

Abstract
Cutaneous and systemic plasmacytosis (CSP) is a rare lymphoproliferative disorder that mainly affects middle-aged Asian individuals. Although Castleman disease is often complicated with various renal involvements, glomerulonephritis associated with CSP, which is considered as a variant of Castleman disease, is rare. This report presents the case of a 41-year-old Japanese man with nephrotic syndrome associated with CSP. Renal biopsy findings showed focal segmental glomerulosclerosis (FSGS) and diffusely mild segmental mesangial proliferation. Plasma cell infiltration in the interstitium was not observed. Electron microscopic findings showed diffuse foot process effacement, localized involvement of subendothelial space widening with amorphous materials, and endothelial cell swelling. Lymph node biopsy findings denied Castleman disease. His skin regions and proteinuria were successfully treated with prednisolone and cyclosporine. The causal relationship between CSP and FSGS is unknown. However, increased serum levels of IL-6 and VEGF and decreased VEGF expression in the podocyte may contribute to renal lesions in patients with CSP. To our best knowledge, this is the first case of a patient with FSGS associated with CSP.
Keywords: Focal segmental glomerulosclerosis, Nephrotic syndrome, Cutaneous and systemic plasmacytosis, Castleman disease
Introduction
Cutaneous and systemic plasmacytosis (CSP) is a rare lymphoproliferative disorder, characterized as peculiar multiple skin eruptions, lymphadenopathy, and polyclonal hypergammaglobulinemia [1]. CSP affects mainly middle-aged Asian patients, particularly Japanese individuals. In most cases, the skin, bone marrow, and lymph nodes are involved in this disease [2]. Although the etiology of CSP has not been clearly elucidated, increased secretion of interleukin 6 (IL-6) from mature plasma cells in the skin to circulation plays a pivotal role in its pathogenesis [3]. Based on comparable clinical and laboratory chemical findings, some investigators considered that CSP is a variant of multicentric Castleman disease, known as angiofollicular lymph node hyperplasia [4]. Castleman disease is often accompanied by various renal lesions, including amyloidosis, thrombotic microangiopathy, and some forms of glomerulonephritis [5, 6]. However, CSP is rarely accompanied by renal lesions [3]. To the best of our knowledge, no report showed that glomerular disease is associated with CSP. Herein, we report a rare case of focal segmental glomerulosclerosis (FSGS) associated with CSP.
Case report
A 41-year-old Japanese man was admitted to our hospital due to nephrotic syndrome. He noticed a sudden onset of bilateral leg edema before admission. The proteinuria was not detected until approximately 10 months previously. He experienced skin eruptions that initially appeared on his face and then gradually spread to his trunk over approximately 10 years. The skin biopsy results revealed CSP. He was treated with 100 mg/day cyclosporine or 10 mg/day oral glucocorticoid, but the skin eruptions did not improve. Immunosuppressive therapy was discontinued 3 years before admission. He took no medication and supplement and had no family history of kidney disease.
Physical examination showed a height of 171 cm, weight of 65.7 kg (weight gain of 2 kg within a month), body temperature of 36.7 °C, pulse rate of 93 beats/min, blood pressure of 143/90 mm Hg, and respiratory rate of 12/min. Multiple 0.5–1-cm-diameter, well-demarcated, brownish-red eruptions were distributed on his face and trunk. Both lower limbs had pitting edema. A slightly soft and enlarged lymph node was found in the right armpit.
Results of serological studies revealed hypoalbuminemia, normal renal function, hypercholesterolemia, and positive C-reactive protein. He had no signs of viral infection and no obvious autoantibodies. Monoclonal protein was not detected by serum protein immunoelectrophoresis. Serum IL-6 and vascular endothelial growth factor (VEGF) were markedly elevated [IL-6 6.7 pg/mL (normal limit < 3.7 pg/mL), and VEGF 1290 pg/mL (normal limit < 38.3 pg/mL)]. His urinalysis showed microscopic hematuria and a severe degree of proteinuria (Table 1).
Table 1.
Laboratory data at the time of administration
| WBC | 7260/μL | IgG | 1215 mg/dL |
| stab | 4% | IgA | 612 mg/dL |
| seg | 60% | IgM | 182 mg/dL |
| lym | 27% | ANA | < 1:40 |
| mono | 8% | anti-DNA-Ab | < 2.0 U/mL |
| eos | 1% | C3 | 97 mg/dL |
| baso | 0% | C4 | 26 mg/dL |
| RBC | 503 × 104/μL | CH50 | 38 U/mL |
| Hb | 14.3 g/dL | MPO-ANCA | < 1.0 U/mL |
| Hct | 42.9% | PR3-ANCA | < 1.0 U/mL |
| Plt | 39.9 × 104/μL | anti-GBM-Ab | < 2.0 U/mL |
| TP | 6.0 g/dL | HBS-Ag | < 0.03 IU/mL |
| Alb | 1.9 g/dL | HBC-Ab | < 1.0 |
| ALT | 16 U/L | HCV-DNA | < 1.0 |
| AST | 8 U/L | HIV-Ag/Ab | < 1.0 |
| LDH | 156 U/L | HHV-8-DNA | < 2.0 × 101 copy |
| CRP | 0.76 mg/dL | ||
| BUN | 6.6 mg/dL | VEGF | 1290 pg/mL |
| Cre | 0.67 mg/dL | IL-6 | 6.7 pg/mL |
| UA | 5.0 mg/dL | ||
| TC | 293 mg/dL | Proteinuria | 7009 mg/day |
| Urinary RBC | 20.0/μL |
ANA antinuclear antibody, Ab antibody, CH50 total complement level, MPO-ANCA myeloperoxidase–antineutrophil cytoplasmic antibody, PR3-ANCA proteinase 3-antineutrophil cytoplasmic antibody, HBS hepatitis B surface, Ag antigen, HBC hepatitis B core, HCV hepatitis C virus, HIV human immunodeficiency virus, HHV-8 human herpesvirus 8, VEGF vascular endothelial growth factor, IL-6 interleukin 6
The renal histological examination showed segmental sclerosis in 3 of 28 glomeruli. Each glomerulus showed diffusely mild segmental mesangial proliferation and hypertrophied podocytes. No obvious changes were seen in the glomerular capillary walls on using periodic acid silver methenamine stain. Plasma cell infiltration in the interstitium was not observed on CD 38 staining (Fig. 1). Immunofluorescence studies indicated faint to mild deposition of IgG, IgA, κ, and λ in the mesangial to paramesangial regions. However, complement components, such as C3, C4, and C1q, were not detected. Electron microscopic findings showed diffuse foot process effacement, localized involvement of subendothelial space widening with amorphous materials, and endothelial cell swelling. No amyloid fibril was observed. Electron-dense deposits were absent in the subendothelial, intramembranous, subepithelial, and mesangial regions (Fig. 2). FSGS (not otherwise specified variant) was diagnosed.
Fig. 1.

Light microscopic findings and immunohistochemical demonstration of CD 38. a Mild segmental mesangial proliferation and podocyte hypertrophy and proliferation are observed in the glomerulus. Arrows and asterisk indicate the mesangial proliferation and podocyte hypertrophy and proliferation, respectively (periodic acid-Schiff stain: original magnification × 400). b Segmental sclerosis that is indicated by sharp is observed in the glomerulus (periodic acid-Schiff stain: original magnification × 400). c Infiltrating cells positive for immunostaining of CD38 as a marker of plasma cell are rarely found in the tubulointerstitial region. The findings of interstitial nephritis and tubulitis are not observed. Arrow head indicates the plasma cell (immunostaining of CD 38: original magnification × 400)
Fig. 2.
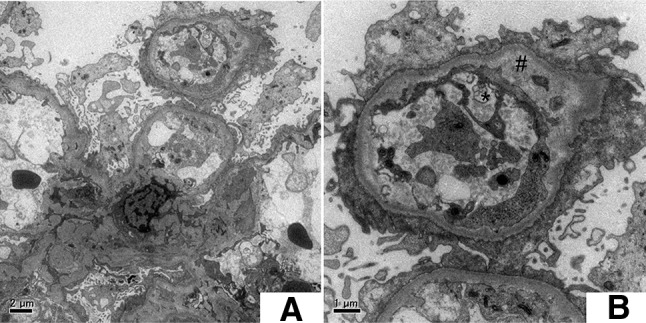
Findings of electron microscopy. a Findings of electron microscopy at low magnification indicate diffuse foot process effacement. Electron-dense deposits are absent in the subendothelial, intramembranous, subepithelial, and mesangial regions. b Subendothelial space widening and endothelial cell swelling are observed in some limited capillaries. Sharp and asterisk indicate subendothelial space with amorphous materials and endothelial cell swelling, respectively
Right axial lymph node biopsy revealed the involvement of plasma cells in the lymph sinus and lymphoid hyperplasia, but no striking interfollicular plasmacytosis and/or trabecular and capsular fibrosis are characteristic findings of Castleman disease (Fig. 3). Therefore, Castleman disease was ruled out.
Fig. 3.
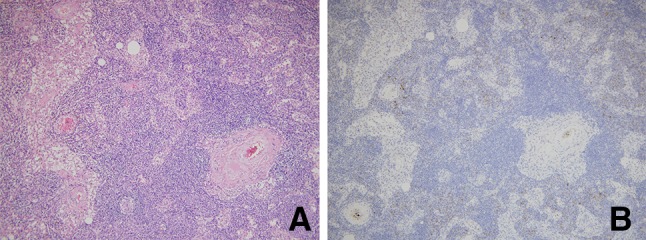
Light microscopic findings of axially lymph node. a Lymphadenitis with sinus histiocytosis without obvious follicular structures is observed. However, no obvious findings are observed, including proliferation of capillary vessels in germinal centers of lymphatic follicles or hyperplasic follicles and an interfollicular region containing prominent increased plasma cell (periodic acid-Schiff stain: original magnification × 400). b Infiltrating cells positive for immunostaining of CD138 as a marker of plasma cell are somewhat increased (immunostaining of CD 138: original magnification × 400)
Treatment with 50 mg/day prednisolone (0.8 mg/kg/day) and 100 mg/day cyclosporine was initiated (Fig. 4). Although the pigmentation persisted, his skin gradually improved. Moreover, complete remission of proteinuria was achieved within 6 months after the treatment.
Fig. 4.

Clinical course after admission. The patient was administered 50 mg/day prednisolone. Subsequently, cyclosporine was combined with prednisolone therapy, and prednisolone was tapered to 40 mg/day at 1 month after the initiation of prednisolone therapy. His proteinuria and serum albumin gradually improved. He could achieve partial or complete remission within 3 or 6 months, respectively. PSL prednisolone, CyA cyclosporine
Discussion
CSP is a rare lymphoproliferative disorder, and mature plasma cells infiltrate into various organ systems, including the skin, lymph nodes, and bone marrow. The elevation of serum IL-6 level produced by the dermal structures is suggested to be the pathogenesis of CSP [3]. Some patients with CSP sometimes have interstitial pneumonia. Only three cases with CSP have been reported to be complicated with nephro-urological disease, one case developed interstitial nephritis, and two cases showed thickening of both ureters [3, 7, 8]. To the best of our knowledge, this is the first case of glomerular disease accompanied by CSP.
It is unknown whether CSP contributes to the development of FSGS. FSGS is a heterogeneous phenotype associated with various conditions, including neoplastic conditions, HIV infections, obesity, and so on [9]. Plasma cell proliferative disorders might also be linked with FSGS. Dingli et al. indicated the epidemiological link between FSGS and plasma cell proliferative disorders, such as myeloma and monoclonal gammopathy of undetermined significance [10]. Although dysproteinemia induced by plasma cell disorders per se might contribute to the development of FSGS, both dysproteinemia and FSGS caused by common immune-dysregulated conditions might be possible, such as plasma cell proliferative disorders.
CSP is thought to be a variant of Castleman disease. IL-6 is one of the common factors for their pathophysiology, and some overlapping cases of both diseases were reported [3, 4, 11, 12]. Castleman disease has sometimes been reported to have renal complications. The analysis of renal histology in 75 cases of Castleman disease revealed that a large number of patients had renal amyloidosis (26 cases, 35%), and a small subset had FSGS (5 cases, 7%) [13]. The causal relationship between FSGS and Castleman disease was also unknown, but IL-6 and VEGF, which may play a pathophysiological role in CSP and/or Castleman disease, might contribute to renal lesions.
Animal models of IL-6 overexpression showed mesangial hyperplasia [14], and human mesangial cells stimulated by IL-6 also showed mesangial proliferation and increase of cell size in vitro study [15]. These data suggest that serum IL-6 may promote mesangial proliferation. In our case, segmental mesangial proliferation without electron-dense deposition was seen.
High permeability of endothelial cells caused by overproduction of VEGF may aggravate endothelial cell damages. In contrast, although the levels of serum VEGF were increased, VEGF expression in the podocyte was reported to be decreased in patients with Castleman disease, wherein endothelial cell damages, such as thrombotic microangiopathy, were found [5]. We could not indicate the downregulation of podocyte VEGF expression in this case. However, the data suggested that downregulation of VEGF expression in podocyte may contribute to endothelial cell damages via endothelial–podocyte interaction [16] and FSGS progression. The subendothelial cell swelling on glomerular capillaries in our case might implicate that the VEGF pathway was modulated to maintain glomerular capillary walls. Other circulating factors, which can cause FSGS, could be possibly produced by polyclonal B cell activation [17].
In summary, we reported the first case with FSGS accompanied by CSP, although the precise mechanisms that cause glomerular disease have not been clearly indicated.
Compliance with ethical standards
Sources of funding
None.
Conflict of interest
All the authors have declared no competing interest.
Human rights
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
Written informed consent was obtained from the patient.
References
- 1.Yashiro A. A kind of plasmacytosis: primary cutaneous plasmacyoma? Jpn J Dermatol. 1987;86:910. [Google Scholar]
- 2.Watanabe S, Ohara K, Kukita A, et al. Systemic plasmacytosis. A syndrome of peculiar multiple skin eruption, generated lymphadenopathy, and polyclonal hypergammaglobulinemia. Arch Dermatol. 1986;122:1314–1320. doi: 10.1001/archderm.1986.01660230106022. [DOI] [PubMed] [Google Scholar]
- 3.Haque M, Hou JS, Hisamichi K, et al. Cutaneous and systemic plasmacytosis vs. cutaneous plasmacytic Castleman disease: review and speculations about pathogenesis. Clin Lymphoma Myeloma Leuk. 2011;11:453–461. doi: 10.1016/j.clml.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Kayasut K, Le Tourneau A, Rio B, et al. Are multicentric Castleman’s disease with cutaneous plasmacytosis and systemic plasmacytosis the same entity? Histopathology. 2006;49:557–558. doi: 10.1111/j.1365-2559.2006.02532.x. [DOI] [PubMed] [Google Scholar]
- 5.El Karoui K, Vuiblet V, Dion D, et al. Renal involvement in Castleman disease. Nephrol Dial Transplant. 2011;26:599–609. doi: 10.1093/ndt/gfq427. [DOI] [PubMed] [Google Scholar]
- 6.Xu D, Lv J, Dong Y, et al. Renal involvement in a large cohort of Chinese patients with Castleman disease. Nephrol Dial Transplant. 2012;27(Supple 3):iii119–iii125. doi: 10.1093/ndt/gfr245. [DOI] [PubMed] [Google Scholar]
- 7.Toda Y, Komine M, Suzuki S, et al. Plasmacytosis: systemic or cutaneous, are they distinct? Acta Dermato Venereol. 2000;80:233–235. doi: 10.1080/000155500750043203. [DOI] [PubMed] [Google Scholar]
- 8.Kodama A, Tani M, Hori K, et al. Systemic and cutaneous plasmacytosis with multiple skin lesion and polyclonal hypergammaglobulinaemia: significant serum interleukin-6 levels. Br J Dermatol. 1992;127:49–53. doi: 10.1111/j.1365-2133.1992.tb14827.x. [DOI] [PubMed] [Google Scholar]
- 9.Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. 2015;11:76–87. doi: 10.1038/nrneph.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dingli D, Larson DR, Plevak MF, et al. Focal and segmental glomerulosclerosis and plasma cell proliferative disorder. Am J Kidney Dis. 2005;46:278–282. doi: 10.1053/j.ajkd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Bower M, Stebbing J. Exploring interleukin 6 in multicentric Castleman’s disease. Lancet Oncol. 2014;15:910–912. doi: 10.1016/S1470-2045(14)70333-X. [DOI] [PubMed] [Google Scholar]
- 12.Higashi Y, Kanekura T, Sakamoto R, et al. Multicentriic Castleman disease with cutaneous manifestations: report of 2 cases and comparison with systemic plasmacytosis. Dermatology. 2007;214:170–173. doi: 10.1159/000098578. [DOI] [PubMed] [Google Scholar]
- 13.Yuan XG, Hu W, Chen FF, et al. Renal complications of Castleman’s disease: report of two cases and analysis of 75 cases. Clin Exp Nephrol. 2011;15:921–926. doi: 10.1007/s10157-011-0499-9. [DOI] [PubMed] [Google Scholar]
- 14.Brandt SJ, Bodine DM, Dunbar CE, et al. Dysregulation interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J Clin Invest. 1990;86:592–599. doi: 10.1172/JCI114749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen WP, Chen A, Lin CY. In vitro effects of interleukins on human mesangial cells: implications for glomerulonephritis. J Pathol. 1995;175:327–337. doi: 10.1002/path.1711750311. [DOI] [PubMed] [Google Scholar]
- 16.Henao DE, Saleem MA, Cadavid AP. Glomerular disturbances in preeclampsia: disruption between glomerular endothelium and podocyte symbiosis. Hypertens Pregnancy. 2010;29:10–20. doi: 10.3109/10641950802631036. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Bu D, Yang Y, et al. Castleman’s tumors and production of autoantibody in paraneoplastic pemphigus. Lancet. 2004;363:525–531. doi: 10.1016/S0140-6736(04)15539-6. [DOI] [PubMed] [Google Scholar]