

Abstract
Viral infections of the central nervous system (CNS) are often associated with blood-brain barrier (BBB) disruption, yet the impact of virus replication and immune cell recruitment on BBB integrity are incompletely understood. Using two-photon microscopy, we demonstrate that Venezuelan equine encephalitis virus (VEEV) strain TC83-GFP, a GFP expressing, attenuated strain with a G3A mutation within the 5′ UTR that is associated with increased sensitivity to type I interferons (IFNs), does not directly impact BBB permeability. Following intranasal infection of both wild-type and IFN-induced protein with tetratricopeptide repeats 1 (IFIT1)-deficient mice, which fail to block TC83-specific RNA translation, virus spreads to the olfactory bulb and cortex via migration along axonal tracts of neurons originating from the olfactory neuroepithelium. Global dissemination of virus in the CNS by 2 days post-infection (dpi) was associated with increased BBB permeability in the olfactory bulb, but not in the cortex or hindbrain, where permeability only increased after the recruitment of CX3CR1+ and CCR2+ mononuclear cells on 6 dpi, which corresponded with tight junction loss and claudin 5 redistribution. Importantly, despite higher levels of viral replication, similar results were obtained in IFIT1-deficient mice. These findings indicate that TC83 gains CNS access via anterograde axonal migration without directly altering BBB function and that mononuclear and endothelial cell interactions may underlie BBB disruption during alphavirus encephalitis.
1. Introduction
Venezuelan equine encephalitis virus (VEEV) is a member of the Togaviridae family of positive-sense RNA alphaviruses, and naturally cycles between mosquitoes and rodents (enzootic cycle), or mosquitoes and horses (epizootic cycle) (Salimi et al., 2016). VEEV is considered an important zoonotic pathogen with several reported outbreaks in South and Central Americas, the latter of which have spread to North America (Morrison et al., 2008; Weaver et al., 1996). Natural or laboratory-acquired infections have been documented in humans with all epizootic and many enzootic VEEV strains. After a 1 to 6 day incubation period, patients develop high fever, headache, malaise, and myalgia. The case fatality rate is < 1%, but can be as high as 20% in patients that develop encephalitis. Although the number of human cases reported is small, the possibility for disease emergence is high due to expansion and spread of mosquito vectors. Furthermore, VEEV can also be spread via aerosolization and thus has significant potential as a bioterrorist agent (Sergeev et al., 1991). Currently, there are no FDA-approved antiviral agents or vaccines to mitigate or prevent VEEV encephalitis. Moreover, our understanding of the mechanisms of VEEV neuroinvasion and virologic control within the central nervous system (CNS) are incompletely understood.
The CNS is normally protected from pathogens by the blood brain barrier (BBB), an anatomical specialization composed of tight junction (TJ)-linked brain microvascular endothelial cells (BMECs), associated pericytes, astrocyte end-feet and extracellular matrix. In previous studies, we have demonstrated that barrier function during neurotropic viral infections is dynamic and that viral sensing by BMECs may improve BBB integrity via mechanisms in which TAM receptor Mertk and the IFN receptor (IFNAR) synergistically impact on the activity of Rac1, a RhoGTPase family member that promotes TJ stabilization, while inhibiting the expression of interleukin (IL)-1, a barrier destabilizing cytokine (Daniels et al., 2014; Miner et al., 2015). IFNAR signaling is also critical for controlling VEEV and other alphavirus infections via upregulation of numerous IFN-stimulated genes (ISGs) including the IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), which normally acts to block translation of virion-delivered virus-specific RNAs (Habjan et al., 2013). While virulent VEEV strains evade IFIT1 activity, the attenuated VEEV strain TC83 encodes a G3A mutation within the 5′ UTR that is associated with increased sensitivity to IFIT1 (Hyde et al., 2014; White et al., 2001). Importantly, TC83 virulence is restored in Ifit1−/− mice, which succumb to infection with TC83. Given that IFNAR signaling is required for stabilization of TJs at the BBB, VEEV infection could potentially increase BBB permeability via direct interactions with BMECs. Indeed, prior studies indicate that VEEV replication after intranasal inoculation induces an inflammatory cascade that promotes BBB opening (Schafer et al., 2011). However, analysis of both BBB permeability and monocyte infiltration using intracranial inoculation of VEEV replicon particles determined that these events coincide (Schafer et al., 2009). Thus, it is unclear whether VEEV effects on the BBB are direct or result from interactions between immune cells and the CNS vasculature.
In this study, we examined VEEV TC83 neuroinvasion and replication in conjunction with alterations in BBB integrity in wild-type and Ifit1−/− mice after intranasal inoculation. We detected viral replication 2 days post-infection (dpi) that peaked in most CNS regions by 4 dpi. Analysis of sodium fluorescein penetration throughout the CNS of wildtype mice revealed a peak increase in BBB permeability at 6 dpi, with no differences between wild-type and Ifit1−/− mice observed at this time-point. Freeze-fracture electron microscopy directly demonstrated loss of TJs at the ultrastructural level at 6 dpi. Using two-photon (2P) microscopy with GFP-expressing TC83, we detected replicating virus as early as 1 dpi in the main olfactory epithelium and in the olfactory bulb. At 2 dpi, TC83-GFP was detected throughout the CNS, and coincided with increased BBB permeability in the olfactory bulb (OB), but not in the cortex or hindbrain, whose permeability increased at time-points coinciding with infiltration of CX3CR1+ and CCR2+ mononuclear cells. Although levels of TC83-GFP expression were increased at 2 dpi in Ifit1−/− mice, we did not find an increase in BBB permeability as compared to wild-type mice. These data suggest that VEEV does not directly alter BBB integrity in vivo, but leads to the recruitment of monocytes that alter BBB integrity as they extravasate from the brain vasculature.
2. Methods
2.1. Animals
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and Ifit1−/− mice were the generous gift of M. Diamond (Washington University School of Medicine, St. Louis, MO). CX3CR1GFP/+ and CCR2RFP/+ mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Claudin5-GFP mice were obtained from D. Agalliu (New York, NY). Animals were housed under pathogen-free conditions in Washington University School of Medicine animal facilities. All experiments were performed in compliance with Washington University animal studies guidelines.
2.2. Mouse model of VEEV TC83 encephalitis
Four-week old male mice were inoculated intranasally (10 μl per nostril) with an attenuated VEEV strain TC83 or TC83-GFP (107 PFU) under light anesthesia. Both TC83 and TC83-GFP were generous gifts of Michael Diamond (St. Louis, MO). TC83-GFP was generated by subgenomic insertion of GFP under an exogenous subgenomic promoter, described previously (Atasheva et al., 2010). Mice were monitored daily for weight loss and scored daily for encephalitic sequelae. Encephalitic score represents a progressive range of behaviors: 1) hunched, ruffled fur, 2) altered gait, slow movement, 3) not moving but responsive, 4) moribund, 5) death.
2.3. Virologic analysis and in vivo assessment of BBB permeability
At various post-infection intervals, TC83-infected mice received intraperitoneal injections of sodium fluorescein salt (100 μl, 100 mg/ ml, Sigma-Aldrich) in sterile phosphate-buffered saline (PBS). After 45 min, mice were anesthetized, and blood was collected by cardiac bleed, followed by extensive cardiac perfusion with PBS and harvesting of CNS tissues. Viral titers were determined using standard plaque assay techniques by serial dilution of tissue homogenates over Vero cells, as described previously (Brien et al., 2013). For sodium fluorescein assay, tissue homogenates were incubated overnight at 4 °C after dilution 1:1 with 2% trichloroacetic acid (Sigma-Aldrich) to precipitate protein. Protein precipitates were pelleted by centrifugation at 3000 rpm at 4 °C. Supernatants were diluted 1:1 in borate buffer, pH 11 (Sigma-Aldrich). Sample fluorescence was measured (EX. 480 nm; EM: 538 nm) by Synergy H1 microplate fluorometer (BioTek Instruments, Inc.), and normalized to standard curves, serum fluorescein concentration, and tissue weight.
2.4. Cell cultures and reagents
Baby hamster kidney (BHK) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Primary murine BMECs were isolated from the cortex of 8 week-old C57BL/6 mice, as previously described (Daniels et al., 2014). Following isolation, 3 μg/ml puromycin was added to the culture medium during the first 72 h post-isolation to eliminate contaminating cell types. Isolated BMECs were maintained in DMEM supplemented with 20% plasma derived bovine serum (Animal Technologies, FBP-186), 1% penicillin and streptomycin, and 10 ng/ml recombinant mouse fibroblast growth factor (ThermoFisher, PMG0035). Primary murine astrocytes (ACs) were isolated from P2–P4 C57BL/6 pups as previously described (Daniels et al., 2014). Isolated astrocytes were cultured in DMEM containing 10% FBS, 10% horse serum, 1% penicillin and streptomycin, and 10 ng/ml recombinant mouse epidermal growth factor (Invitrogen, #PMG0062).
2.5. In vitro BBB model
An in vitro BBB model was constructed as previously described (Daniels et al., 2014). Briefly, BMECs were seeded at 3 × 104 cells/well on fibronectin-coated transwell inserts (BD Falcon, 24-well, 8 μm pores). Concurrently, primary murine ACs were cultured at 4 × 104 cells/well in a fibronectin-coated 24-well plate until they reached confluence, at which point BMEC inserts were transferred to astrocyte-containing wells. Culture medium was replaced after 72 h with fresh medium containing 110 μM hydrocortisone (HC) to induce differentiation of BMECs and upregulate expression of tight junction proteins. Culture medium was replaced 24 h later with serum-free media (SFM) supplemented with 110 μM HC, 250 μM cAMP (Sigma-Aldrich, C3912), and 17.5 μM RO 20-1724 (Sigma-Aldrich, B8279) to further promote barrier formation between BMECs. The cells were maintained in culture for an additional 24 h before being used for experiments. TC83 (MOI 2) was added either to BMEC inserts (top chamber) or to ACs in the bottom chamber. Endothelial permeability was assessed by measuring transendothelial electric resistance (TEER) in ohms (Ω) at various time-points post-infection using an automated TEER measuring system (REMS AutoSampler, World Precision Instrument). Positive and negative controls include transwell inserts containing BMECs treated with either 100 ng/ml tumor necrosis factor (TNF)-α or phosphate-buffered saline (PBS), respectively. Inserts with medium alone were used for blank resistance measurements.
2.6. Leukocyte isolation and flow cytometry
Wild-type mice were inoculated with TC83 (107 PFU), anesthetized, and cardiac perfused with PBS, at 2, 4, 6, and 8 dpi. Leukocytes were isolated from dissociated olfactory bulbs and cortices and stained with fluorescently conjugated antibodies as described previously (McCandless et al., 2006). For flow cytometry, cells were stained with CD45, CD11b, CD11c, Ly6C, CD4 antibodies from BioLegend, and CD8a antibodies (BD Pharminogen). Samples were analyzed using a LSRII flow cytometer (BD Biosciences). Gating and data analysis were performed using FlowJo (FlowJo, LLC). Leukocyte single cells were first gated for CD45 expression. The various cell populations were quantified as a percentage of this CD45+ population.
2.7. RNA isolation and quantitative RT-PCR
Olfactory bulbs and cortices were isolated from cardiac-perfused wildtype mice at 2 and 6 days after TC83 infection (107 PFU i.n.). RNA was isolated from tissues using RNeasy kit (Qiagen) according to manufacturer’s instructions, and quantified using a NanoDrop (Thermo Scientific). Following DNAse I treatment (Invitrogen) of RNA samples (1 μg) was reverse transcribed using Taqman Reverse Transcriptase kit (Applied Biosystems). qRT-PCR was performed using Power SYBR Green (Applied Biosystems) on a ViiA7 Real-Time PCR system (Applied Biosystems) using manufacturer’s recommended cycle parameters. ΔCt values are reported as the Ct values for target genes normalized to Ct values of GAPDH (Ctgene−CtGAPDH). Primers (5′−3′) used are as follows: GAPDH: (F) GGCAAATTCAACGGCACAGT, (R) AGATGGTGATGGGCTTCCC; TNFα: (F) GCACAGAAAGCATGATCCG, (R) GCCCCCCATCTTTTGGG; IL-1β: (F) ACCTGTCCTGTGTAATGAAAGACG, (R) TGGGTATTGCTTGGGATCCA; IFNγ: (F) AACGCTACACACTGCATCTTGG, (R) GCCGTGGCAGTAACAGCC.
2.8. Intravital 2P imaging
Time-lapse imaging was performed with a custom built 2P microscope equipped with a 1.0 NA 20 × water dipping objective (Olympus) running ImageWarp acquisition software (A & B Software) as previously described (Kreisel et al., 2010; Zinselmeyer et al., 2009). In vivo imaging of the brain was performed using a thinned skull preparation and a custom imaging chamber. Mice were anesthetized with isoflurane and body temperature was maintained with a warming plate. Mice were given subcutaneous (s.c.) saline for hydration for imaging experiments lasting > 1.5 h. For explant imaging, either fixed or fresh tissues were glued to plastic cover slips using VetBond adhesive and submerged in CO2 independent medium for imaging. Video-rate scanning was used to identify microcirculation approximately 40–100 μm below the dura. Microglial dynamics, reporter virus and blood vessel leak were analyzed using 3D time-lapse imaging for up to 2 h for each time-point. Tissue perfusion was verified by injecting blood labels i.v. after the imaging preparation was completed. Florescence was excited with a Chameleon Vision II Ti:Sapphire laser (Coherent) tuned to 915 nm and fluorescence emission detected by PMTs simultaneously using 495 nm and 560 nm dichroic filters: Blue (< 495 nm, SHG collagen), green (495–560 nm, eGFP) and red (> 560 nm, RFP, or 70 kD tetramethylrhodamine-dextran (TMR-dex)). Auto fluorescence appears as mix of color (495–600 nm) and thus can be discriminated from eGFP, RFP and TMR-dex signals. For time-lapse imaging, we acquired a 220×240×75μm volume as 31 sequential 2.5 μm z-steps with a time resolution of approximately 32 s. The X,Y resolution was 0.75 μm/pixel, which is adequate to resolve individual cells and small capillaries. Intravital imaging focused on pial vessels extending < 100 μm below the subarachnoid space. Ex plant imaging was used to analyze vessels and infected cells in the main olfactory epithelium (MOE), olfactory bulb (OB) and deeper regions of the cortex that are inaccessible in the thinned skull preparation. Mice were sacrificed, MOE, OB and cortex were harvested and placed in CO2 independent media (Gibco®) to control for pH and imaged at room temperature, which arrests cellular dynamics, but preserves tissue viability for several hours in our hands. For in vivo vessel leak measurements, 70 kD fluorescent TMR- or 10 kD Alexa594-dextran was injected via retro orbital sinus immediately before imaging and leak assessed continuously for 30–60 min. Both dextrans gave similar results. For ex plant experiments, dextran was injected ~ 30 min before mice were sacrificed and imaged. Although, the emission of Alexa594 and RFP overlap somewhat, RFP has a more prominent green tail and so the cells appear slightly orange shifted in our videos relative to the dextran signal. Multi-dimensional data sets were rendered in 3D using Imaris (Bitplane). Cell tracking and data analysis were performed using Imaris (Bitplane) and Motility Lab (2ptrack.net). TC83-GFP reporter virus expression is very high and in some cells the GFP signal saturated our detectors and bled through into other channels to produce white cells in the image (R + B + G = W). Decreasing the laser power and PMT gain to reduce signal saturation made it difficult to detect fine membrane structures and thin axons that were important for documenting cell morphology. To address this issue, we used non-linear enhancements in some cases to minimize channel bleed through and optimize contrast to resolve detailed cell morphology. These images were not used for quantitative measurements or colocalization analysis.
2.9. Thinned skull preparation for in vivo imaging
Mice were anesthetized with an intraperitoneal injection (20 μl/g body weight) of 310 mg/kg Avertin. A cotton pillow was placed underneath the chin to support the head and a heating pad underneath the body to maintain body temperature of ~ 37 °C. Additional injections of Avertin (halved doses) were given as needed to maintain anesthesia during surgery. The scalp was shaved with an electronic razor, residual hair removed and cleaned with a sterile alcohol prep pad. A midline scalp incision was made extending from between the ears to between the eyes. A 2–2.5 mm diameter circular area of the skull was thinned using a dentist drill or surgical hand drill equipped with a round carbide bur drill bit as described by others (Li et al., 2014). We avoided areas located directly over cranial sutures, as the skull is less stable in these areas. A small amount of Vetbond tissue adhesive was applied around the edges of the upper plate of the imaging chamber, and gently held against the skull to secure it with the thinned region centered in the hole in the upper chamber. The animal was then placed on the lower warming plate of the chamber and the upper plate immobilized to two spring loaded posts using thumb screws. The thinned region was covered with saline for 2P imaging.
2.10. Transmission electron microscopy
Mice were anesthetized by an intraperitoneal injection of ketamine and perfused with 20 ml PBS followed by 20 ml of 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2). The perfused mouse brain was minced into 1 mm3 pieces, and cerebral cortical pieces were selected out and stored at 4 °C overnight in fixative solution. Following fixation, brain pieces were rinsed with three exchanges of cacodylate buffer over one hour period and then osmicated for 1 h in 1% osmium tetroxide prepared in cacodylate buffer, rinsed two times over 20 min in cacodylate buffer and then washed with dH2O for 10 min followed by en bloc staining with 1% uranyl acetate/H2O for 60 min. After three dH2O rinses, brain pieces were dehydrated with ethanol and embedded in Araldite resin. The 75 nm ultrathin sections were cut on a Leica UC7 ultramicrotome, picked up on formvar coated grids and post stained with 1% uranyl acetate/dH2O and Reynolds lead citrate. Sections were viewed on a JEM-1400 transmission microscope (JEOL) at 80KV with an AMT XR111 4k digital camera.
2.11. Quick-freeze deep-etch electron microscopy
Quick-freeze Deep-etch EM was performed according to published protocol, with minor modifications (Heuser and Kirschner, 1980). Dissected mouse cerebral cortexes were frozen by abrupt application of the sample against a liquid helium cooled copper block with a Cryopress freezing machine. Frozen samples were transferred to a liquid nitrogen cooled Balzers 400 vacuum evaporator, fractured and etched at minus 104 °C for 2.5 min, then rotarily replicated with ~ 2 nm platinum deposited from a 20° angle above the horizontal plane, followed by an immediate ~ 10 nm stabilization film of pure carbon deposited from an 85° angle. Replicas were floated onto a dish of bleach and transferred through several rinses of dH2O, picked up onto formvar coated grids, and imaged on a JEM1400 transmission microscope (JEOL) at 80KV with attached AMT XR111 4k digital camera.
2.12. Statistical analyses
Reported values are mean values ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism 5 software. Wild-type in vivo fluorescein experiments and flow cytometry were compared via one-way analysis of variance (ANOVA), Bonferroni’s post hoc test was subsequently used for comparison of individual means. Cytokine expression and fluorescein comparisons in IFIT1-deficient mice were analyzed via unpaired Student t-test. In vitro TEER experiments were analyzed by two-way ANOVA with correction for repeated measures. P values P < 0.05 were considered significant. Statistical values are indicated as follows *, P < 0.05; **, P < 0.01; ***, P < 0.001, unless otherwise stated.
3. Results
3.1. VEEV TC83 infects neurons and astrocytes
Virulence of VEEV TC83 is attenuated in subcutaneously infected wild-type mice, but restored in Ifit1−/− mice, which exhibit increased CNS titers of replicating virus and 100% mortality compared with wildtype animals (Hyde et al., 2014). Similarly, intranasal (i.n.) inoculation of VEEV TC83 (107 PFU) in 4 week-old Ifit1−/− mice resulted in 100% lethality by 6 dpi, loss of 37% of body weight and severe clinical signs prior to death, while similarly infected age-matched, wild-type animals, exhibited 20% lethality with minimal weight loss and morbidity (Fig. 1A–D). Lethality of VEEV TC83-infected Ifit1−/− mice occurred a day later when the inoculum was reduced 3 log-fold (104 with similar weight loss and morbidity (Fig. 1A–D). Evaluation of viral burdens in wild-type mice after intranasal inoculation of VEEV TC83 showed peak viral loads at 2–4 dpi in the forebrain and 4–6 dpi in the hindbrain (Fig. 1E–I), suggesting that virus tracks through the CNS via neuronal infection.
Fig. 1.
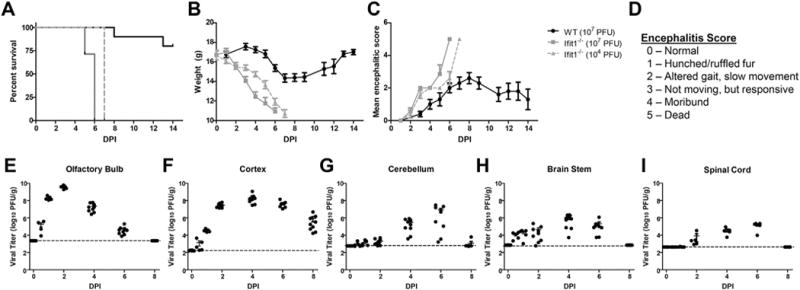
VEEV TC83 infection of wild-type and Ifit1−/− mice. Four week-old wild-type and Ifit1−/− mice (N = 5–9/group) were intranasal inoculated with VEEV TC83 at indicated PFUs and followed for survival (A), weight loss (B) morbidity (C), the latter via encephalitis scoring (D), and viral loads (E–I). Mice were i.n. inoculated with 107 PFU TC83 and viral burden was measured via plaque assay from olfactory bulb, cortex, cerebellum, brain stem, and spinal cord. Data presented as mean ± SEM for N = 5 mice/group.
Several potential mechanisms could lead to viral invasion of the CNS and encephalitis in our model. Others have reported that VEEV can move in a retrograde manner up olfactory neurons crossing the cribriform plate and infecting glomeruli in the olfactory bulb, which could seed infection of the cortex and ultimately the hindbrain. Alternatively, virus could enter the brain directly during epithelial disruption at the choroid plexus or by crossing the BBB after moving via lymphatic drainage into the circulation (Phillips et al., 2016). We used in vivo 2P imaging of wild type and Ifit1−/− mice to analyze the spatiotemporal distribution of virus following intranasal infection. Because Ifit1−/− mice exhibit defective IFN responses to TC83 infection, they recapitulate key features of virulent VEEV infection (i.e., rapid virus replication and dissemination), while allowing us to work under BSL2 containment procedures. Our goal was to determine whether viral infection of the nasal mucosa leads to BBB disruption during invasion of the CNS and encephalitis. We first investigated the possibility that VEEV disseminates along neuronal tracks by infecting wild-type mice with TC83-GFP and then examining explanted fresh tissues with 2P imaging at increasing times after infection. As early as 1 dpi, we detected virus positive cells in the main olfactory epithelium (MOE) of wild type mice (Fig. 2A), but not the ventral nasal organ (data not shown). The infected cells appeared to be olfactory sensory neurons based on their distinct morphology (Fig. 2A, D), axonal labeling and location in the nasal epithelium (blue autofluorescence). At 1 dpi, clusters of infected neurons were also prominent in the olfactory bulb (OB) (Fig. 2B, Supplemental Fig. 1) suggestive of olfactory projections into glomeruli associated with mitral cells. At 2 dpi, numerous infected cells were observed in the cerebral cortex (CTX) (Fig. 2C). These infected cells had distinct morphologies (Supplemental Fig. 1) with most resembling neurons (Fig. 2B, cyan arrow), and others with the characteristic appearance of astrocytes (Fig. 2B, yellow arrow). By 6 dpi, during peak disease, infected cells with both neuronal (Fig. 2C, cyan arrow) and astrocyte morphology (Fig. 2C, yellow arrow) were widespread in the cortex (Supplemental Fig. 1). Immunofluorescent microscopy (IFM) experiments were performed to confirm the identity of the infected cells and the majority of TC83-GFP positive cells were neurons with the remaining cells staining positive for astrocytes (data not shown). We imaged Ifit1−/− mice on 1–3 dpi with 107 intranasal VEEV TC83-GFP (Fig. 2D–F) to examine the distribution of reporter virus. Infected cells in the MOE of Ifit1−/− mice had olfactory neuron morphology on 1 dpi (Fig. 2D, cyan arrows) similar to wild-type mice, but in contrast the number of infected cells in the MOE increased dramatically on 2–3 dpi. In addition to numerous olfactory neurons, we also frequently observed infected neuronal axons projecting towards the cribriform plate (Fig. 2D, right panel, cyan arrow). In the IFIT1-deficient OB at 1 dpi (Fig. 2E, Supplemental Fig. 1) infected cells with neuronal (cyan arrow) and astrocyte (yellow arrow) morphology were observed. Infected cells were first observed in the IFIT1-deficient cortex on 2 dpi with astrocyte morphology (Fig. 2F), but infection was more extensive in the cortex by 3 dpi (Fig. 2F, right panel), with widespread infection of neurons and astrocytes (cyan and yellow arrows). TC83-GFP levels were demonstrably higher in Ifit1−/− mice (Fig. 2F) compared to wild-type animals (Fig. 2C) at 2–3 dpi, as well as on 6 dpi (data not shown), consistent with the important role IFIT1 plays in limiting viral replication.
Fig. 2.
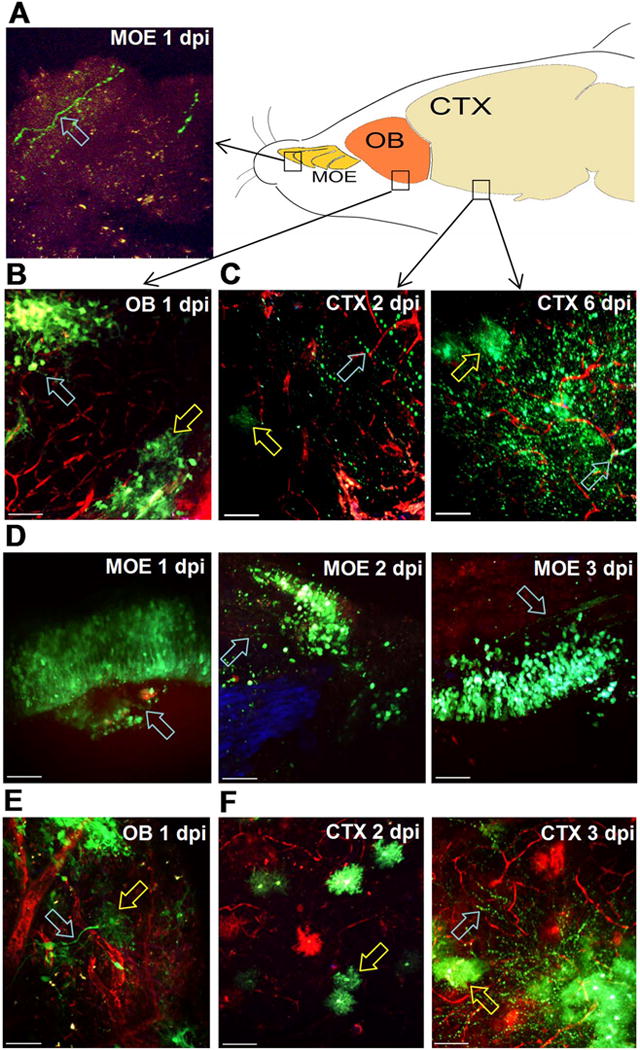
Intranasal VEEV neural invasion and dissemination in wild-type versus Ifit1−/− mice. A–C. Wild-type and Ifit1−/− mice were challenged with 107 PFU.n. VEEV TC83-GFP and tissues explanted for 2P microscopy at the indicated times. 70 kD TMR-dextran was injected i.v. to visualize the brain vasculature (red). In wild-type animals, infected cells (green) were detected in (A) the main olfactory epithelium (MOE) with olfactory neuron morphology (cyan arrow) 1 dpi and (B) olfactory bulb (OB) clustering in glomerular structures (cyan arrow) and nearby cells with astrocyte morphology (yellow arrow) on 1 dpi. C. Infected cells (green) in the cortex (CTX) 2 dpi have both neuronal (cyan arrow) and microglial or astrocyte morphology (yellow arrow). At 6 dpi, TC83-GFP reporter virus is widespread in the cortex and infected cells are observed with both neuronal (cyan arrow) and microglial morphology (yellow arrow) D. In Ifit1−/− mice, infected cells (green) have olfactory sensory neuron morphology (cyan arrows) and are wide spread in the MOE over the first three days post infection. Infected neuronal tracks projecting towards the cribriform plate were also observed (right panel, cyan arrow). TC83-GFP cells with different neuronal and astrocyte/microglia (cyan, yellow arrows) were widespread in Ifit1−/− mice (E) in the OB at 1 dpi and (F) the cortex at 2 and 3 dpi. Scale bar = 50 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Neuroinvasion with VEEV does not directly impact BBB function
We examined BBB integrity during TC83 infection by measuring the extravasation of intraperitoneal administered sodium fluorescein in whole brains. The BBB in the OB exhibited increased permeability on 1 dpi (Fig. 3A), when virus was detected by both PFU assay (Fig. 1) and 2P microscopy (Fig. 2). Increased BBB permeability in the OB became significant by 2 dpi, recovering to baseline by 8 dpi. In contrast, BBB disruption in all other CNS regions was not significant until 6 dpi, after viral dissemination had occurred and coinciding with the start of viral clearance (Fig. 3B–E). Despite the increased replication of TC83 in the CNS of Ifit1−/− mice (Fig. 2D–E), BBB permeability was not significantly different at 6 dpi compared with infected wild-type animals (Fig. 3F). These data indicate that while VEEV TC83 neuroinvasion of the OB may decreases BBB integrity, viral infection of other brain regions does not lead to rapid changes in BBB permeability regardless of viral load.
Fig. 3.
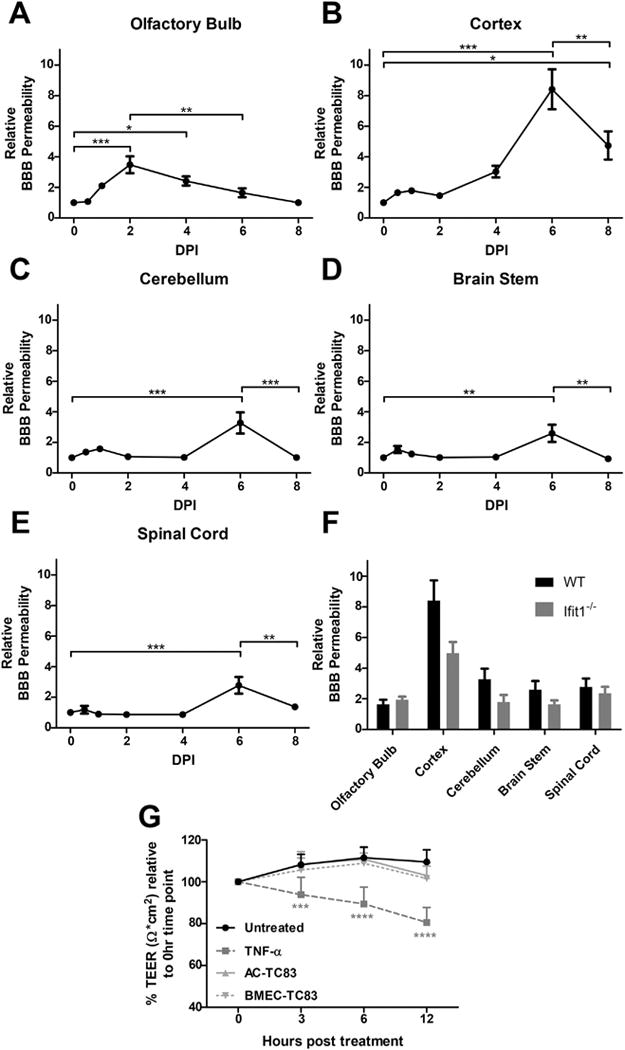
VEEV TC83 does not alter BBB function during neuroinvasion. BBB permeability was assessed in TC83-infected wild-type mice by measuring accumulation of sodium fluorescein in olfactory bulb (A), cortex (B), brainstem (C), cerebellum (D), and spinal cord (E) following i.p administration with tissue fluorescence normalized to plasma fluorescence from each animal at various time-points. Comparison between wild-type (WT) and Ifit1−/− F. BBB permeability at 6 dpi after i.n. infection with TC83 (104 PFU). Data presented as mean fluorescence ± SEM for N = 5–9 mice/group. *, P < 0.05; **, P < 0.01; ***, P < 0.001 via 1-way ANOVA. G. BMECs grown on transwell inserts were exposed to TC83 (BMEC-TC83, MOI 2), TNF-α or PBS and TEER values (Ω × cm2) were measured at indicated time-points. To examine the effect of infected astrocytes on BBB integrity, TC83 (MOI 2) was added to astrocytes in the bottom chamber (AC-TC83) in indicated wells. TEER values for each replicate are presented as normalized values, relative to their respective TEER obtained at 0 h. Results are representative of two independent experiments, each with 6 replicates. Error bars indicate mean ± SEM. Statistically significant differences were determined via 2-way ANOVA and are indicated as ***, P < 0.0005; ****, P < 0.0001.
The effect of TC83 on endothelial barrier integrity was further assessed using in vitro BBB model. Addition of TC83 (MOI 2) either to BMECs grown on porous transwell inserts or to astrocytes in the bottom chamber of transwell dishes did not impact BBB permeability, as determined by measuring transendothelial electrical resistance (TEER; Ω × cm2) at indicated time-points post infection (Fig. 3G). In contrast, treatment of BMECs with TNF-α, which is known to compromise barrier integrity, resulted in a significant and continuous reduction in TEER over the course of the experiment.
We also tested the hypothesis that VEEV infection promotes acute BBB disruption at focal sites, which then leads to CNS invasion and widespread BBB breakdown. We used in vivo 2P imaging to assess vessel permeability relative to sites of TC83-GFP replication. Our rationale was that if virus crossed the BBB to infect the CNS, then BBB disruption should precede the detection of TC83-GFP positive cells and moreover, that foci of TC83-GFP infection should be associated with leaky vasculature. We assessed vessel permeability in vivo by injecting 70kD tetramethylrhodamine-dextran (TMR-dex) to label the cerebral vasculature immediately before intravital imaging and then measuring dextran signal intensity and distribution from blood vessel lumen to the adjacent parenchyma. Vessels branching off of pial venules that invade the brain parenchyma were imaged up to 150 μm deep, where they became post-capillary venules as well as smaller capillaries that branched off of these vessels at ~ 200 μm. In uninfected mice, vessel edges were sharp and little TMR-dex signal was detected in the brain parenchyma (Fig. 4A, Movie 1). In mice infected i.n. with TC83-GFP the BBB remained intact at 2 dpi (Fig. 4B), despite the fact that virus was detected readily at this time in perfused brains by plaque assays (Fig. 1) and by 2P microscopy (Fig. 2B). However, on 6 dpi at the peak of TC83-GFP infection, focal BBB disruption was observed (Fig. 4B, boxed area and lower panel) and was often associated with evidence of leukocyte recruitment (Movie 2). We also imaged claudin5-GFP transgenic mice to assess claudin 5 expression and localization during infection. In uninfected mice, claudin 5 formed well defined streaks in the endothelium at bi- and tri-cellular junctions. At 2 dpi with TC83 (Fig. 4D), claudin 5 expression and localization were similar, suggesting that no overt disruption of the BBB had occurred. However, by 6 dpi, claudin 5 expression appeared clumpy and disorganized, indicating the BBB was significantly disrupted during the later stages of infection, consistent with results of the in vivo 2PM and sodium fluorescein analyses.
Fig. 4.
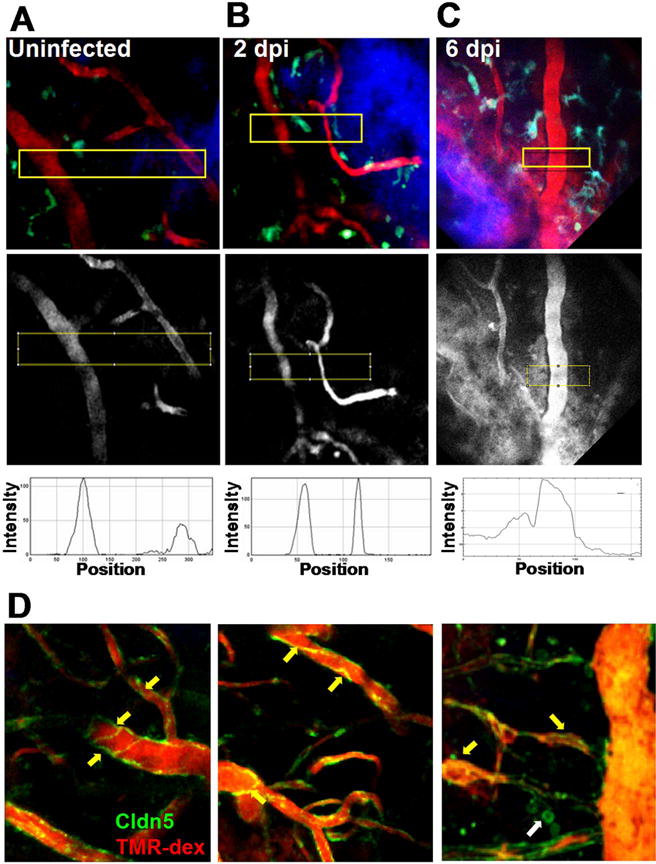
2P microscopy of regional BBB integrity and VEEV dissemination. CX3CR1-GFP reporter mice were mock infected (A) or challenged with 107 PFU i.n. VEEV TC83 and BBB integrity examined at 2 (B) and 6 (C) dpi. Microglia (green or cyan) are seen near blood vessels (70 kD TMR-dex, red), in the cortex approximately 125 μm beneath thinned skull (blue). No disruption of the BBB is detectable until 6 dpi (middle panel boxed regions and kymographs). Claudin5-GFP transgenic mice mock infected (D, left panel) and at 2 dpi. (D, middle panel) have similar claudin 5 expression and distribution, which becomes more disorganized and globular at 6 dpi (D, right panel, yellow arrows). In some cases, claudin5-GFP positive cells were observed that appeared to by leukocytes based on their size and morphology (D, right panel, white arrow). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We then used electron microscopy to assess tight junction ultrastructural integrity at 2 dpi and 6 dpi time-points. While TJ morphology appeared normal in mice infected 2 dpi with TC83, several prominent alterations indicative of TJ disintegration were observed on 6 dpi and summarized as follows. Compared with the well-organized luminal cell junction structures (Fig. 5A, top panel) in naïve mouse cortex, endothelial cell swelling and clotting can be readily identified (Fig. 5A, bottom panel) in TC83-infected mouse cortex. Notably, adherens junctions (AJ), marked by the electron dense sub-junctional F-actin can still be seen (Fig. 5A, bottom panel: arrow), but TJ, marked by membrane fusion appeared to be absent. Instead, multivesicular body (Fig. 5A, bottom panel: arrow head) and membrane vesiculation (Fig. 5A, bottom panel: star) became predominant. Freeze fracture electron microscopy (Severs, 2007) was used to generate an en face view of the subcellular organization of endothelial TJ structures at nanometer resolution. In uninfected mouse cerebral endothelium, intact tight junction can be seen as continuous protein particle strands on the P-face of cleaved membrane (Fig. 5B, top panel). In contrast, in TC83 infected mice the endothelial TJ strands, became discontinuous (Fig. 5B, bottom panel). Together, these results indicate that TJ are disrupted at the BBB during VEEV infection and this is a molecular hallmark of severe infection and neuroinflammation.
Fig. 5.
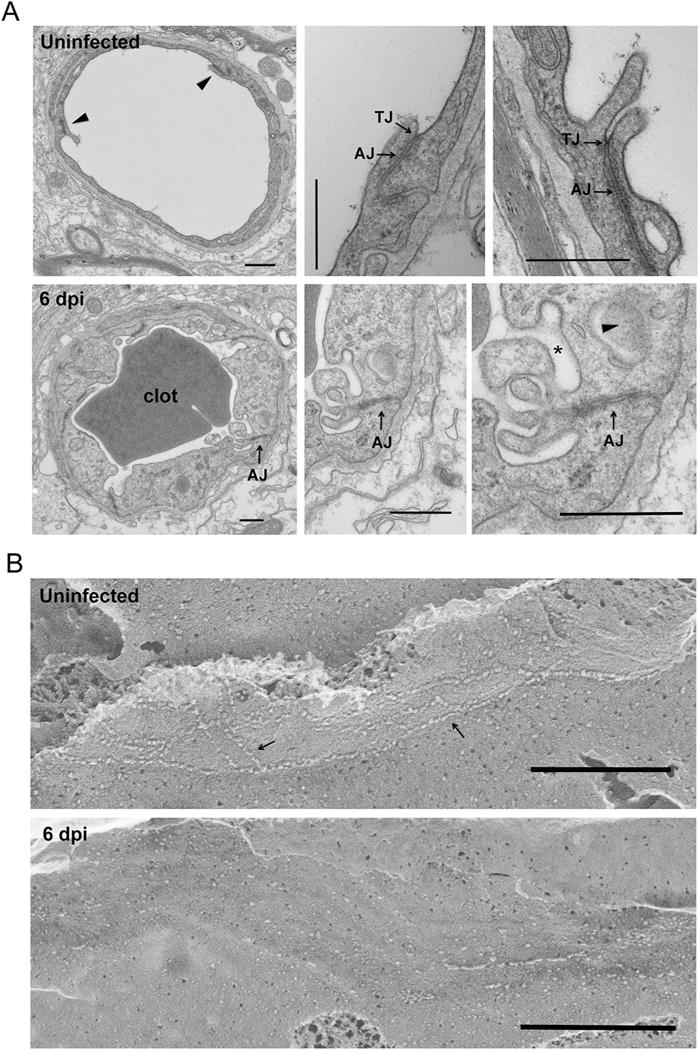
BBB tight junction ultrastructural alteration during VEEV TC83 infection. A. Transmission EM micrographs showing cerebral capillary microvessels in naïve or infected (6 dpi of TC83) mice. Top panel: naïve animal. Bottom panel: infected animal. Bar: 500 nm. TJ: tight junction. AJ: adherens junction. Note that infected mouse BBB endothelium showed cell swelling and membrane vesiculation (star). Multivesicular body also identified (arrow head). B. Freeze-fracture EM micrographs showing cerebral endothelial cell junction (arrows) in naïve or infected (6 dpi of TC83) mice. The protoplasmic (P) face of fractured membrane was shown. Bar: 500 nm. Note that infected endothelial cell junction became discontinuous.
3.3. BBB disruption occurs in conjunction with monocyte infiltration
Given the lack of evidence that TC83 directly modulates the BBB, we speculated that BBB disruption could be a consequence of leukocyte recruitment and extravasation. To identify candidate, infiltrating leukocytes that could impact BBB permeability within the VEEV-infected brain, we phenotyped CD45+ leukocytes in cell suspensions derived from the OB and the cerebral cortex at 2, 4, 6 and 8 dpi. Percentages of infiltrating CD45hiCD11bhi Ly6C+ cells were highest in the OB at 2 dpi, decreasing significantly thereafter (Fig. 6A–B). In contrast, percentages of infiltrating CD45hiCD11bhi Ly6C+ cells within the cortex peaked at 6 dpi (Fig. 6C–D). In both brain regions, percentages of CD45hiCD11bloCD11c+ cells peaked from 4 to 6 dpi, while percentages of infiltrating CD4+ and CD8+ cells were highest at 8 dpi (Fig. 6B, D). Of interest, the timing of peak infiltration of CD45hiCD11bhi Ly6C+ cells corresponds to peak increases in BBB permeability for both CNS regions (Fig. 2). Consistent with this, mRNA levels of barrier destabilizing cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β and interferon (IFN)γ (Daniels et al., 2014), which are not expressed in naive animals (Durrant et al., 2013; Klein et al., 2005), were also elevated at this time-point (Fig. 7).
Fig. 6.
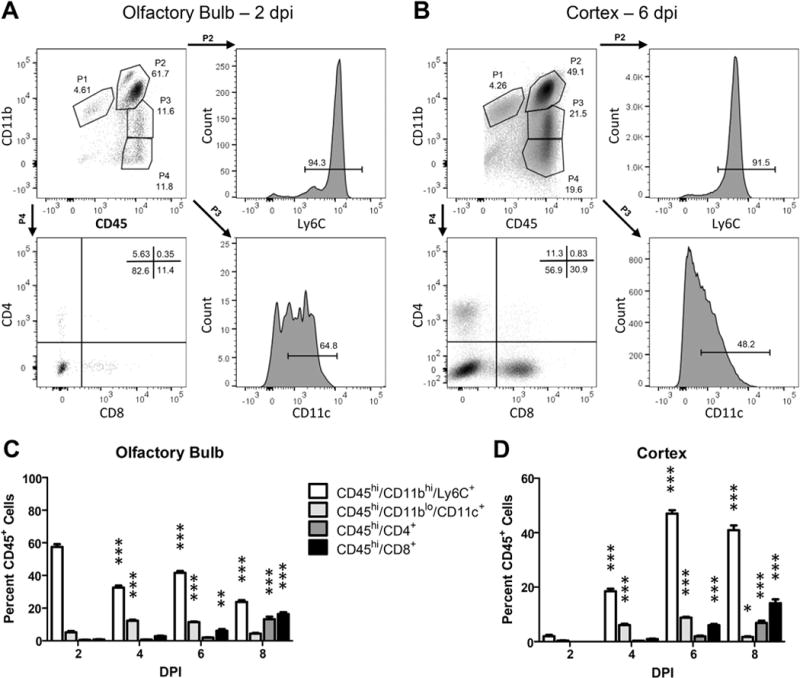
Kinetics of inflammatory immune cell infiltration during intranasal TC83 infection. Representative gating strategies for flow cytometric analysis of leukocytes isolated from wildtype olfactory bulb (A) or cortex (B) at peak BBB permeability, 2 dpi and 6 dpi respectively (TC83, 107 PFU i.n.). Single leukocytes were initially gated by FSC and SSC followed by CD45+ expression. Gates for additional antibodies were set by comparison to unstained controls. Quantification of various gated cell populations isolated from wild-type olfactory bulb (C) or cortex (D) 2, 4, 6, or 8 days after TC83 infection (107 PFU i.n.). Data presented as mean percent CD45+ cells ± SEM for N = 5 mice/group. One-way ANOVA followed by Bonferroni’s post hoc test was used to compare cell numbers relative to 2 dpi, *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig. 7.
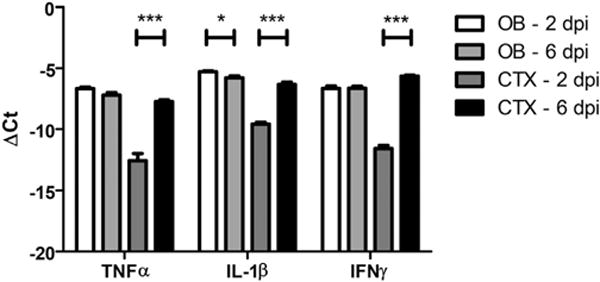
Expression of inflammatory cytokines at peak BBB permeability during TC83 infection. Expression of inflammatory cytokines, TNFα, IL-1β, and IFNγ, during peak BBB permeability of olfactory bulb and cortex (TC83, 107 PFU i.n.). Expression was analyzed by quantitative RT-PCR of whole tissue homogenates. Data presented as mean ΔCt+ SEM for N = 5 mice/group. *, P < 0.05; ***, P < 0.001 via unpaired student t-test.
We next used intravital 2P imaging of the cerebral cortex to directly assess the BBB relative to sites of monocyte extravasation during TC83 infection (Fig. 8) as well as explant imaging to assess monocyte recruitment and BBB disruption more broadly (Supplemental Figs. 2, 3). CCR2RFP/+ CX3CR1GFP/+ mice were given 10 kD Alexa594-dex by retro orbital injection and imaged continuously for 30–90 min (Fig. 8). In uninfected mice, monocytes were rare and microglia were distributed evenly, and the BBB remained intact as shown by a sharp transition in Alexa594-dex fluorescence intensity from the vessel lumen to the parenchyma (Fig. 8A, middle and right panels). However, at 6 dpi, microglia accumulated near regions of BBB disruption and extravascular Alexa594-dex signal was present in a perivascular pattern and leaking into the cortical parenchyma (Fig. 8B, middle and right panels), consistent with results of the in vivo imaging and the sodium fluorescein extravasation assays (Figs. 3 and 4). In addition, we imaged the OB of infected CCR2RFP/+ CX3CR1GFP/+ reporter mice found that monocyte recruitment to the OB occurs on 2 dpi (Supplemental Fig. 2), which precedes recruitment to the cortex by several days, but corresponds with increased sodium fluorescein extravasation (Figs. 3). Moreover, Alexa594-dex leak was observed at sites of active monocyte recruitment, which occurred primarily in superficial medium size vessels (mean 38.6 μm ± 8.19) that lacked perivascular elastin (detected by the 2P second harmonic generation signal), rather than deeper smaller capillaries characteristic of the brain microcirculation (Supplemental Fig. 3).
Fig. 8.
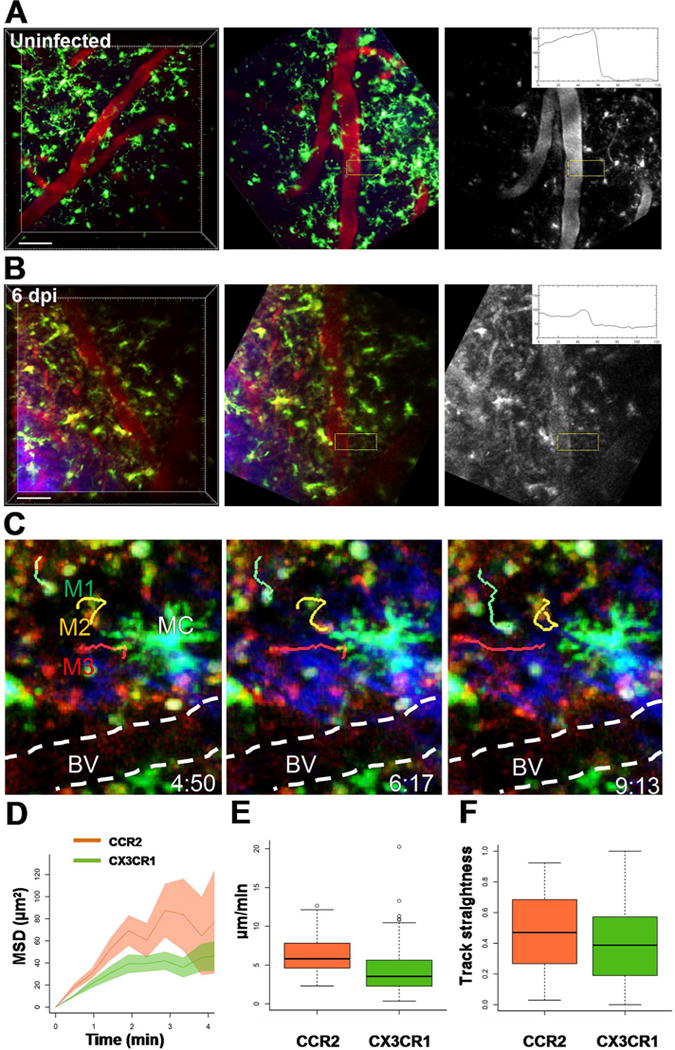
In vivo imaging of monocyte recruitment and trafficking during VEEV infection. A. Intravital 2P imaging of uninfected CX3CR1GFP/+ mice (upper panels) showed evenly distributed microglia (green, large cells with fine membrane projections), but few monocytes (small round cells) and no apparent BBB disruption (monochrome image, boxed region and kymograph). Immediately before imaging, 10 kD Alexa594-dex (red) was injected i.v. to visualize brain vasculature and assess vessel leak (red). B. Intravital 2P imaging of CX3CR1GFP/+ reporter mice challenged with 107 PFU i.n. TC83-GFP at the peak of infection on 6 dpi (lower panels). CX3CR1+ microglia (green) with multiple processes were common in the cortex, but are readily distinguished from monocytes that were more round, compact and motile. Sites of monocyte recruitment were associated with increased vessel permeability to dextran (monochrome image, boxed region and kymograph). Scale bar = 50 μm. C. Monocytes extravasation and trafficking in the cortex of CCR2RFP/+ CX3CR1GFP/+ dual reporter mice on 6 dpi with TC83. CX3CR1GFP/+ (M1, green) and CCR2RFP/+ (M3, red) monocyte migration were tracked near a region of BBB disruption in the cortex. The blood vessels were highlighted with a dashed white line. The orange track (M3) shows the migration of a CCR2RFP/+ CX3CR1GFP/+ double positive cell near a large microglial cell. The elapsed time is shown as min:sec. The motility of CX3CR1GFP/+ and CCR2RFP/+ monocytes was analyzed by plotting (D) mean squared displacement over time (MSD), (E) average track speed and (F) track straightness. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We also performed intravital 2P microscopy to verify that monocytes had indeed extravasated from venules and trafficked into the parenchyma (Fig. 8C). CX3CR1GFP/+CCR2RFP/+ dual reporter mice were challenged with 107 i.n. VEEV TC83-GFP, and imaged on 6 dpi. We found large numbers of both CX3CR1+ and CCR2+ monocytes recruited into the cortex. Resident CX3CR1+ microglia were readily distinguishable from recruited monocytes because microglia have more complex morphology and lower motility. We focused on sites of monocyte extravasation at 6 dpi and found that recruitment occurred primarily in superficial medium sized venules and was associated with increased venule leak (Fig. 8C), as compared to uninfected controls (Fig. 8A) and small capillaries in infected mice, which lacked active monocyte recruitment. We tracked the motility of CX3CR1+ and CCR2+ monocytes in the cortex near regions of BBB disruption (Fig. 8C) and confirmed that cells had indeed extravasated, as CX3CR1GFP/+ and CCR2RFP/+ and dual expressing monocytes displayed cell migration that was not confined to the lumen of the vessel, but rather explored the brain parenchyma without a clear directional bias (Fig. 8D–F). In particular, CCR2+ monocytes (CCR2RFP/+) patrolled more of the brain parenchyma after leaving blood vessels (Fig. 8D) and displayed increased motility compared to CX3CR1+ monocytes (CX3CR1GFP/+) (6.36 ± 2.63 μm/min vs. 4.31 ± 3.23 μm/min, Fig. 8E). Track straightness for both monocyte subsets was similar (Fig. 8F) and consistent with a lack of strong chemotactic guided migration (0.5 typical for random migration and values close to 1 for strong chemotaxis). Taken altogether, these results show that infection of the cortex occurs before BBB disruption; challenging the notion that infection of the CNS is due to VEEV crossing the BBB directly and further suggest that BBB disruption might be a consequence of immune cell recruitment at later stages of infection.
4. Discussion
Our results indicate that VEEV TC83 does not exhibit direct effects on the BBB as it infects the cortex via the olfactory route, even in IFIT1- deficient mice with high levels of viral replication. Increased BBB permeability and loss of TJ integrity were observed predominately in venules within the cortex 5–6 dpi intranasal VEEV TC83 infection, which coincided with the infiltration of monocytes. In a recent study, intranasal inoculation with VEEV replicon particles was associated with an increase in BBB permeability within cortical regions at two days post-infection that permitted the entry of intravenously administered GFP-expressing virus (Schafer et al., 2011). Given that those experiments examined replicons that undergo a single round of replication without propagation and spread (Pushko et al., 1997), it is possible that this system doesn’t recapitulate the full range of BBB modulation that occurs during disease progression. Other studies suggest that endothelial cell expression of inflammatory molecules involved in the recruitment and retention of mononuclear cells, including CCL2 and ICAM-1, are critical for BBB disruption, which occurs at sites with perivascular accumulations of lymphocytes and monocytes (Sharma et al., 2011). However, the cellular and molecular mechanisms governing mononuclear cell interactions with the CNS vasculature during VEEV infection are not well defined. Therefore, we infected wild-type and Ifit1−/− mice intranasally with GFP-expressing TC83 virus and used 2P microscopy to determine if sites of vascular leak were associated with virus replication in the neurovascular unit (NVU), which include endothelial cells, pericytes, astrocytes and neurons, or at sites of active monocyte recruitment. The TC83 model provided an opportunity to look at CNS infection independent of a large circulating viral load. We found that widespread viral infection on 2 dpi preceded BBB disruption and that TC83 infected neurons and astrocytes, but not endothelial cells or pericytes, the latter of which are key regulators of the NVU. Moreover, vascular permeability increased dramatically at later stages of infection when immune cell infiltration was evident. We demonstrate that vascular leak occurs at both medium sized venules and small capillaries deep within the brain parenchyma. The most robust opening observed by 2P microscopy and higher molecular weight dextran (70 kD) leakage occurred predominately in venules, rather than small capillaries. However, leakage of sodium fluorescein, a smaller, more permeable dye, throughout the CNS tissue and TJ disruption in small capillaries demonstrate that vascular leak also occurs in small parenchyma vessels during late infection.
The BBB occurs at the levels of capillaries and post-capillary venules, and results from the selectivity of TJs and AJs that restrict the passage of solutes, low expression of leukocyte adhesion molecules, and an additional layer of barrier support provided by ensheathing pericytes and astrocyte endfeet (Reinhold and Rittner, 2016). In our model VEEV entry into the CNS occurs along axons of olfactory sensory neurons, however modulation of the BBB may occur through mechanisms that are independent of the inoculation route. Indeed, recent studies have raised questions regarding direct mechanisms of BBB disruption through virus-mediated interactions with cellular constituents of the NVU. In the study by Schafer et al., VEEV infection and replication within the OB preceded BBB disruption within cortical regions (Schafer et al., 2011). The authors’ suggested that VEEV infection in the OB promotes BBB opening, which allows virus to enter the brain. However, the endothelium of the OB exhibits direct conduits with the perivascular spaces of cerebral vessels (Lochhead et al., 2015), suggesting that alternative mechanisms also exist. In our study, viral neuroinvasion and replication within the cortex did not coincide with BBB disruption and VEEV TC83 exerted no direct effects on BBB permeability in vitro. These results suggest that BBB disruption is not required for VEEV entry into the CNS at locations distant from its initial migration along olfactory neuronal pathways into the OB. Of interest, similar findings have been observed in murine models of lethal infection with rabies virus, which exhibits viral spread within the CNS without alterations in the BBB (Roy and Hooper, 2008).
IFIT1 is an important IFN-stimulated protein that normally acts to block translation of virion-delivered, virus-specific RNAs (Reynaud et al., 2015). Published studies have shown that virulent VEEV strains, such as ZPC-738, evade IFIT1 activity while the attenuated VEEV strain TC83 encodes a G3A mutation within the 5′ UTR that is associated with increased sensitivity to IFN (Kulasegaran-Shylini et al., 2009). Consistent with this, studies have identified host IFIT1 as the agent responsible for TC83 IFN-mediated attenuation (Hyde et al., 2014). The authors showed that the G3A mutation destabilizes a stem-loop at the beginning of the 5′ UTR and exposes the non 2′-O methylated 5′ cap to recognition by host IFIT1. Thus, virulence of TC83 is restored during infections of mice with IFIT1 deficiency, which succumb to peripheral inoculation within 6–7 dpi. Intranasal infection of Ifit1−/− mice with TC83-GFP showed that axonal tracking into cortical regions, similar to that observed in wild-type animals, follows infection of the OB via olfactory sensory neuron axons. However, infection of the OB and cortex of Ifit1−/− mice exhibited more extensive GFP expression by 1 dpi compared with their wild-type counterparts without increased BBB permeability. These findings support the notion that virus does not mediate direct effects on the BBB. By 6 dpi however, BBB permeability was significantly increased in both genotypes to the same extent. In wild-type mice the BBB response returned to baseline permeability levels by at 8 dpi, a time-point that coincided with viral clearance. These data are consistent with reported effects of infiltrating mononuclear cells on both virologic control and BBB permeability (Schafer et al., 2009), and suggest that the transient opening of the BBB may be a necessary consequence of the immune response.
Studies utilizing in vitro BBB models demonstrate significant increases in permeability of soluble molecules and an enhanced rate of T cell migration following monocyte migration across human brain-derived endothelial cells (Seguin et al., 2003). However, in vivo studies of mice with CNS autoimmune disease have suggested that alterations in BBB permeability and entry of mononuclear cells may be two distinct processes (Floris et al., 2004). However, using 2P approaches, vascular leakage of fibrinogen was shown to occur during leukocyte infiltration and induce microglial activation and clustering (Davalos et al., 2012), similar to our observations in the setting of TC83 infection. In previous studies, we showed that in vivo BBB permeability increased after subcutaneous infection with West Nile virus (WNV), a neurotropic flavivirus, prior to initial neuroinvasion and during inflammatory infiltration (Daniels et al., 2014). Indeed, both WNV and VEEV induce increased expression of vascular adhesion molecules at endothelial barriers that recruit lymphocytes and monocytes (Schafer et al., 2009; Verma et al., 2009), which, via expression of BBB destabilizing cytokines including interleukin (IL)-2, IL-1 and TNF-α has recently been shown to decrease TJ integrity at the BBB via effects on RhoG-TPases and/or TJ expression (Daniels et al., 2014; Rochfort et al., 2016; Rochfort et al., 2014; Wylezinski and Hawiger, 2016). In contrast, CNS infection with rabies virus is associated with maintenance of BBB integrity, which has been shown to limit immune cell infiltration and virologic control, contributing to lethality (Roy and Hooper, 2007). While the exact mechanisms of virus and/or immune cell effects on BBB function during VEEV, WNV or Rabies virus remain to be elucidated, our findings suggest that increased BBB permeability may occur during immune cell-mediated clearance of certain neurotropic viruses.
Understanding mechanisms for controlling the integrity of the BBB is relevant not only for identifying new strategies to modulate disease caused by encephalitic alphaviruses, but for targeting other pathogens that infect and cause disease in the brain and spinal cord. Identifying molecular mechanisms that promote BBB disruption may also lead to the development of novel therapeutics to aide in delivering anticancer agents or other drugs that normally do not cross the BBB.
Supplementary Material
Acknowledgments
This work was supported by NIH grants U19 AI083019 and R01 NS052632 (to RSK), R01DK084059 (to JH) HDTRA11510032, R01 DK097 and U01 AI095550, R01 AI077600 (to MJM) and (to RSK, MJM, JH). MC was supported by T32-AI007172.
Experimental support was provided by the Speed Congenics Facility of the Rheumatic Diseases Core Center. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number P30AR048335. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jneuroim.2017.04.008.
References
- Atasheva S, Krendelchtchikova V, Liopo A, Frolova E, Frolov I. Interplay of acute and persistent infections caused by Venezuelan equine encephalitis virus encoding mutated capsid protein. J Virol. 2010;84:10004–10015. doi: 10.1128/JVI.01151-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD, Lazear HM, Diamond MS. Propagation, quantification, detection, and storage of West Nile virus. Curr Protoc Microbiol. 2013;31 doi: 10.1002/9780471729259.mc15d03s31. (15D 3 1-D 3 8) [DOI] [PubMed] [Google Scholar]
- Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio. 2014;5:e01476–14. doi: 10.1128/mBio.01476-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant DM, Robinette ML, Klein RS. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med. 2013;210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris S, Blezer EL, Schreibelt G, Dopp E, van der Pol SM, Schadee-Eestermans IL, et al. Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain. 2004;127:616–627. doi: 10.1093/brain/awh068. [DOI] [PubMed] [Google Scholar]
- Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, et al. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Kirschner MW. Filament organization revealed in platinum replicas of freeze-dried cytoskeletons. J Cell Biol. 1980;86:212–234. doi: 10.1083/jcb.86.1.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, et al. A viral RNA structural element alters host recognition of nonself RNA. Science. 2014;343:783–787. doi: 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasegaran-Shylini R, Thiviyanathan V, Gorenstein DG, Frolov I. The 5′UTR-specific mutation in VEEV TC-83 genome has a strong effect on RNA replication and subgenomic RNA synthesis, but not on translation of the encoded proteins. Virology. 2009;387:211–221. doi: 10.1016/j.virol.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Baran U, Wang RK. Application of thinned-skull cranial window to mouse cerebral blood flow imaging using optical microangiography. PLoS One. 2014;9:e113658. doi: 10.1371/journal.pone.0113658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab. 2015;35:371–381. doi: 10.1038/jcbfm.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol. 2006;177:8053–8064. doi: 10.4049/jimmunol.177.11.8053. [DOI] [PubMed] [Google Scholar]
- Miner JJ, Daniels BP, Shrestha B, Proenca-Modena JL, Lew ED, Lazear HM, et al. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat Med. 2015;21:1464–1472. doi: 10.1038/nm.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AC, Forshey BM, Notyce D, Astete H, Lopez V, Rocha C, et al. Venezuelan equine encephalitis virus in Iquitos, Peru: urban transmission of a sylvatic strain. PLoS Negl Trop Dis. 2008;2:e349. doi: 10.1371/journal.pntd.0000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AT, Rico AB, Stauft CB, Hammond SL, Aboellail TA, Tjalkens RB, et al. Entry sites of Venezuelan and western equine encephalitis viruses in the mouse central nervous system following peripheral infection. J Virol. 2016;90:5785–5796. doi: 10.1128/JVI.03219-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- Reinhold AK, Rittner HL. Barrier function in the peripheral and central nervous system-a review. Pflugers Arch. 2016 doi: 10.1007/s00424-016-1920-8. [DOI] [PubMed] [Google Scholar]
- Reynaud JM, Kim DY, Atasheva S, Rasalouskaya A, White JP, Diamond MS, et al. IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLoS Pathog. 2015;11:e1004863. doi: 10.1371/journal.ppat.1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One. 2014;9:e101815. doi: 10.1371/journal.pone.0101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Tumour necrosis factor-alpha-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. 2016;136:564–572. doi: 10.1111/jnc.13408. [DOI] [PubMed] [Google Scholar]
- Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. J Virol. 2007;81:7993–7998. doi: 10.1128/JVI.00710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Hooper DC. Immune evasion by rabies viruses through the maintenance of blood-brain barrier integrity. J Neuro-Oncol. 2008;14:401–411. doi: 10.1080/13550280802235924. [DOI] [PubMed] [Google Scholar]
- Salimi H, Cain MD, Klein RS. Encephalitic arboviruses: emergence, clinical presentation, and neuropathogenesis. Neurotherapeutics. 2016;13:514–534. doi: 10.1007/s13311-016-0443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Whitmore AC, Konopka JL, Johnston RE. Replicon particles of Venezuelan equine encephalitis virus as a reductionist murine model for encephalitis. J Virol. 2009;83:4275–4286. doi: 10.1128/JVI.02383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Brooke CB, Whitmore AC, Johnston RE. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol. 2011;85:10682–10690. doi: 10.1128/JVI.05032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin R, Biernacki K, Rotondo RL, Prat A, Antel JP. Regulation and functional effects of monocyte migration across human brain-derived endothelial cells. J Neuropathol Exp Neurol. 2003;62:412–419. doi: 10.1093/jnen/62.4.412. [DOI] [PubMed] [Google Scholar]
- Sergeev AN, Ryzhikov AB, Bulychev LE, Stepkina EO, Tkacheva NV. The course of airborne infection in rabbits infected with the Venezuelan encephalomyelitis virus. Vopr Virusol. 1991;36:492–495. [PubMed] [Google Scholar]
- Severs NJ. Freeze-fracture electron microscopy. Nat Protoc. 2007;2:547–576. doi: 10.1038/nprot.2007.55. [DOI] [PubMed] [Google Scholar]
- Sharma A, Bhomia M, Honnold SP, Maheshwari RK. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol J. 2011;8:197. doi: 10.1186/1743-422X-8-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology. 2009;385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Salas R, Rico-Hesse R, Ludwig GV, Oberste MS, Boshell J, et al. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet. 1996;348:436–440. doi: 10.1016/s0140-6736(96)02275-1. [DOI] [PubMed] [Google Scholar]
- White LJ, Wang JG, Davis NL, Johnston RE. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5′ untranslated region. J Virol. 2001;75:3706–3718. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylezinski LS, Hawiger J. Interleukin 2 activates brain microvascular endothelial cells resulting in destabilization of adherens junctions. J Biol Chem. 2016;291:22913–22923. doi: 10.1074/jbc.M116.729038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinselmeyer BH, Dempster J, Wokosin DL, Cannon JJ, Pless R, Parker I, et al. Two-photon microscopy and multidimensional analysis of cell dynamics. Methods Enzymol. 2009;461:349–378. doi: 10.1016/S0076-6879(09)05416-0. (Chapter 16) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.