

Abstract
Fabry disease is an X-linked lysosomal storage disorder caused by a lack of α-galactosidase A activity, which leads to the accumulation of globotriaosylceramide in various organs. A complete lack of α-galactosidase A activity in a hemizygous male is the classical phenotype, and some hemizygous males show primarily cardiac and/or renal symptoms that appear in adulthood; this is called the variant type or the late-onset type. The kidney and heart are the major target organs, with damage to these organs related to mortality. Thus, in Fabry patients, early detection and early treatment are critical to longevity. Here, we present a 55-year-old Japanese male patient who was diagnosed with late-onset Fabry nephropathy with cardiomyopathy but with no abnormal urinary findings except for urinary mulberry cells and mulberry bodies. In spite of the absence of abnormal urinary findings, the light microscopic and electron microscopic pathological findings showed extensive deposition of globotriaosylceramide to podocytes. In this paper, we propose that the presence of mulberry cells and mulberry bodies can be used for the earlier detection of Fabry nephropathy, especially the late-onset type.
Keywords: Fabry nephropathy, Late-onset, Mulberry bodies, Mulberry cells
Introduction
Fabry disease is an X-linked inherited lysosomal storage disorder caused by a deficiency of α-galactosidase A activity, resulting in the intracellular accumulation of glycosphingolipids, especially globotriaosylceramide (Gb3) [1]. A complete lack of α-galactosidase A activity in a hemizygous male is the classical phenotype, which manifests with angiokeratomas, acroparesthesia, hypohidrosis, and gastrointestinal symptoms in childhood, together with cardiovascular, cerebrovascular, and renal disease in adulthood. In contrast, some hemizygous males show primarily cardiac and/or renal symptoms that appear in adulthood; this is called the variant type or the late-onset type, in which the residual α-galactosidase A activity is slightly higher than that in the classical type. The existence of these different phenotypes is partly explained by the levels of residual α-galactosidase A activity, but the precise pathogenic mechanism is not yet clear.
Although the early detection and the early initiation of enzyme replacement therapy are the most important factors in the treatment of Fabry nephropathy [2], it is difficult to identify the late-onset type of Fabry disease, because there are no overt symptoms such as angiokeratomas, acroparesthesia, corneal opacity, or hypohidrosis in childhood.
Urinary mulberry cells are regarded as distal tubular epithelial cells in which Gb-3 has accumulated; they are the characteristic feature of Fabry disease. Moreover, urinary mulberry bodies are a component of mulberry cells that can be distinguished easily from fat particles by their inner lamellar appearance. At present, however, urinary mulberry cells and bodies are not utilized much in the diagnosis of Fabry disease.
Here, we present a case of late-onset type Fabry nephropathy with cardiomyopathy but with no abnormal urinary findings except for urinary mulberry cells and mulberry bodies. This case showed severe Gb-3 deposition to podocytes in a histological examination. This case demonstrates the usefulness of these urinary markers in the earlier detection late-onset type Fabry nephropathy.
Case report
A 55-year-old-Japanese male had been treated for cryptogenic cardiomyopathy for several years. After his first cousin, who had been in dialysis treatment, was diagnosed with Fabry disease, he asked his attending physician to check for the presence of Fabry disease. The examination showed his α-galactosidase A activity in the leucocytes to be 1.1 nmol/mg protein/h (normal range 49.8–116.4 nmol/mg protein/h), and he was suspected to have Fabry disease and referred to our Department of Nephrology to determine whether he had Fabry nephropathy or not. At the time of the consultation, no angiokeratomas, acroparesthesia, hypohidrosis, or corneal opacities were observed. On examination, his blood pressure was 103/63 mmHg, the pulse rate was 62 beats/min (regular sinus rhythm), body height was 167 cm, and body weight was 63 kg. The patient’s laboratory data showed that his serum creatinine and blood urea nitrogen levels were 0.95 and 15.5 mg/dL, respectively. His serum cystatin C level was 0.73 mg/L (0.61–1.0 mg/L) and creatinine clearance was 120 ml/min. His hemoglobin and serum albumin levels were 17 and 4.7 g/dL, respectively, and his lactate dehydrogenase and triglycerides levels were high (262 IU/L, 381 mg/dL). His white blood cells, platelets, C-reactive protein, blood glucose, and electrolytes were all normal. Since the first urinalysis tests showed no abnormalities (urine protein negative, 0.12 g/gcre, urine albumin negative, 23.8 mg/gcre, occult blood negative, and red blood cells 0–1/high power field), and no tubulointerstitial damage was observed [N-acetyl-β-d-glucosaminidase 6.4 U/L (<11.5 U/L), β-2 microglobulin <70 μg/L (<200 μg/L)], we considered that this patient did not have Fabry nephropathy. Soon thereafter, however, a medical technologist reported the presence of mulberry cells and mulberry bodies in the patient’s urine (Fig. 1a, b). Thus, we conducted a percutaneous renal biopsy to investigate the presence of Fabry nephropathy. Surprisingly, all glomeruli showed strikingly enlarged and vacuolated podocytes under light microscopy (Fig. 2), and myelin-like bodies were detected in the podocytes by electron microscopy (Fig. 3). We could not find foot process effacement in electron microscopy (Fig. 3). There was no accumulation of Gb-3 in the tubulointerstitial area. After the renal biopsy, we performed mutation analysis and identified L403S in exon 7.
Fig. 1.
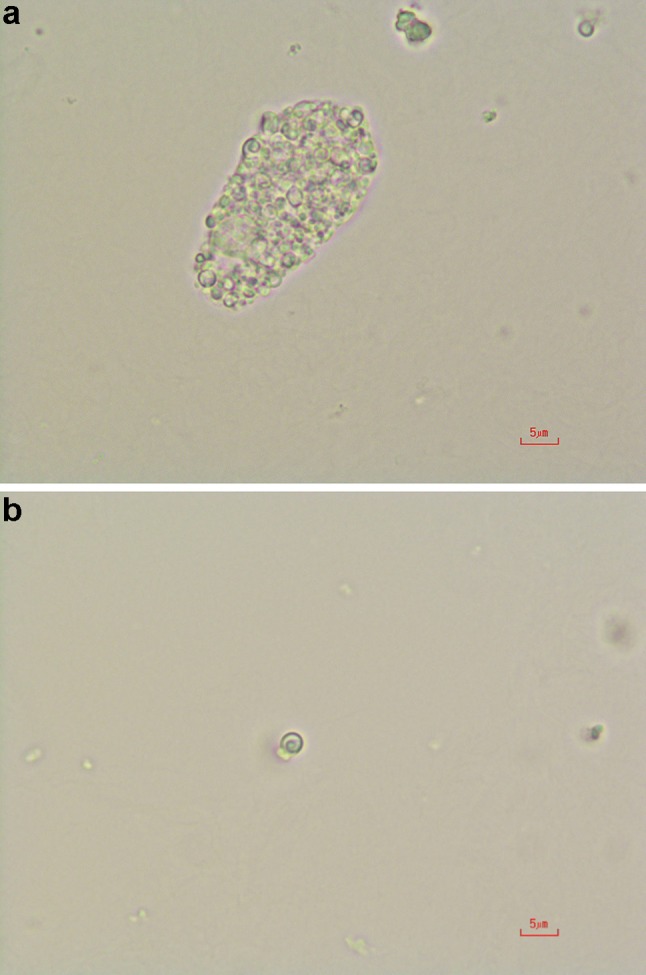
a Mulberry cells in the urine sediment. Magnification is ×400. b Mulberry body in the urine sediment. A lamellar appearance is the characteristic feature of mulberry bodies. Magnification is ×400
Fig. 2.
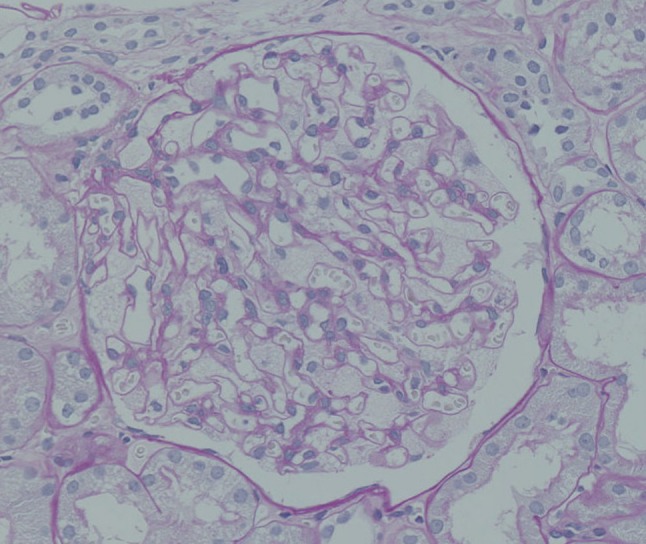
Light microscopic image of a kidney biopsy sample. Enlarged and vacuolated podocytes are shown (periodic acid–Schiff stain ×400)
Fig. 3.
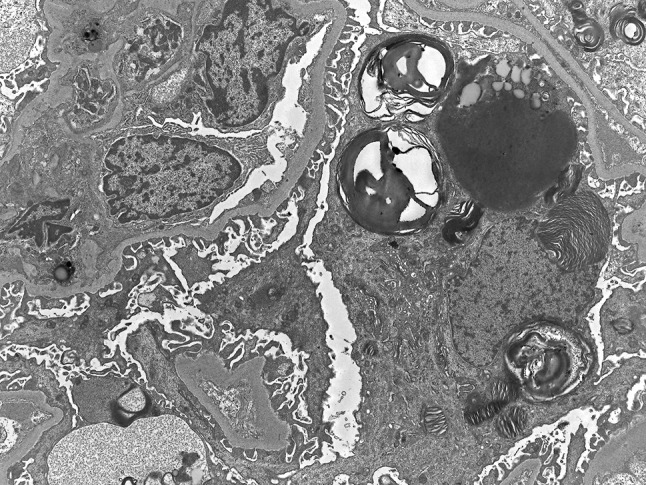
Electron microscopic image of a kidney biopsy sample. Myelin-like bodies are present in the podocytes. Foot process effacement is not observed. Magnification is ×2500
From the results of the renal biopsy, together with the presence of cardiomyopathy without general symptoms, the patient’s low level of α-galactosidase A activity, and the results of genetic testing, we diagnosed our patient with late-onset Fabry disease, specifically, a cardiac variant with no clinical renal symptoms but renal histological damage. After the diagnosis was made, enzyme replacement therapy with recombinant alpha-galactosidase A was initiated and has continued to be administered every 2 weeks.
Discussion
Fabry nephropathy is the representative life-threatening complication of Fabry disease. At the third to fifth decade, almost all untreated hemizygous males and some heterozygous females develop end-stage renal disease. To prevent the progressive decrease of renal function, the early detection and the early initiation of enzyme replacement therapy are recommended [3–5].
Hemizygous Fabry male patients are divided into two categories: the classical and atypical types. The atypical type mainly shows cardiac symptoms [6] or renal symptoms [7] or both without classical general symptoms such as angiokeratoma, hypohidrosis, and acroparesthesia. The phenotype of the atypical type presents at a late stage of life; thus, this type is also referred to as the late-onset type and is often overlooked.
To detect Fabry nephropathy earlier, a variety of methods have been proposed. One of these is the urinary detection of podocytes. In a previous report, Tondel et al. showed that podocyte foot process effacement was an early sign of Fabry nephropathy without proteinuria or a decreased glomerular filtration rate (GFR) [8]. Consequently, podocyte loss into the urine occurs in the course of disease progression [9]. Teramachi et al. described a case of podocyturia without proteinuria and with normal renal function in a young Fabry patient and demonstrated that podocyturia is a useful biomarker for the early detection of Fabry nephropathy. However, as the author stated, using podocyturia for detection of Fabry nephropathy is time-consuming, because immunofluorescence techniques using synaptopodin antibody are needed to identify urine podocytes [10]. In our case, we were able to detect mulberry cells and mulberry bodies in the urine before the onset of renal manifestations only under light microscopy and thus were able to diagnose Fabry nephropathy. Furthermore, since we could not detect foot process effacement in electron microscopy findings, our case might be more early stage of Fabry nephropathy. Although mulberry cells are specific to Fabry disease, using them as a diagnostic tool is uncommon throughout the world [11]. On the other hand, Maltase cross particles are a representative feature of Fabry nephropathy; however, they are not specific to Fabry nephropathy and are often seen in other types of glomerulonephritis.
Recently, many Japanese researchers have found mulberry cells to be an inexpensive, noninvasive, and useful diagnostic tool [12–14]. Mulberry cells and mulberry bodies are the characteristic features of Fabry nephropathy, and are distinguishable from oval fat bodies and fat particles based on the inner lamellar appearance of mulberry bodies. Our group more recently reported a case of a renal variant of Fabry disease diagnosed on the basis of the presence of urinary mulberry cells [14]. In that case, the patient showed proteinuria as well as mulberry cells and bodies, but the present case showed only mulberry cells without proteinuria. That is, we were able to detect Fabry nephropathy before the presence of renal clinical symptoms through the identification of urinary mulberry cells or mulberry bodies. According to a previous report, the extent of Gb-3 deposition to the glomeruli correlates with urinary protein excretion rates [15]. However, renal intracellular Gb-3 deposits are thought to be present even in young children with a normal GFR and absent microalbuminuria [16]. In our case, many Gb-3 depositions to podocytes were observed in spite of the lack of proteinuria or microhematuria. Urinary mulberry cells and mulberry bodies were the only abnormal findings indicating renal damage.
In general, although mulberry cells are regarded as distal tubular epithelial cells in which Gb-3 has accumulated, we could not find any accumulation of Gb-3 in distal tubular epithelial cells but only in podocytes, so the origin of the mulberry cells might have been podocytes in our case. Moreover, it is unclear whether the amount of mulberry cells or mulberry bodies present is correlated to disease progression. These topics are the next subjects of future research in our case.
In the present case, since we could start the enzyme replacement therapy before the presence of proteinuria and renal impairment, his renal function seemed to keep stable during enzyme replacement therapy.
In conclusion, we presented a case of late-onset Fabry disease, specifically of the cardiac variant type without renal clinical manifestation. Our patient was found to have Fabry nephropathy, which was revealed by renal biopsy. In our case, the decisive factor in the renal biopsy was the presence of urinary mulberry cells and mulberry bodies. These urinary markers can be a useful tool for the earlier detection of late-onset Fabry disease.
Compliance with ethical standards
Conflict of interest
The authors have declared that no conflict of interest exists.
Human and animal rights
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Kint JA. Fabry’s disease: alpha-galactosidase deficiency. Science. 1970;167:1268–1269. doi: 10.1126/science.167.3922.1268. [DOI] [PubMed] [Google Scholar]
- 2.Tondel C, Bostad L, Larsen KK, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–138. doi: 10.1681/ASN.2012030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pisani A, Visciano B, Imbriaco M, et al. The kidney in Fabry’s disease. Clin Genet. 2014;86:301–309. doi: 10.1111/cge.12386. [DOI] [PubMed] [Google Scholar]
- 4.Terryn W, Cochat P, Froissart R, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant. 2013;28:505–517. doi: 10.1093/ndt/gfs526. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz A, Oliveira JP, Wanner C, Brenner BM, Waldek S, Warnock DG. Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nat Clin Pract Nephrol. 2008;4:327–336. doi: 10.1038/ncpneph0806. [DOI] [PubMed] [Google Scholar]
- 6.Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293. doi: 10.1056/NEJM199508033330504. [DOI] [PubMed] [Google Scholar]
- 7.Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801–807. doi: 10.1046/j.1523-1755.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 8.Tondel C, Kanai T, Larsen KK, et al. Foot process effacement is an early marker of nephropathy in young classic Fabry patients without albuminuria. Nephron. 2015;129:16–21. doi: 10.1159/000369309. [DOI] [PubMed] [Google Scholar]
- 9.Eng CE, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:182. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- 10.Trimarchi H, Canzonieri R, Muryan A, et al. Copious podocyturia without proteinuria and with normal renal function in a young adult with Fabry disease. Case Rep Nephrol. 2015;257628. [DOI] [PMC free article] [PubMed]
- 11.Becher GJ, Nicholls K. Lipiduria-with special relevance to Fabry disease. Clin Chem Lab Med. 2015;53:s1465–s1470. doi: 10.1515/cclm-2015-0499. [DOI] [PubMed] [Google Scholar]
- 12.Nakamichi T, Miyazaki M, Nakayama K, et al. Fabry’s disease discovered with chance urinary mulberry cells: a case report. CEN Case Rep. 2013;2:49–52. doi: 10.1007/s13730-012-0038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honda T, Komatsu E, Furuse S, Mise N. Fabry disease diagnosed based on the detection of urinary mulberry bodies. Int Med. 2016;55:2903. doi: 10.2169/internalmedicine.55.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimohata H, Ogawa Y, Maruyama H, Hirayama K, Kobayashi M. A case of renal variant of Fabry disease diagnosed by the presence of urinary mulberry cells. Int Med. 2016;55:3475–3478. doi: 10.2169/internalmedicine.55.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Najafian B, Svarstad E, Bostad L, et al. Progressive podocyte injury and globotriaosylceramide (Gb-3) accumulation in young patients with Fabry disease. Kidney Int. 2010;79:663–670. doi: 10.1038/ki.2010.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51:767–776. doi: 10.1053/j.ajkd.2007.12.032. [DOI] [PubMed] [Google Scholar]