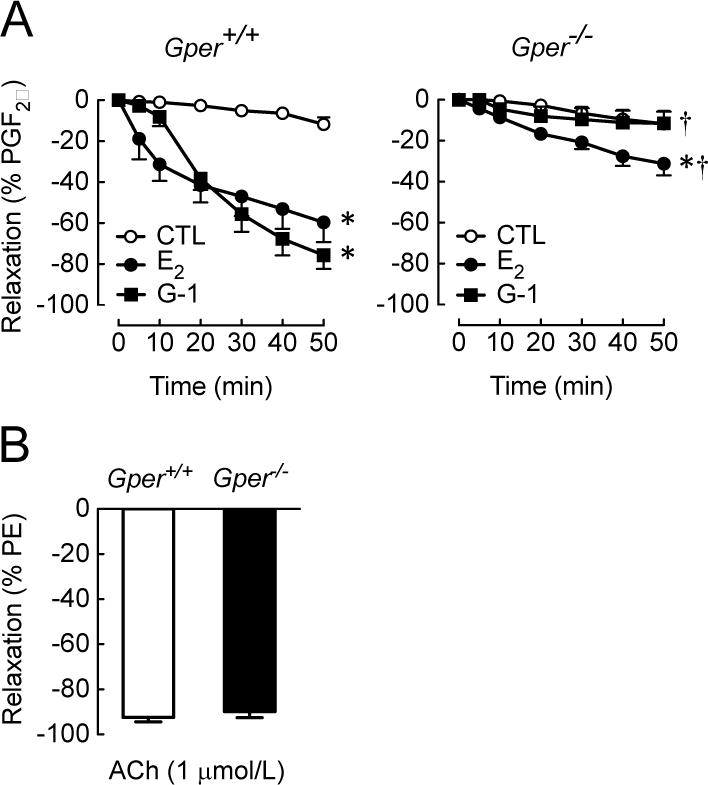
Direct vasodilator responses to the non-selective ER agonist E2 and the GPER-selective agonist G-1 (3 μM each) were induced in the aorta from wild-type (Gper+/+) and GPER-deficient (Gper−/−) mice (A). For comparison, ER-independent vasodilation to the muscarinic M3 receptor agonist acetylcholine (ACh,1 μM) is shown (B). Data (n=3–8) were analyzed by two-way ANOVA with repeated measures followed by Bonferroni’s post-hoc test and graphed as mean±s.e.m.; *P<0.001 vs. vehicle (CTL, EtOH 0.1%), †P<0.001 vs. Gper+/+. PGF2α, prostaglandin F2α; PE, phenylephrine.
DISCUSSION
The present study demonstrates the following insights: (i) that activation of GPER by SERMs and SERDs, as well as E2, is substantially involved in eNOS-dependent NO production, (ii) that NO production by E2 and G-1 require the activation of c-Src and EGFR as well as PI3K and ERK1/2, (iii) approximately 50% of the endothelium-dependent vasodilator response to E2 is dependent upon GPER and (iv) the GPER-selective agonist G-1 does not induce relaxation in Gper-deficient mice. Previous studies are consistent with our findings that eNOS activation by GPER involves the PI3K/Akt and ERK1/2 pathways and that vasodilation in response to G-1 involves a c-Src/EGFR-mediated pathway [52, 54, 55]. Although multiple forms of ERα have also been associated with E2-mediated eNOS activation [25], our current findings point towards an essential contribution of GPER to the overall endothelium-dependent, NO-mediated vascular response to E2 and reveal new insights regarding GPER in mediating the effects of clinically relevant ER-targeting therapeutics. GPER agonists are known to activate multiple signaling pathways that are also involved in E2-mediated NO production, including c-Src, EGFR transactivation, PI3K, and ERK1/2 [21, 22, 61, 62]. Our results show that inhibition of these signaling pathways reduced NO formation in response to both nonselective ER activation (E2) as well as GPER-selective activation (G-1), consistent with recent studies of GPER-mediated activation of eNOS [54]. These findings extend previous observations that plasma membrane-associated subpopulations of ERα are capable of mediating E2-induced eNOS activation via c-Src/PI3K [22], revealing an alternative pathway via GPER.
This study enhances our understanding of the role of GPER in mediating the effects of clinically used SERMs 4OH-tamoxifen and raloxifene, as well as the SERD ICI182,780 in endothelial generation of NO. Since these compounds have previously been shown to bind and activate GPER [14, 15, 17, 30], our results reinforce the notion that SERMs or SERDs may have additional effects outside of modifying ERα/ERβ signaling. SERMs impart pharmacologic effects by activation or inhibition of classic ERα/ERβ; however, SERM classification is specifically defined by their effects on ERα, regardless of their effects on GPER signaling. Conversely, ICI182,780, commonly known for its inhibitory effects on ERα/ERβ, has been shown to induce vasodilation in porcine epicardial arteries, an observation compatible with GPER-dependent mechanisms [38]. Interestingly, a common clinical side-effect of ICI182,780 is arterial hypotension [67], possibly resulting from GPER-mediated endothelial NO formation. Furthermore, there is a reduced incidence of adverse clinical events related to coronary artery disease in randomized trials of SERMs in the treatment of breast cancer and osteoporosis [60, 68, 69], potentially resulting from GPER-mediated increases in NO formation that contribute to the inhibition of atherosclerosis [52]. Thus, the discovery that clinically approved drugs are able to activate GPER may require reconsideration of their therapeutic rational.
Vasodilator effects to E2 in men were first described in 1939 [70] and have since been confirmed experimentally in numerous studies, with estrogen being particularly involved in the regulation of endothelium-dependent, NO-mediated modulation of vascular tone. In intact arteries, ERα may be a principal mediator of rapid E2-induced, NO-mediated, endothelium-dependent vasodilation [71], although more recent evidence suggests that selective pharmacological activation of GPER may potently recapitulate these effects [38, 72]. In line with previous reports [56, 73], we observed no difference between vasodilatory responses to E2 (non-selective ER activation) and G-1 (GPER-selective agonist). Furthermore, using a genetically modified mouse model, we found ~50% reduction of the relaxation response to E2 in GPER-deficient aortae. Collectively, these results indicate that GPER is capable of mediating a substantial portion of E2-induced vasodilatory responses independent of ERα.
Although it may seem intuitive that the benefits of estrogen receptor targeting would be greater in females, males may also benefit from ER-targeted therapeutics. In this study, we utilized endothelial cells from male fetal umbilical tissue and arteries from male mice, reinforcing previous observations indicating that E2 potently affects vascular function in males [74]. The findings of the present study also extend previous observations of a potent regulatory role of GPER on vasomotor tone in males [53, 59, 75]. Given the absence of feminizing effects of G-1 [36, 76], there may be pharmacologic utility in GPER-selective agonism via compounds such as G-1 in promoting cardiovascular health in men as well as women.
The substantial contribution of GPER in E2-mediated eNOS activation further supports the development of GPER-targeted compounds that may yield salutary vascular effects by promoting eNOS activity, increasing NO bioavailability and reducing vascular tone. Although large-scale clinical trials on hormone replacement therapy with conjugated equine estrogens and medroxyprogesterone acetate in postmenopausal women reported adverse effects on cardiovascular risk [77, 78], subsequent re-analyses revealed decreased risk if therapy was initiated soon after menopause [79, 80]. GPER activation has been shown to induce the greatest vasodilation responses in the arteries of postmenopausal women [53], suggesting that older women may benefit from GPER-targeted therapy. In support of this, our group has shown that G-1 treatment in mice partially abrogated diet-induced atherosclerosis [52], a disease that is closely linked to reduced NO generation by the endothelium [12]. Thus, pharmacologic modulation of GPER activity with small molecules such as G-1 could aid in promoting or maintaining cardiovascular function in post-menopausal women, given the lack of feminizing side effects of GPER-selective compounds on uterine growth observed in response to E2 [52]. We conclude therefore that GPER may mediate multiple protective cardiovascular effects by promoting eNOS activation and NO production, and that this mechanism may contribute to the salutary vascular effects of estrogen signaling.
Highlights.
Estrogen and G-1 activate eNOS to similar extents
SERMs and SERDs promote NO formation via GPER
GPER produces NO via c-Src, EGFR, PI3K and ERK1/2
GPER-deficient mice demonstrate G-1 specificity
Estrogen-mediated vasodilation mediated in part by GPER
Acknowledgments
We thank Dr. Chelin Hu and Dan Cimino for expert technical assistance. This study was supported by the National Institutes of Health (NIH R01 CA127731, CA163890 and CA194496 to E.R.P.), Dedicated Health Research Funds from the University of New Mexico School of Medicine allocated to the Signature Program in Cardiovascular and Metabolic Diseases (to E.R.P.), and the Swiss National Science Foundation (grants 135874 & 141501 to M.R.M.). N.C.F. was supported by NIH training grant HL07736.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS
M.R.M. and E.R.P. are inventors on a U.S. patent application for the therapeutic use of compounds targeting GPER. E.R.P. is an inventor on U.S. patents 7,875,721 and 8,487,100. N.C.F. declares no competing financial interests.
References
- 1.Barrett-Connor E. Menopause, atherosclerosis, and coronary artery disease. Curr Opin Pharmacol. 2013;13:186–91. doi: 10.1016/j.coph.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenck-Gustafsson K, Brincat M, Erel CT, Gambacciani M, Lambrinoudaki I, Moen MH, Tremollieres F, Vujovic S, Rozenberg S, Rees M, Emas EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas. 2011;68:94–7. doi: 10.1016/j.maturitas.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Meyer MR, Barton M. Estrogens and Coronary Artery Disease: New Clinical Perspectives. Adv Pharmacol. 2016;77:307–60. doi: 10.1016/bs.apha.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Menazza S, Murphy E. The expanding complexity of Estrogen Receptor signaling in the cardiovascular system. Circ Res. 2016;118:994–1007. doi: 10.1161/CIRCRESAHA.115.305376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm A, Nilsson BO. Identification and characterization of new mechanisms in vascular oestrogen signalling. Basic Clin Pharmacol Toxicol. 2013;113:287–93. doi: 10.1111/bcpt.12118. [DOI] [PubMed] [Google Scholar]
- 6.Prabhushankar R, Krueger C, Manrique C. Membrane estrogen receptors: their role in blood pressure regulation and cardiovascular disease. Curr Hypertens Rep. 2014;16:408. doi: 10.1007/s11906-013-0408-6. [DOI] [PubMed] [Google Scholar]
- 7.Chakrabarti S, Morton JS, Davidge ST. Mechanisms of estrogen effects on the endothelium: an overview. Can J Cardiol. 2014;30:705–12. doi: 10.1016/j.cjca.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 9.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furchgott RF. Studies on relaxation of rabbit aorta by sodium nitrite: the basis for the proposal that acid-activable inhibitory factor from bovine retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide In Vasodilation: Vascular Smooth Muscle, Peptides, Autonomic Nerves and Endothelium (P M Vanhoutte, ed) Raven Press; New York: 1988. pp. 401–414. [Google Scholar]
- 11.Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension. 1994;23:1121–31. doi: 10.1161/01.hyp.23.6.1121. [DOI] [PubMed] [Google Scholar]
- 12.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–37. 837a–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caulin-Glaser T, García-Cardeña G, Sarrel P, Sessa WC, Bender JR. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ Res. 1997;81:885–92. doi: 10.1161/01.res.81.5.885. [DOI] [PubMed] [Google Scholar]
- 14.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–60. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 15.Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, Hathaway HJ, Prossnitz ER. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int. 2013;2013:472720. doi: 10.1155/2013/472720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scaling AL, Prossnitz ER, Hathaway HJ. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer. 2014;5:146–60. doi: 10.1007/s12672-014-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 18.Sharma G, Prossnitz ER. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic beta-cells. Endocrinology. 2011;152:3030–9. doi: 10.1210/en.2011-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Hisamoto K, Kim KH, Haynes MP, Bauer PM, Sanjay A, Collinge M, Baron R, Sessa WC, Bender JR. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc Natl Acad Sci U S A. 2007;104:16468–73. doi: 10.1073/pnas.0704315104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–82. doi: 10.1161/01.res.87.8.677. [DOI] [PubMed] [Google Scholar]
- 22.Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003;278:2118–23. doi: 10.1074/jbc.M210828200. [DOI] [PubMed] [Google Scholar]
- 23.Florian M, Lu Y, Angle M, Magder S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids. 2004;69:637–45. doi: 10.1016/j.steroids.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem. 2005;280:19704–10. doi: 10.1074/jbc.M501244200. [DOI] [PubMed] [Google Scholar]
- 25.Kim KH, Young BD, Bender JR. Endothelial estrogen receptor isoforms and cardiovascular disease. Mol Cell Endocrinol. 2014;389:65–70. doi: 10.1016/j.mce.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton M, Meyer MR, Prossnitz ER. Alike but not the same: anatomic heterogeneity of estrogen receptor-mediated vasodilation. J Cardiovasc Pharmacol. 2013;62:22–5. doi: 10.1097/FJC.0b013e31829709d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) 2011;203:259–69. doi: 10.1111/j.1748-1716.2010.02237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takada Y, Kato C, Kondo S, Korenaga R, Ando J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–41. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 29.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem. 2002;277:3388–96. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 30.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 31.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. 2015;153:114–26. doi: 10.1016/j.jsbmb.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–12. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 34.Burai R, Ramesh C, Shorty M, Curpan R, Bologa C, Sklar LA, Oprea T, Prossnitz ER, Arterburn JB. Highly efficient synthesis and characterization of the GPR30-selective agonist G-1 and related tetrahydroquinoline analogs. Org Biomol Chem. 2010;8:2252–9. doi: 10.1039/c001307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421–7. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–66. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer MR, Haas E, Prossnitz ER, Barton M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol Cell Endocrinol. 2009;308:9–16. doi: 10.1016/j.mce.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86:58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barton M, Prossnitz ER. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol Metab. 2015;26:185–92. doi: 10.1016/j.tem.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. 2014;389:71–83. doi: 10.1016/j.mce.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindsey SH, Chappell MC. Evidence that the G protein-coupled membrane receptor GPR30 contributes to the cardiovascular actions of estrogen. Gend Med. 2011;8:343–54. doi: 10.1016/j.genm.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol. 2011;55:17–25. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindsey SH, Yamaleyeva LM, Brosnihan KB, Gallagher PE, Chappell MC. Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension. 2011;58:665–71. doi: 10.1161/HYPERTENSIONAHA.111.175174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One. 2010;5:e15433. doi: 10.1371/journal.pone.0015433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–8. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: a transmembrane estrogen receptor. J Steroid Biochem Mol Biol. 2008;109:350–3. doi: 10.1016/j.jsbmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007:265–266. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prossnitz ER, Sklar LA, Oprea TI, Arterburn JB. GPR30: a novel therapeutic target in estrogen-related disease. Trends Pharmacol Sci. 2008;29:116–23. doi: 10.1016/j.tips.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7:715–26. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 51.Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154:4136–45. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, Amann K, Arterburn JB, Barton M, Prossnitz ER. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep. 2014;4:7564. doi: 10.1038/srep07564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arefin S, Simoncini T, Wieland R, Hammarqvist F, Spina S, Goglia L, Kublickiene K. Vasodilatory effects of the selective GPER agonist G-1 is maximal in arteries of postmenopausal women. Maturitas. 2014;78:123–30. doi: 10.1016/j.maturitas.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Pang Y, Thomas P. Additive effects of low concentrations of estradiol-17β and progesterone on nitric oxide production by human vascular endothelial cells through shared signaling pathways. J Steroid Biochem Mol Biol. 2017;165:258–267. doi: 10.1016/j.jsbmb.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 55.Jang EJ, Seok YM, Arterburn JB, Olatunji LA, Kim IK. GPER-1 agonist G1 induces vasorelaxation through activation of epidermal growth factor receptor-dependent signalling pathway. J Pharm Pharmacol. 2013;65:1488–99. doi: 10.1111/jphp.12113. [DOI] [PubMed] [Google Scholar]
- 56.Lindsey SH, Carver KA, Prossnitz ER, Chappell MC. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J Cardiovasc Pharmacol. 2011;57:598–603. doi: 10.1097/FJC.0b013e3182135f1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prossnitz ER, Arterburn JB. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev. 2015;67:505–40. doi: 10.1124/pr.114.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol. 2006;80:4833–46. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer MR, Fredette NC, Barton M, Prossnitz ER. Regulation of vascular smooth muscle tone by adipose-derived contracting factor. PLoS One. 2013;8:e79245. doi: 10.1371/journal.pone.0079245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinkovich S, Shah D, Planey SL, Arnott JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interv Aging. 2014;9:1437–52. doi: 10.2147/CIA.S66690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J Biol Chem. 2005;280:35943–52. doi: 10.1074/jbc.M504606200. [DOI] [PubMed] [Google Scholar]
- 62.Salerno JC, Ghosh DK, Razdan R, Helms KA, Brown CC, McMurry JL, Rye EA, Chrestensen CA. Endothelial nitric oxide synthase is regulated by ERK phosphorylation at Ser602. Biosci Rep. 2014;34 doi: 10.1042/BSR20140015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim KH, Bender JR. Rapid, estrogen receptor-mediated signaling: why is the endothelium so special? Sci STKE. 2005;2005:pe28. doi: 10.1126/stke.2882005pe28. [DOI] [PubMed] [Google Scholar]
- 64.Kim KH, Moriarty K, Bender JR. Vascular cell signaling by membrane estrogen receptors. Steroids. 2008;73:864–9. doi: 10.1016/j.steroids.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim KH, Bender JR. Membrane-initiated actions of estrogen on the endothelium. Mol Cell Endocrinol. 2009;308:3–8. doi: 10.1016/j.mce.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shaul PW. Rapid activation of endothelial nitric oxide synthase by estrogen. Steroids. 1999;64:28–34. doi: 10.1016/s0039-128x(98)00105-6. [DOI] [PubMed] [Google Scholar]
- 67.Vergote I, Abram P. Fulvestrant, a new treatment option for advanced breast cancer: tolerability versus existing agents. Ann Oncol. 2006;17:200–4. doi: 10.1093/annonc/mdj047. [DOI] [PubMed] [Google Scholar]
- 68.Cummings SR, Ensrud K, Delmas PD, LaCroix AZ, Vukicevic S, Reid DM, Goldstein S, Sriram U, Lee A, Thompson J, Armstrong RA, Thompson DD, Powles T, Zanchetta J, Kendler D, Neven P, Eastell R, Investigators, P. S. Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med. 2010;362:686–96. doi: 10.1056/NEJMoa0808692. [DOI] [PubMed] [Google Scholar]
- 69.Ensrud K, LaCroix A, Thompson JR, Thompson DD, Eastell R, Reid DM, Vukicevic S, Cauley J, Barrett-Connor E, Armstrong R, Welty F, Cummings S. Lasofoxifene and cardiovascular events in postmenopausal women with osteoporosis: Five-year results from the Postmenopausal Evaluation and Risk Reduction with Lasofoxifene (PEARL) trial. Circulation. 2010;122:1716–24. doi: 10.1161/CIRCULATIONAHA.109.924571. [DOI] [PubMed] [Google Scholar]
- 70.Reynolds SR, Foster FI. Peripheral vascular actions of estrogen in the human male. J Clin Invest. 1939;18:649–55. doi: 10.1172/JCI101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Traupe T, Stettler CD, Li H, Haas E, Bhattacharya I, Minotti R, Barton M. Distinct roles of estrogen receptors alpha and beta mediating acute vasodilation of epicardial coronary arteries. Hypertension. 2007;49:1364–70. doi: 10.1161/HYPERTENSIONAHA.106.081554. [DOI] [PubMed] [Google Scholar]
- 72.Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. GPER-novel membrane oestrogen receptor. Clin Sci (Lond) 2016;130:1005–16. doi: 10.1042/CS20160114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–91. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meyer MR, Haas E, Barton M. Need for research on estrogen receptor function: importance for postmenopausal hormone therapy and atherosclerosis. Gend Med. 2008;5(Suppl A):S19–33. [Google Scholar]
- 75.Meyer MR, Amann K, Field AS, Hu C, Hathaway HJ, Kanagy NL, Walker MK, Barton M, Prossnitz ER. Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension. 2012;59:507–12. doi: 10.1161/HYPERTENSIONAHA.111.184606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang WB, Cong Y, Ru JY, Ying SQ, Zhu T, Wang DS, Liu XW, Liu G, Zhao JN. Osteoprotective effect of combination therapy of low-dose oestradiol with G15, a specific antagonist of GPR30/GPER in ovariectomy-induced osteoporotic rats. Biosci Rep. 2015;35 doi: 10.1042/BSR20150146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605–13. doi: 10.1001/jama.280.7.605. [DOI] [PubMed] [Google Scholar]
- 78.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 79.Giordano S, Hage FG, Xing D, Chen YF, Allon S, Chen C, Oparil S. Estrogen and Cardiovascular Disease: Is Timing Everything? Am J Med Sci. 2015;350:27–35. doi: 10.1097/MAJ.0000000000000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khalil RA. Estrogen, vascular estrogen receptor and hormone therapy in postmenopausal vascular disease. Biochem Pharmacol. 2013;86:1627–42. doi: 10.1016/j.bcp.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]